

Abstract
Structure-based drug discovery (SBDD) is becoming an essential tool in assisting fast and cost-efficient lead discovery and optimization. The application of rational, structure-based drug design is proven to be more efficient than the traditional way of drug discovery since it aims to understand the molecular basis of a disease and utilizes the knowledge of the three-dimensional structure of the biological target in the process. In this review, we focus on the principles and applications of Virtual Screening (VS) within the context of SBDD and examine different procedures ranging from the initial stages of the process that include receptor and library pre-processing, to docking, scoring and post-processing of topscoring hits. Recent improvements in structure-based virtual screening (SBVS) efficiency through ensemble docking, induced fit and consensus docking are also discussed. The review highlights advances in the field within the framework of several success studies that have led to nM inhibition directly from VS and provides recent trends in library design as well as discusses limitations of the method. Applications of SBVS in the design of substrates for engineered proteins that enable the discovery of new metabolic and signal transduction pathways and the design of inhibitors of multifunctional proteins are also reviewed. Finally, we contribute two promising VS protocols recently developed by us that aim to increase inhibitor selectivity. In the first protocol, we describe the discovery of micromolar inhibitors through SBVS designed to inhibit the mutant H1047R PI3Kα kinase. Second, we discuss a strategy for the identification of selective binders for the RXRα nuclear receptor. In this protocol, a set of target structures is constructed for ensemble docking based on binding site shape characterization and clustering, aiming to enhance the hit rate of selective inhibitors for the desired protein target through the SBVS process.
Keywords: Computer-aided drug design, docking and scoring, ensemble docking, lead optimization, library design, PI3Kα, RXRα, virtual screening
1. INTRODUCTION
The identification of lead compounds showing pharmacological activity against a biological target and the progressive optimization of the pharmacological properties and potency of these compounds are the focal points of early-stage drug discovery. To this end, the pharmaceutical industry has adopted the experimental screening of large libraries of chemicals against a therapeutically-relevant target (high-throughput screening or HTS) as a means to identify new lead compounds. Through HTS, active compounds, antibodies or genes, which modulate a particular biomolecular pathway, may be identified; these provide starting points for drug discovery and for understanding the role of a particular biochemical process in biology. Although HTS remains the method of choice for drug discovery in the pharma industry, the various drawbacks of this method, namely the high cost, the time-demanding character of the process as well as the uncertainty of the mechanism of action of the active ingredient have led to the increasing employment of rational, structure-based drug design (SBDD) with the use of computational methods.
SBDD is nowadays central to the efficient development of therapeutic agents and to the understanding of metabolic processes. SBDD is proven to be more efficient than the traditional way of drug discovery since it aims to understand the molecular basis of a disease and utilizes the knowledge of the three-dimensional (3D) structure of the biological target in the process. By using computational methods and the 3D structural information of the protein target, we are now able to investigate the underlying molecular interactions involved in ligand-protein binding and thus interpret experimental results in atomic-level detail. The use of computers in drug discovery bears the additional advantage of delivering new drug candidates more quickly and cost-efficiently.
State of the art structure-based drug design methods include virtual screening (VS) and de novo drug design; these serve as an efficient, alternative approach to HTS. In virtual screening, large libraries of drug-like compounds that are commercially available are computationally screened against targets of known structure, and those that are predicted to bind well are experimentally tested [1, 2]. However, database screening does not provide molecules that are structurally “novel” as these molecules have been previously synthesized by commercial vendors. Existing molecules can only be patented with a “method of use” patent covering their use for a unique application and not their chemical structure. In the de novo drug design approach, the 3D structure of the receptor is used to design structurally novel molecules that have never been synthesized before using ligand-growing programs and the intuition of the medicinal chemist [3].
Computer-aided drug discovery has recently had important successes: new biologically-active compounds have been predicted along with their receptor-bound structures and in several cases the achieved hit rates (ligands discovered per molecules tested) have been significantly greater than with HTS [1, 4-6]. Moreover, while it is rare to deliver lead candidates in the nM regime through VS, several reports in the recent literature describe the identification of nM leads directly from VS; these strategies will be discussed herein [7-9]. Therefore, computational methods play a prominent role in the drug design and discovery process within the context of pharmaceutical research.
In this review, we focus on the principles and applications of VS in the SBDD framework, starting from the initial stages of the process that include receptor and library pre-processing, to docking, scoring, and post-processing of top-scoring hits. We also highlight several successful studies and protocols that led to nM leads, discuss novel applications of Structure-Based VS (SBVS) such as substrate identification for the discovery of novel metabolic pathways, and provide recent trends in library design. Limitations of SBVS are also examined. Finally, we present two developed VS protocols that aim to enhance inhibitor selectivity for the target protein structure.
2. VIRTUAL SCREENING IN STRUCTURE-BASED DRUG DISCOVERY
The general scheme of a SBVS strategy is shown in Fig. (1) [1, 2, 5]. SBVS starts with processing the 3D target structural information of interest. The target structure may be derived from experimental data (X-ray, NMR or neutron scattering spectroscopy), homology modeling, or from Molecular Dynamics (MD) simulations. There are numerous fundamental issues that should be examined when considering a biological target for SBVS; for example, the druggability of the receptor, the choice of binding site, the selection of the most relevant protein structure, incorporating receptor flexibility, suitable assignment of protonation states, and consideration of water molecules in a binding site, to name a few. In fact, the identification of ligand binding sites on biological targets is becoming increasingly important. The need for novel modulators of protein/gene function has recently directed the scientific community to pursue druggable allosteric binding pockets. Another consideration for SBVS includes the careful choice of the compound library to be screened in the VS exercise according to the target in question, and the preprocessing of libraries in order to assign the proper stereochemistry, tautomeric, and protonation states.
Fig. (1).
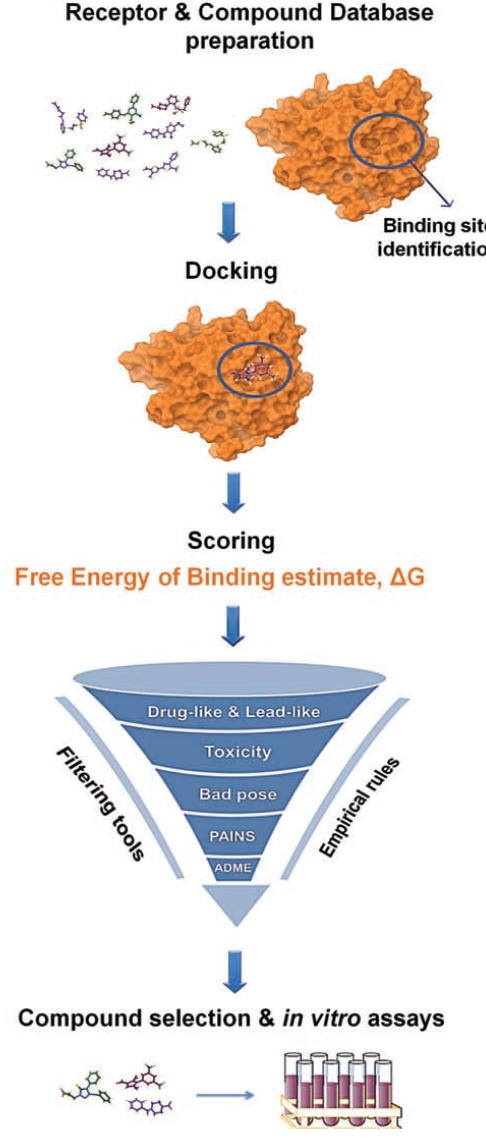
Structure-Based Virtual Screening work-flow.
Following library and receptor preparation, each compound in the library is virtually docked into the target binding site with a docking program. Docking aims to predict the ligand-protein complex structure by exploring the conformational space of the ligands within the binding site of the protein. A scoring function is then utilized to approximate the free energy of binding between the protein and the ligand in each docking pose. Docking and scoring produce ranked compounds, which are then post-processed by examining calculated binding scores, validity of generated pose, undesirable chemical moieties, metabolic liabilities, desired physicochemical properties, lead-likeness, and chemical diversity. Post-processing results in a small number of selected compounds, which proceed to experimental assaying [1, 2, 5, 10].
2.1. Protein Preparation Schemes for SBVS
The success of a SBVS campaign largely depends on reasonable starting structures for both the protein and the ligand. A typical PDB structure file consists only of heavy atoms (if the input is an X-ray structure) and may contain water molecules, cofactors, activators, ligands, and metal ions as well as several protein subunits. Moreover, the structure has generally no information on bond orders, topologies, or formal atomic charges. Terminal amide groups and asparagine residues may be misarranged as X-ray structures cannot unambiguously distinguish between O and NH2 groups. Ionization and tautomeric states are also unassigned in most cases, residue side chains or larger loops may be missing because of low resolution of a particular protein area, and steric clashes may exist.
To efficiently address the above-mentioned structural issues, several protein preparation schemes have been proposed [5, 11, 12]. The general proposed strategy is to first determine the protonation states of the aminoacids in the protein using available software. Popular freely available software include PROPKA [13], H++ [14], SPORES [15]. The next step is to assign hydrogen atoms and optimize protein hydrogen bonds according to an optimal hydrogen bond network. A widely-used software for these tasks is the PDB2PQR software [16]. The next steps are assignment of partial charges, capping of residues, treating metals, filling in missing loops and missing side chains, and minimizing the protein structure to relieve steric clashes. In addition, a decision needs to be made regarding whether water molecules will be left in or removed from the binding site. Several methods have been developed to tackle this challenging problem, such as 3D RISM [17-19], SZMAP [20], JAWS [21], WaterMap [18, 22], which are utilized in commercial software [23-25]. In case the protein is co-crystallized with substrates, cofactors, etc., these ligands must be prepared to create 3D geometries, assign proper bond orders, and generate accessible tautomer and ionization states prior to VS. These tasks may be performed within available free software packages such as the “Protein Preparation Wizard” of Maestro [26] or scripts that use a variety of different programs to prepare a protein structure for SBVS, e.g. WebPDB (see Table 1 for a full list of freely-available programs).
Table 1.
Free available software and tools for performing a SBVS workflow.
| Software | Free Academic License | Website |
|---|---|---|
| GLARE [130] | Yes | http://glare.sourceforge.net/ |
| CDK-Taverna [131] | Yes | http://cdktaverna.wordpress.com/ |
| CLEVER [71] | Yes | http://datam.i2r.a-star.edu.sg/clever/ |
| e-LEA3D [72] | Yes | http://chemoinfo.ipmc.cnrs.fr/ |
| SmiLib v2.0 [132] | Yes | http://gecco.org.chemie.uni-frankfurt.de/smilib/ |
| Library synthesizer (Tripod) [133] | Yes | http://tripod.nih.gov/?p=370 |
| Swissbioisostere [75] | Yes | http://www.swissbioisostere.ch/ |
| VAMMPIRE [76] | Yes | http://vammpire.pharmchem.uni-frankfurt.de/#!home |
| Virtual Library [77] | Yes | Contact authors |
| Pipeline pilot (Accelrys) [68] | No | http://www.accelrys.com |
| Reactor [134](ChemAxon) | No | http://www.chemaxon.com |
| OELib library enumeration [135] | No | http://www.eyesopen.com |
| CombiLibMaker and Legion (Tripos) [136] | No | http://www.tripos.com |
| QuaSAR-CombiGen [137] | No | http://www.chemcomp.com |
| ChemOffice CombiChem [138] | No | http://www.cambridgesoft.com/ |
| ICM-Chemist [139] | No | http://www.molsoft.com |
| LUCIA [140] | No | http://www.eidogen-sertanty.com/ |
The importance of protein preparation in docking performance has been recently reported [11]. Docking performance is significantly improved when employing a best-practice preparation scheme over using minimally-prepared structures from the PDB structure. In the only systematic study available in the literature regarding the influence of the protein preparation procedure in the success of a SBVS workflow, Sastry et al. have explored each of the steps involved in preparing a system for VS [11]. In their study, a large number of parameters in protein preparation is examined using the Glide validation set of 36 crystal structures and 1,000 decoys. Several protocols are considered and applied on the Directory of Useful Decoys (DUD) database [27, 28], showing that database enrichment is improved after proper receptor preparation and that neglecting certain steps of the preparation process produces a systematic degradation in enrichments, which may be large for some targets.
2.2. Binding Site Identification
Binding site identification is often an additional prerequisite for performing SBVS, when the binding site is not known or when new, allosteric modulators of protein function are sought. Ideally, the target binding site is a pocket, typically a concave, having a variety of probable hydrogen bond donors and acceptors and hydrophobic characteristics. Currently four approaches in the identification of putative binding sites exist in the literature: 1) Static approaches, where computational solvent mapping with chemical probes (small organic molecules) is utilized to identify binding hot spots on a 3D structure (from Xray, MD, etc). These approaches determine the hot spot druggability and provide information for drug design. Examples of such an approach include SiteMap [29], FTMap [30], Fpocket [31], MDpocket [32], QsiteFinder [33], MED-SUMO [34] and SiteHound-web [35]. Selecting a different chemical probe each time may result in the identification of a different type of binding site. 2) Dynamic approaches, where the probes and the protein evolve dynamically in time and more than one probe may be used in one simulation. Analysis of these simulations provides direct access to interaction free energies between the protein and small organic molecules, which can then be used to detect binding sites and predict the maximal affinity that a drug-like fragment could attain. Programs following this approach are MDMix [36], SILCS [37], MixMD [38]. In this context, studies that aim to understand ligand binding mechanisms using microsecond MD simulations or enhanced sampling techniques [39-41] may also be used for revealing new binding pockets using small organic molecules as probes, although this approach is not computationally-efficient. 3) In the mixed approach, the detection of ligand binding sites is performed with a chemical probe as in the static approach and in addition, the putative binding site is evaluated in terms of flexibility. Flexibility can be determined by performing normal mode analysis or by identifying flexible residues within the binding site and probing residue alternative conformations using a rotamer library. FTFlex, which is an extension of the FTMap server, uses the latter approach [42]. 4) The last approach uses water as a probe to identify putative binding sites on proteins. Approaches following the inhomogeneous solvation theory [43] include JAWS [21], WaterMap [22], and WATMD (Novartis), but also other methods have been developed such as 3D-RISM [17] and SZMAP [20].
2.3. Compound Database Preparation
The construction of compound databases is the next important step in the SBVS process. Databases for SBVS contain drug-like small molecules, often freely available or available via purchase or synthesis, which possess desirable characteristics such as stability and solubility in aqueous media, existence of appropriate functional groups to interact with biological targets and absence of toxic and undesirable moieties. Several rules have been applied to ensure ‘drug-likeness’, with the most popular being the “Lipinski Rule of Five" [44], which states that drug-like compounds should have molecular weight lower than 500, lipophilicity (logP) lower than 5, less than five hydrogen bond donors, and less than 10 hydrogen bond acceptors. As increasingly more compounds break some of these rules and enter the market (e.g. natural product drugs as well as 50% of marketed drugs do not comply with the “Rule of Five”) [45], attempts to improve the predictions of druglikeness have spawned many extensions to the Lipinski Rule of Five. Molecules may be
considered as drug-like within a range of log P in −0.4 to +5.6, molar refractivity from 40 to 130, molecular weight from 180 to 500, number of atoms from 20 to 70 (includes H-bond donors (e.g.; OH's and NH's) and H-bond acceptors (e.g.; N's and O's), polar surface area no greater than 140 Å2, and/or fewer than ten rotatable bonds [46]. Moreover, the “Rule of Three” is an extension to the Rule of Five specifically for fragments (molecular weight < 300, logP < 3, number of hydrogen bond donors and acceptors < 3, number of rotatable bonds < 3) [47]. However, in a recent study by Klebe and coworkers, it was shown that only four of their 11 discovered fragments were compliant with the Rule of Three [48]. Strict compliance to this rule would have strongly limited the variety of chemotypes among the fragment hits, indicating that it is advisable to follow these rules with a certain degree of flexibility, i.e. allow one violation to the rule each time [49]. The overly rigid application of strict cut-off points may introduce artificial distinctions between similar compounds and runs the risk of missing valuable opportunities [50]. A more recent rule accounting for the physico-chemical properties relating to preclinical toxicology outcomes is the “Pfizer's Rule of 3/75", which is based on the values of calculated partition coefficient (ClogP) and topological polar surface area (TPSA). According to this rule, compounds with ClogP lower than 3 and TPSA higher than 75 are approximately 2.5 times more likely to be safe in in vivo assays [51]. The “Jorgensen Rule-of-Three” is another widely-followed rule for lead like properties and states that the aqueous solubility measured as logS should be greater than -5.7, the apparent Caco-2 cell permeability should be faster than 22 nm/s, and the number of primary metabolites should be less than 7; these limits are based on the properties of 90% of 1700 oral drugs [52]. Bickerton et al. have proposed a measure of drug-likeness based on the concept of desirability called “the quantitative estimate of drug-likeness” (QED) [53]. The QED functions rank chemical structures by their merit relative to the properties of oral drugs. Moreover, QED functions have the additional advantage that they are based on the underlying distribution data of drug properties and may identify cases in which a generally unfavorable property may be tolerated when the other parameters are close to ideal. Other metrics of promise include ligand efficiency (LE = potency/heavy atom count) and lipophilic LE (LLE = potency - cLogP), which is a measure of lipophilicity that leads to the observed potency. These metrics show how well a molecule engages in receptor interactions; taking these rules into account provides excellent insights for lead optimization [49].
It should be noted that the majority of commercial compounds found in chemical libraries have a larger molecular weight and higher hydrophobicity compared to orally available drugs. This shift to higher hydrophobicity and lower solubility is also reflected in compounds with significant biological activity found in the ChEMBL database, indicating a trend towards larger, more hydrophobic compounds. In particular, drugs that have been recently approved were found to possess much lower median logP values than those previously suggested as optimal [54].
It has been demonstrated that docking huge compound collections such as PubChem (30M compounds) and ChemSpider (26M compounds), in other words ‘blind docking’, not only is time-consuming and computationally demanding, but also results in redundant possibilities and poses a great burden for compound selection. Apart from filtering for lead-like properties, one should exclude known toxicophores or metabolically liable moieties [55]. Moreover, several novel filters have been recently developed, aiming at the quality enhancement of database content. The Pan Assay Interference Compounds (PAINS) [56] and the ALARM-NMR [57] filters, contain compounds found to be chemically reactive and assay-interfering, appear as frequent hitters, and are not identified by toxicophoric filters. Therefore, a combination of filtering for desired pharmacological and adsorption, distribution, metabolism, excretion, and toxicological (ADMET) properties is advisable early in the drug design process. To efficiently filter a library of compounds against such criteria, several online tools have been developed.
Chembioserver is a publicly available online application specializing in filtering and selection of small molecules [58]. The objective of this application is to facilitate compound preparation prior to (or after) VS computations by utilizing its many sections, such as (i) basic search, (ii) filtering (steric clashes and toxicity), (iii) advanced filtering based on custom chosen physicochemical properties, (iv) clustering (according to structure and compound physicochemical properties providing representative compounds for each cluster), (v) customized pipeline and (vi) visualization of compound' properties through property graphs and thus, increase the efficiency and the quality of compounds that proceed to in vitro assaying. The FAF-Drugs2 server is a public tool for computationally filtering compounds by considering ADMET and physicochemical properties, and identifying key functional or undesirable moieties [59]. Moreover, users have the opportunity to select filtering thresholds and rules among 23 physicochemical and 204 substructure searching rules. Last but not least, this tool provides numerous distribution diagrams of the properties of the filtered compounds in a web-server version [60].
Finally, compound datasets should be preprocessed in realistic 3D representations. Therefore, the compound set to be used for SBVS should have realistic bond lengths and angles as these may not change during docking. Also, it must be devoid of accompanying fragments such as counter-ions, metals, and solvent molecules, all compounds need to have assigned bond order and filled valences, partial charges, an appropriate protonation state at physiological pH or at the pH of the interest and proper tautomeric states [61, 62]. However, it was shown that consideration of all possible tautomers (stable and unstable) yields slightly poorer results than including only the most stable form in water [63]. Docking programs usually include ligand preparation software such as Autodock Tools [64], LigPrep within the Schrödinger suite [65], MOE [23] and the MAPS platform [66]. Moreover, stand-alone programs exist for ligand preparation such as DISI [67], Pipeline pilot [68], or Hyperchem [69]. Grinter et al. have created a workflow through custom-made script files for preparing large libraries for docking, including scripts for ligand preparation, which are offered freely to the academic community [70].
2.4. Library Design
Although numerous drug-like compound libraries are freely available online [1, 5], users may often need to create a custom-made library. Libraries may be divided in a) generic virtual high throughput (vHTS) libraries, which contain large sets of compounds, b) diversity-oriented, containing highly chemically diverse compounds, c) target-oriented, which are designed with a specific target in mind, d) molecular property diversity libraries, which are designed with specific molecular property profiles (i.e. solubility, lipophilicity etc.), e) natural product libraries.
Online tools for library design include CLEVER [71] that supports chemical library manipulation, combinatorial chemical library enumeration using user-specified chemical components, chemical format conversion, as well as chemical compound analysis and filtration with respect to drug-likeness, lead-likeness, and fragment-likeness based on the physicochemical properties computed from the derived molecules. Also provided is an integrated property-based graphing component that visually depicts the diversity, coverage and distribution of selected compound collections. Another tool for library design is e-LEA3D, which performs combinatorial library design that is based on a user-drawn scaffold and reactants coming, for example, from a chemical supplier [72]. More freely-available as well as commercial software packages are listed in Table 2. A review on how to design target-focused compound libraries, e.g. for kinases, ion channels, GPCRs, protein-protein interfaces can be found in Ref. [73]. In another approach, by using a genetic algorithm, Lisurek et al. have identified substructures typically occurring in bioactive compounds using the World Drug Index; subsequently available compounds containing the selected substructures were collected from vendor libraries [74].
Table 2.
Free and commercial software packages for library design.
| Software | Free Academic License | Website |
|---|---|---|
| GLARE [130] | Yes | http://glare.sourceforge.net/ |
| CDK-Taverna [131] | Yes | http://cdktaverna.wordpress.com/ |
| CLEVER [71] | Yes | http://datam.i2r.a-star.edu.sg/clever/ |
| e-LEA3D [72] | Yes | http://chemoinfo.ipmc.cnrs.fr/ |
| SmiLib v2.0 [132] | Yes | http://gecco.org.chemie.uni-frankfurt.de/smilib/ |
| Library synthesizer (Tripod) [133] | Yes | http://tripod.nih.gov/?p=370 |
| Swissbioisostere [75] | Yes | http://www.swissbioisostere.ch/ |
| VAMMPIRE [76] | Yes | http://vammpire.pharmchem.uni-frankfurt.de/#!home |
| Virtual Library [77] | Yes | Contact authors |
| Pipeline pilot (Accelrys) [68] | No | http://www.accelrys.com |
| Reactor [134](ChemAxon) | No | www.chemaxon.com |
| OELib library enumeration [135] | No | http://www.eyesopen.com |
| CombiLibMaker and Legion (Tripos) [136] | No | http://www.tripos.com |
| QuaSAR-CombiGen [137] | No | http://www.chemcomp.com |
| ChemOffice CombiChem [138] | No | http://www.cambridgesoft.com/ |
| ICM-Chemist [139] | No | http://www.molsoft.com |
| LUCIA [140] | No | http://www.eidogen-sertanty.com/ |
In order to build a compound library with a range of possible replacements for a single substructure, Wirth et al. have developed a useful platform for the applicability of Matched Molecular Pairs (MMPs) to optimize the binding affinity of a small-molecule ligand to a target protein, namely the SwissBioisostere database [75]. In this scheme, MMPs are defined as two molecules, which differ in one particular substituent and exhibit different properties. The underlying assumption of MMPs is that the difference in properties can be extrapolated to another pair of molecules exhibiting the same substitution pattern. Weber et al. have improved the bridging between MMPs and structural data by developing the Virtually Aligned Matched Molecular Pairs Including Receptor Environment (VAMMPIRE) database [76]. In their work, they have proposed that the involved atoms in the ligand-protein complexes play a critical role in the exchange of the substituent and therefore in the binding affinity change, providing the possibility to extrapolate activities from one biological system to another. In seek of designing diverse libraries to enrich the screening collection, Vainio et al., have constructed “Virtual Library”, a virtual automatic system that uses validated synthetic protocols and available starting materials to generates a large number of virtual compound libraries [77].
An informative analysis and novel insights on preparing a high-quality database of lead-like compounds for SBVS are presented in Ref. [78]. The application of functional group, lead-like, PAINS, and Tanimoto similarity exclusion filters is discussed resulting in a library of ~350,000 compounds that represent 80% of available leadlike space based on vendor information in what is proposed to be the globe’s highest quality collection of available screening compounds. The article is a nice review of filtering process steps also providing detailed information on the filters and pointing out several issues as resupply, minimum concentration to be tested, etc., which is of practical use to medicinal chemists.
Constructing libraries with inherent chemical diversity such as in Ref. [78] may be desired when investigating underexplored targets with few known ligands. However, the construction of targeted libraries based on properties of known ligands may also be required in order to identify hits with improved potency. Xing et al., designed combinatorial libraries to search for novel soluble epoxide hydrolase (sEH) inhibitors based on a benzoxazole template forming conserved hydrogen bonds with the catalytic machinery of sEH [79]. Consequently, screening of these libraries resulted to 90% hit rate and more than 300 submicromolar sEH inhibitors were finally discovered.
2.5. Docking & Scoring
A large number of docking programs have been developed recently, including AutoDock [64], Dock [80], FlexX [81], Glide [82], Gold [83], Surflex [84], ICM [85], LigandFit [86], Drugster [87], and eHiTS [88] (also reviewed in [5]). Docking entails predicting the protein-ligand complex structure and is followed by scoring in SBVS in order to rank the compounds. Docking programs utilize various methods of conformational search in order to explore the ligand conformational space; these are categorized as following: a) Systematic methods, which place ligands in the predicted binding site after considering all degrees of freedom, b) Random or stochastic torsional searches about rotatable bonds, such as Monte Carlo and genetic algorithms to “evolve” new low energy conformers, (c) Molecular Dynamics simulation methods and energy minimization for exploring the energy landscape of a molecule [reviewed in Ref. 89].
In order to rank compounds, docking programs utilize scoring functions that aim to estimate the free energy of binding of a ligand to a specific target based on a generated docked pose after docking different ligands of a database. A variety of scoring functions have hitherto been developed (reviewed in [1, 5]). Commonly-used scoring functions can be categorized as follows: (a) Force field-based functions that estimate the binding free energy by summing the strength of intermolecular van der Waals, electrostatic interactions and hydrogen bonding between all atoms of the two binding partners in the complex. Solvation and entropy contributions are also taken into account. (b) Empirical scoring functions that are based on counting the number of various types of interactions between the two binding partners, i.e. hydrophobic contacts, number of hydrogen bonds and number of rotatable bonds immobilized in complex formation. These functions have proven to be successful for many protein-ligand complexes. LUDI, FlexX, F-Score, ChemScore and Fresno are a few examples of functions that are contained in this group. (c) Knowledge-based functions that use statistical observations of intermolecular contacts in receptor-ligand complexes with known structural conformations. For instance, some functions of this category are Potential of Mean Force (PMF), DrugScore and SMoG (Small Molecule Growth). Furthermore, there exist scoring functions that combine two or more of the above mentioned scoring function categories.
It is commonly accepted that while docking results to successful binding pose prediction, scoring usually fails to correctly rank different compounds with the difficulty increasing in congeneric series [95, 96]. Hence, identifying correctly the right binding pose as the top-ranked one still remains a challenge [1]. Addressing the correspondence between predicted and experimental binding affinities in empirical scoring functions still remains unresolved. Based on that critical issue, Zilian et al. have developed a new scoring function, SFCscoreRF, with significantly improved performance in comparison to previously developed SFCscore functions, by using SFCscore descriptors and a PDBbind set of 1005 complexes as a training set in combination with random forest for regression [97]. HYDE is another novel scoring function for protein-ligand complexes which is based on HYdration and DEsolvation terms. It consistently describes hydrogen bonds, the hydrophobic effect and desolvation. Recently, Schneider et al. have validated the HYDE scoring function through large-scale docking experiments, which resulted to successful prediction of the correct binding mode in 93% of complexes when checked with the Astex diverse set [98]. Alternatively, Ravindranathan et al. have defined a physics based scoring function and more precisely, a variable dielectric model based on residue types for better description of protein–ligand electrostatics in MM-GBSA scoring, which results in a higher correlation with affinity data [99]. Certainly, while much progress has been made in delivering more accurate scoring functions, further improvements in this direction are desirable.
The assessment of the efficiency and accuracy of scoring functions has been addressed in a number of studies. Cross et al., have assessed six molecular docking programs (DOCK, FlexX, GLIDE, ICM, PhDOCK, and Surflex) in order to evaluate docking pose, scoring and thus VS accuracy [90]. The results revealed that ICM, GLIDE, and Surflex reproduced accurately the X-ray poses, while GLIDE and Surflex showed superior receiver operating characteristic (ROC) areas under the curve (AUC) and ROC enrichments after performing a VS exercise on the 40 protein targets in the Directory of Useful Decoys (DUD). McGann et al. have compared the performance of FRED and HYBRID docking programs on known datasets, where the second utilizes a modified version of FRED that uses both ligand- and structure-based information for docking [91]. The results have shown that HYBRID scoring function increases identified actives. Especially, when using multiple crystal structures considering protein flexibility, HYBRID efficiency increases even more without increasing significantly the docking time (~ 15 %). Liebeschuetz et al. have tested the four GOLD scoring functions in docking and scoring computations and demonstrated that the ChemPLP scoring function comes first for both pose and affinity prediction [92].
In an attempt to improve docking and scoring performance, Spitzer et al. used Surflex-Dock to incorporate protein flexibility in the docking process. Ligand-protein optimization before VS, resulted in large differences in the VS performance, indicating that ligand re-docking for pose prediction assessment may have limitations [93]. Docking protocols, which incorporate protein flexibility, performed better in terms of pose prediction. Applying various VS protocols (docking, 2D molecular similarity, and 3D molecular similarity) resulted in achieving better hit rates. Neves et al. have investigated the ICM flexible docking and scoring benchmarks for 85 co-crystal structures of the modified Astex data set and derived significant improvements, compared to single rigid docking [94]. Reparsky et al. also have obtained enrichment results for WScore, a new scoring function and sampling methodology incorporating WaterMap results into Glide screening [27]. In this study, WScore results show early enrichment and outerperform the DUD best-practices Glide SP results.
2.6. Improving Pose/Compound Selection After Docking (Post-processing)
A rate-limiting step in SBVS is often the need for a computational chemist expert to post-process compounds that result from a VS/docking exercise before selecting the ones that will pass to the experimental test phase. The implementation of the simplified scoring functions and sometimes the inadequate sampling of the conformational space for the ligand may lead to unrealistic poses, intra-ligand steric clashes, twisted amides, E/Z esters, imperfect hydrogen-bonding network, and poses based on shape complementarity. These poses may result to an unreasonably high score and need to be discarded. Therefore, visual inspection of thousands of docking poses is normally needed by the medicinal chemist in order to select the appropriate compound set for assaying. To this end, significant efforts have been dedicated to increase the efficiency and the quality of compound selection [58, 106].
In the process of post-processing the results, one may resort to rescoring. To examine the reliability of VS results without post-processing, Malmstrom et al. have investigated a set of known binders and non-binders of L99A T4 lysozyme using both free energy of binding (FEB) rescoring and other empirical docking scores and concluded that FEB rescoring may assist lead identification resulting from a SBVS exercise [107]. Ding et al. have introduced a directional approach, MIECs (molecular interaction energy components), which successfully predicted the binding affinities of protein-ligand complexes or distinguished binding from non-binding ligands [108]. This methodology was extended to SBVS. HIV-1 protease was used as a model system to assess the performance of the method on ranking docking poses and docked ligands and a support vector machine (SVM) was trained in evaluating the energetic and geometric characteristics that discriminate binding ligands from the non-binding ones. As previously-mentioned, several tools exist for the pre-processing of ligand libraries prior to the VS procedure. These tools may be also used for post-processing of SBVS results are readily available. The ChemBioServer for example, uses vdW filtering to remove compounds with steric clashes. Poses that are far from the energy minimum are unlikely to be adopted in nature and hence, should be discarded. Compounds that pass vdW filtering may be then subjected to more stringent physicochemical property filtering compared to the initial compound selection for SBVS. Subsequently, hierarchical clustering may be performedin order to group compounds with similar structures/physicochemical properties and derive subsets with maximal chemical diversity [58].
2.7. Improvements in SBVS Efficiency
2.7.1. Ensemble Docking (ED)
Selecting the ideal crystal structure of a receptor target is the first step in the SBVS process. Unfortunately, crystal structures provide a single conformation of the protein, which is influenced by the crystallization conditions, and do not include any information on protein dynamics. Furthermore, crystal structures are highly affected by ligand binding, which leads to conformational changes in both the protein and the ligand. Proteins can exhibit induced fit effects upon binding of a ligand, in which the protein conformation changes significantly. Hence, although crystal structures are a preferable starting point for SBDD, in some cases they provide misleading information. Therefore, many efforts have been made to include receptor flexibility in docking programs, as it confers a more realistic depiction of the modeled system [100]. In order to avoid biasing towards one protein conformation and implicitly including protein flexibility, one may use an ensemble of representative structures to dock candidate ligands. ED comprises docking a single ligand library against multiple rigid receptor conformations, contrary to the standard single rigid receptor docking methods. In this way, ED mimics the realistic structural variation of the protein-target [101].
Currently, a huge amount of protein structures that can be used as biological targets for specific diseases is available. Therefore, the challenge in SBVS has changed from “Is there a promising structure available?” to “How do I appropriately select a single or multiple protein structures for my screening exercise?”. Consequently, standardized protocols that specialize in the selection of suitable conformations for ensemble-based virtual screening and finally result to potent leads of the target are needed. For instance, Rueda et al. conclude that the largest binding sites in X-ray structures lead to the highest enrichments [102]. Osguthorpe et al. apply a shape-based characterization of the binding site using molecular dynamics (MD) to construct ensembles with structural diversity [103]. The ensembles for the performance of ED may be selected through X-ray crystallography, NMR spectroscopy or a combination of both and through computational techniques (molecular dynamics simulations, homology modeling) [100].
There are certain advantages that distinguish ED among other flexible-receptor docking methods, i.e. the capability of accounting for any type of protein motion. The major disadvantage of ED is the computational effort, which scales linearly with the number of receptor conformations in the ensemble. As a result, the ED calculations have a significantly longer duration comparing to standard docking protocols. Moreover, possible inaccurate results being output from ED computations may be due to the possible imperfections in scoring functions, which may lead to inaccurate predictions of certain protein-ligand interactions [101, 103].
2.7.2. Consensus Induced-Fit Docking
Consensus Induced Fit Docking (cIFD) is an alternative docking approach, which allows the protein binding site to adapt to multiple ligands during SBVS. This methodology has been practiced by Kalid et al., firstly by validating the cIFD protocol on COX-2, the estrogen receptor, and HIV reverse transcriptase, which were previously shown to be challenging for docking programs and then by proving the utility of cIFD in discovering novel irreversible Crm1 inhibitors [104]. Houston et al. have introduced Consensus Docking for improving the probability of identifying accurately docked poses [105]. In cIFD, a similar approach to consensus scoring schemes is applied; this methodology combines information about predicted binding modes rather than predicted binding affinities. It results in 82% success rate of pose prediction by using more than one docking program, while the success rate was found to be lower for each docking program alone, e.g. 55% for Autodock, 58% for DOCK, and 64% for Vina.
3. PROPOSED WORKFLOWS-PROTOCOLS
Nasr et al. have introduced a set of “binding site properties-based” guidelines that aim to optimize future prospective drug discovery protocols in presence of several available receptor structures [109]. Several trends in the properties of binding sites, including hydrophobicity, volume, and opening, were identified and tested in SBVS with several programs (Surflex-dock or ICM). Planesas et al. have proposed a three-step VS protocol to improve actives from decoys selection using VEGFR-2 (Kdr-kinase) inhibitors. This protocol includes the conventional docking step, a pharmacophore post-filter step, and a similarity search post-process [110]. When this protocol is retrospectively applied to VEGFR2 inhibitors, both the overall and early VS performance are improved. Osguthorpe et al. have generated a SBVS protocol through which a set of target structures is constructed for ED based on binding site shape characterization and clustering, in order to account for protein flexibility into the VS process [103]. The protocol’s steps are: a) receptor structures selection from X-ray crystallography and MD, b) binding site shape characterization, c) pairwise volume overlap computation, d) hierarchical clustering, e) docking calculations of known actives and decoys to the shape-diverse binding sites, and f) comparison of ED results with the performance of a single rigid receptor.
One approach to achieve nM hits directly from VS is to bias the compound selection toward known chemotypes of high potency. This high potency may be achieved through the design of target-focused libraries for virtual screening. Schröder et al. discovered covalent reversible inhibitors of cathepsin K through virtual screening of a focused library built for this purpose, which included compounds bearing electrophilic groups that act as warheads in mediating covalent reversible inhibition [8]. Substantial effort was invested in developing appropriate electrophilic warheads in their corresponding transition state. Covalent docking of this focused library resulted in identifying 21 inhibitors of cathepsin K, including 3 nanomolar leads, out of the 44 initially-selected putative binders.
In another SBVS for β2-adrenergic receptor ligands, virtual screening was performed on the lead-like subset of the ZINC database using the available crystal structure [9]. 0.05% of the top-ranked compounds (500 molecules) were visualized, and 25 molecules were finally selected based on their chemical diversity, commercial availability, and overall balance between polar and non-polar complementarity to the binding site. These efforts resulted in the identification of a9 nM inverse agonist of the β2-adrenergic receptor. Although the virtual screening procedure was unbiased, it was later found that the lead-like subset of the ZINC database is biased towards GPCR ligands.
A novel approach targeting GPCR inhibition, has unraveled nanomolar leads for the melanin-concentrating hormone-1 receptor (MCH-1R) [7]. This approach integrates GPCR modeling, prediction of antagonist binding site, design, synthesis, in combination with utilizing a focused library for screening. A primary hit compound from a pyranose-based VAST library (pyranose scaffolds mimic active GPCR peptides) was initially utilized to construct a high quality MCH-1R model. Subsequently, the model was validated by a virtual enrichment experiment and the model-driven structure-based expansion of the original hit was used to identify key interactions in the binding site of the protein. Following the model validation, a structure-based virtual screen of a library with ≤ 0.7 Tanimoto similarity to known MCH-1R ligands, provided a 14% hit rate and 10 novel chemotypes of potent MCH-1R antagonists, including two nanomolar leads. Ligand selection also took into consideration AMBER interaction energies in the range of the top 10% of known MCH-1R antagonists.
In all the above-mentioned approaches, a high hit rate and high affinity binders were achieved by using a focused library to identify lead candidates.
Apart from identifying novel lead candidates, other applications of SBVS may be envisaged such as the screening for novel substrates for genetically-engineered proteins. Recently, it has been shown that new specific substrates of genetically-engineered proteins can be used for identifying the direct protein targets of enzymes [111-113]. In this context, SBVS could be applied for the identification of substrates specifically-designed to target the mutated enzymes that are then used to elucidate signal transduction pathways. In this direction, the group of Matt Jacobson has recently unveiled a strategy for assigning valid biological functions to proteins identified in genome projects using SBVS [114]. Computer-aided strategies for functional discovery with ‘metabolite docking’ were used to predict substrate specificities of several enzymes encoded by a bacterial gene cluster. This work led to the correct prediction of the in vitro activity of a structurally characterized enzyme of unknown function (PDB 2PMQ), and also to the correct identification of the catabolic pathway in which Hyp-B 2-epimerase participates. These studies establish the utility of structure-guided functional predictions to enable the discovery of new metabolic and signal transduction pathways.
Another emerging application of SBVS is the search for inhibitors of multifunctional proteins. Stumpfe et al. [115] report a virtual screening approach for finding inhibitors of the cytohesins, a family of cytoplasmic proteins with multiple known functions. Two virtual screening approaches, fingerprint similarity searching, and support vector machine modeling were combined to identify structurally diverse compounds capable of inhibiting the different functions of the cytohesins. Specific inhibitors of guanine nucleotide exchange, insulin signaling, and leukocyte adhesion were identified from the screen.
3.1. Protocol for the Discovery of Mutant-Specific PI3Kα Inhibitors
The PIK3CA gene is one of the most frequently mutated oncogenes in human cancers. It encodes p110α, the catalytic subunit of phosphatidylinositol 3-kinase, α (PI3Kα) isoform. The PI3Kα protein is implicated in signaling cascades, which lead to cell proliferation, survival, and cell growth. One of the most frequent mutations in PI3Kα is a histidine changed to arginine in exon 20 (H1047R) [116]. Here, we describe a VS workflow to identify novel inhibitors of the H1047R mutant form of PI3Kα [95]. The workflow incorporates the use of ChemBioServer [58], a free web-based application, which enables the efficient final selection of compounds to be tested experimentally.
Initially, the crystal structure of the mutant H1047R PI3Kα (PDB ID: 3HIZ) was constructed using a combination of homology and loop modeling and the atomistic model of the full-length H1047R mutant was created using Modeller 9v8 [117]. The resulting model was solvated in water and employed in Molecular Dynamics (MD) simulations using the NAMD package [118] for ~70ns. The final snapshot of the simulation was extracted and used for binding site identification calculations through SiteMap module of Schrödinger v2.4. A non ATP binding site close to the H1047R mutation was identified to be among the top-ranked and was used in the present study. After binding site identification, we performed VS using the docking program Glide 5.7 [82]. The drug-like subset of the HitFinder collection from the Maybridge database (www.maybridge.com) was used for the VS exercise [119]. All structures were docked and scored using the Glide standard precision (SP) mode. The 10,000 top-ranked structures from the SP filter were redocked and rescored using the Glide extra precision (XP) mode. The complexes for the top-ranked 1,000 compounds resulting from the XP processing were submitted to further post-processing with the ChemBioServer. In order to filter out poses far from the energy minimum, we used the van der Waals filter of the server to remove compounds with steric clashes. The remaining compounds were then subjected to physicochemical property filtering based on the “Jorgensen rule of 3” [52] as well as toxicity filtering based on a database available in ChemBioServer, which contains known toxic moieties. Subsequently, a hierarchical clustering was performed for the remaining compounds using the Tanimoto coefficient and the Ward Clustering Linkage in order to maximize chemical diversity but at the same time minimize visual inspection efforts. Finally, the resulting cluster representatives were visually inspected and the ten most promising compounds were purchased and submitted to in vitro assay testing. Four out of the ten purchased molecules inhibited the PI3Kα protein activity in vitro in μm concentrations, indicating that the workflow described herein can be successfully applied in drug discovery [120]. The process is described in Fig. (2).
Fig. (2).
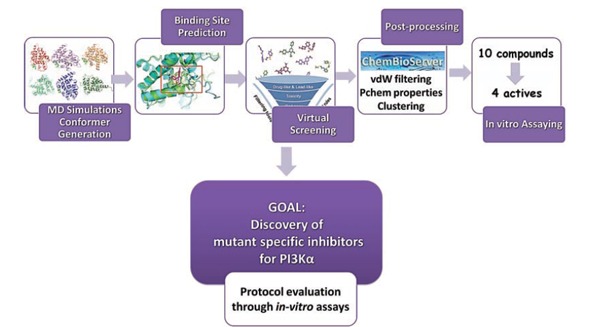
Workflow for the discovery of mutant-specific PI3Kα inhibitors based on a SBVS protocol involving conformer generation, binding site prediction, and compound post-processing.
3.2. A Proposed Method to Increase the Probability of Virtual Screening Hits: Application on the RXRα Nuclear Receptor
The availability of multiple solved protein structures of the same protein may offer the opportunity to increase the hit rate in biological assays of compounds selected in silico. The case of receptors in particular, which represent the majority of targets for pharmaceutical intervention, could be good examples for such approaches since several crystal structures of a particular receptor with agonists or antagonists are available. We propose to use such available structures in combination with the protocol developed below in order to increase the rate of compound identification that can indeed modulate the target in question.
Here, we use the nuclear receptor Retinoid X receptor alpha (RXRα) as an example for a selective SBVS screening protocol, which is based on ensemble docking in order to discover novel selective agonists. Retinoid X receptor (RXR) is a heterodimer partner for about one third of the 48 human nuclear receptor superfamily members, thus, its ligand binding pocket may be altered upon binding to a different heterodimerization partner. Heterodimerization greatly enhances DNA binding and transcriptional activation [121] and RXRα overexpression has been reported to enhance the transcriptional response to ligand binding. RXR-selective retinoids can potentiate the antiproliferative and apoptotic responses of breast cancer cell lines to PPAR ligands. Recently, RXR-selective ligands were discovered that inhibited proliferation of all-trans RA resistant breast cancer cells in vitro and caused regression of the disease in animal models. Therefore, RXRα is potentially a therapeutic target for cancer [121, 122].
Based on the fact that multiple RXRα receptor conformations exist depending on binding of the receptor to different heterodimerization partners, we seek to increase the specificity of identified binders for a given heterodimer partner by carefully choosing the RXRα receptor structure for SBVS. In our method, the choice of RXRα conformations for the SBVS is based on four criteria: a) Pairwise comparison of the receptor conformations according to RMSD calculations, b) analysis and clustering of RXRα structures comparing the binding-site shape and volume using SiteMap (Fig. 3), c) docking of a small-database of known actives for a specific heterodimer partner to the resulting shape-diverse subset of binding sites resulting from (a) and (b) using Glide 5.8 SP and XP [82], d) retrieving representative protein conformations for the structure of interest from MD simulations using GROMACS [123]. VS performed on three different subsets of RXRα receptor conformations, which arise by binding to different heterodimerization partners, selected as mentioned above, may enhance the success rate of the process (Fig. 3).
Fig. (3).
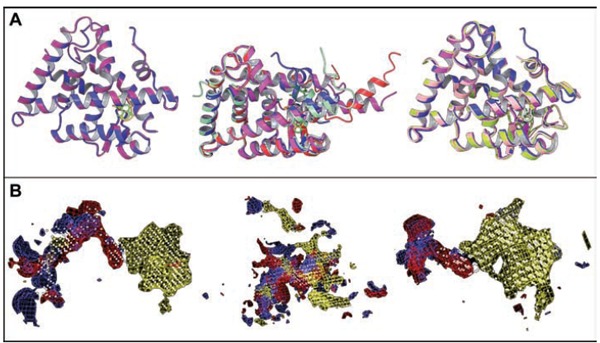
(A) Superposition of different RXRα receptor structures arising from different heterodimer complexes and/or binding to different ligands and (B) shape-based binding site comparison using SiteMap 2.6 (Schrödinger, LLC).
Docking compound databases for this SBVS exercise entails pre-processing to assign protonation and tautomeric states as described in [119]. Compounds for assying are selected as follows: Molecules that score high when docked in the RXRα protein ensemble that binds to the heterodimer partner of interest and at the same time score low for RXRα structures that bind to heterodimer partners of no interest, may be selected in order to achieve selectivity. Finally, a post-processing step is imposed to the top-scoring compounds by using Chembioserver [58] and FAF-Drugs2 [60] filtering tools as well as pharmacological property prediction with the QikProp software [124]. The workflow of this protocol is shown in Fig. (4).
Fig. (4).
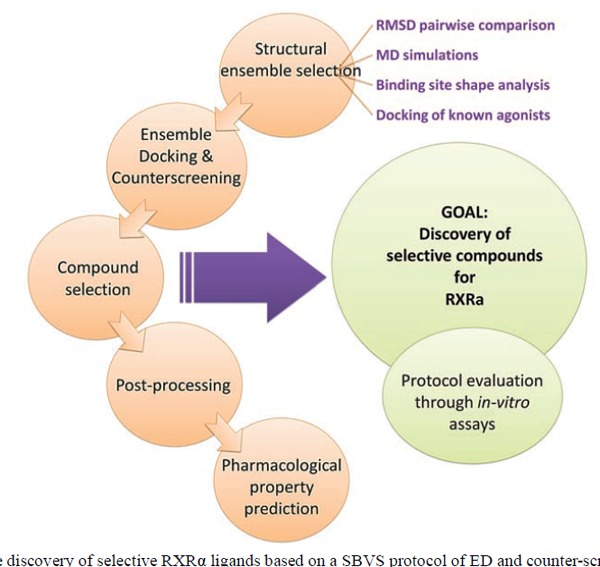
Workflow for the discovery of selective RXRα ligands based on a SBVS protocol of ED and counter-screening.
CONCLUSION
For decades, drug discovery was carried out using trial and error experimental techniques for screening large libraries of chemicals against a biological target. Recent advances in computer-aided drug design allow the tailored design of drugs for a target protein, shortening the development cycle of new drugs. The advent of Structure Based Virtual Screening has undoubtedly changed and improved the drug discovery process and has been established as one of the most promising in silico techniques for drug design. The focal point of this review is the detailed description of the SBVS steps in drug design, in combination with the presentation of recent advances and the introduction of various protocols that facilitate the identification of inhibitors with nM potency.
Furthermore, we also propose herein two novel VS protocols that aim to enhance inhibitor selectivity for the target protein against close homologs. First, a SBVS workflow, which was utilized to discover novel inhibitors of the H1047R mutant form of PI3Kα is presented. In this workflow, MD simulations in aqueous solution were carried out for both WT and mutant PI3Ka proteins. Binding site analysis in the kinase domain identified cavities in the vicinity of the H1047R mutation and the membrane binding regions. SBVS was performed in several binding pockets and top-ranked compounds in terms of predicted binding affinity were carefully post-processed to ensure the validity of docked poses, chemical diversity, desirable physicochemical properties and the absences of toxic or metabolically liable moieties. Finally, ten promising compounds were selected for in vitro assaying, four of which emerged as µM inhibitors of the H1047R mutant PI3Kα protein validating our approach [120]. A second SBVS protocol is contributed for the identification of binders for the RXRα nuclear receptor that are selective depending on the protein’s heterodimer partner. A structural RXRα ensemble was created for this purpose by selecting different RXRα crystal structures based on (i) RMSD calculations, (ii) binding-site shape and volume, (iii) docking of a small database of known actives, and (iv) choosing representative structures from MD simulations. SBVS was then performed on three different subsets of RXRα arising from different heterodimer complexes of RXRα and/or its binding to different ligands. Candidate binders of RXRα were selected for purchase with an eye on their different orientation at the binding site of the various structures and different interactions with specific surrounding residues in order to maximize their selectivity potential. In vitro assying of these compounds is still pending experimental testing.
Although SBVS is widely used nowadays by multiple academic and industrial research groups in the drug discovery process, it suffers from limitations that restrict its effectiveness (reviewed in Table 3). Significant breakthroughs are required in order to address fundamental challenges such as, for example, scoring, target flexibility and appropriately treating water molecules. Such challenges ultimately lead to the query: Is SBVS an indispensable tool for modern drug design? The significant reduction in time and cost compared to the high-throughput screening process, the continuous efforts in improving the efficiency of docking programs and scoring functions, and the plentiful successful case studies that have led to low nM leads are only a few representative examples showing that SBVS is here to stay. However, prospective users of the method should remember that SBVS is not as simple as running a computer program. Careful choices need to be made; appropriately selecting the structural ensemble for the screening exercise, cautious preparation of the biological target and the database to be used, treatment of water molecules in the cavity, and careful post-processing of SBVS results are of utmost importance that ultimately result in enhancing lead identification and selectivity rates.
Table 3.
Advantages and Drawbacks of SBVS.
| Virtual Screening | |
|---|---|
| Advantages | Limitations |
| Time and cost reduction of screening process of millions of small molecules, compared to HTS | Many VS tools are applicable and successful to specific case studies (based on the training set) and not in general cases. |
| There is no need for physically existing compounds to perform the screening process, unlike HTS. |
Compounds being identified by HTS are usually more bioactive than compounds identified by VS. |
| Different approaches of VS have been created for lead discovery depending each time on the availability of experimental information (SBVS Ligand-Based VS, Fragment-Based VS,etc.) |
Weakness in perfect inclusion of receptor structural flexibility and of water in docking computations due to computational-cost and high complexity of its modeling |
| Several successful examples of identifying low nM leads that show the intended biological activity | Very potent leads (i.e. low nM) are rarely identified through VS. |
| A large number of docking programs and scoring functions | Scoring is still challenging in predicting accurately the correct binding pose and ranking of the compounds due to the difficulty in parameterizing the complexity of the ligand-receptor binding interactions and the approximations in calculating desolvation and entropic terms. |
| VS can use as input a desirable target structure complexed with a specific ligand even if there are no experimental data, through molecular modeling. | Predicted protein structures from homology modeling and predicted protein-ligand complexes may result to increased rates of false positive/negative results. |
| Does not perform in congeneric series. | |
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The initial part of this work was co-funded by the NSRF 2007-2013, the European Regional Development Fund and national resources, under the grant “Cooperation" [No. 09 ΣΥΝ 11-09 ΣΥΝ 11-675]. This work has been also supported by a Marie Curie Reintegration Grant (FP7-PEOPLE-2009-RG, No 256533) and an AACR Judah Folkman Fellowship for Cancer Research in Angiogenesis (08-40-18-COUR). EL was supported by an IKY graduate scholarship from funds of the “Education and lifelong learning” of the European Social Fund 2007-2013. Part of the computational work was performed.
Computational analyses presented herein were performed on resources of PRACE-PR and LinkSCEEM/Cy-tera, which are also gratefully acknowledged. Part of the computational work was performed at BRFAA using a cluster funded from European Economic Area Grant No. EL0084.
References
- 1.Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 2013;20(23):2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- 2.Reddy A.S, Pati S.P, Kumar P.P, Pradeep H.N, Sastry G.N. Virtual screening in drug discovery ― a computational perspective. Curr. Protein Pept. Sci. 2007;8(4):329–351. doi: 10.2174/138920307781369427. [DOI] [PubMed] [Google Scholar]
- 3.Jorgensen W.L. The many roles of computation in drug discovery. Science. 2004;303(5665):1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 4.Benod C, Carlsson J, Uthayaruban R, Hwang P, Irwin J.J, Doak A.K, Shoichet B.K, Sablin E.P, Fletterick R.J. Structure-based discovery of antagonists of nuclear receptor LRH-1. J. Biol. Chem. 2013;288(27):19830–19844. doi: 10.1074/jbc.M112.411686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng T, Li Q, Zhou Z, Wang Y, Bryant S.H. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14(1):133–141. doi: 10.1208/s12248-012-9322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andricopulo A.D, Salum L.B, Abraham D.J. Structure-based drug design strategies in medicinal chemistry. Curr. Top. Med. Chem. 2009;9(9):771–790. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]
- 7.Heifetz A, Barker O, Verquin G, Wimmer N, Meutermans W, Pal S, Law R.J, Whittaker M. Fighting obesity with a sugar-based library: discovery of novel MCH-1R antagonists by a new computational-VAST approach for exploration of GPCR binding sites. J. Chem. Inf. Model. 2013;53(5):1084–1099. doi: 10.1021/ci4000882. [DOI] [PubMed] [Google Scholar]
- 8.Schröder J, Klinger A, Oellien F, Marhöfer R.J, Duszenko M, Selzer P.M. Docking-based virtual screening of covalently binding ligands: an orthogonal lead discovery approach. J. Med. Chem. 2013;56(4):1478–1490. doi: 10.1021/jm3013932. [DOI] [PubMed] [Google Scholar]
- 9.Kolb P, Rosenbaum D.M, Irwin J.J, Fung J.J, Kobilka B.K, Shoichet B.K. Structure-based discovery of beta2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. USA. 2009;106(16):6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Köppen H. Virtual screening - what does it give us? Curr. Opin. Drug Discov. Devel. 2009;12(3):397–407. [PubMed] [Google Scholar]
- 11.Sastry G.M, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 12.Pitt W.R, Calmiano M.D, Kroeplien B, Taylor R.D, Turner J.P, King M.A. Structure-based virtual screening for novel ligands. Methods Mol. Biol. 2013;1008:501–519. doi: 10.1007/978-1-62703-398-5_19. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Robertson A.D, Jensen J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61(4):704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 14.Anandakrishnan R, Aguilar B, Onufriev A.V. Onufriev, A.V. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulation. Nucleic Acids Res. 2012;40:W537-41–W541. doi: 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ten Brink T, Exner T.E. pK(a) based protonation states and microspecies for protein-ligand docking. J. Comput. Aided Mol. Des. 2010;24(11):935–942. doi: 10.1007/s10822-010-9385-x. [DOI] [PubMed] [Google Scholar]
- 16.Dolinsky T.J, Czodrowski P, Li H, Nielsen J.E, Jensen J.H, Klebe G, Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35(Web Server issue):W522-5. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovalenko A. Three-dimensional RISM theory for molecular liquids and solid-liquid interfaces. In: Hirata F. , Mezey P.G., editors. 360. Vol. 24. Dordrecht: Kluwer Academic Publishers; 2003. pp. 169–275. [Google Scholar]
- 18.Young T, Abel R, Kim B, Berne B.J, Friesner R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc. Natl. Acad. Sci. USA. 2007;104(3):808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abel R, Young T, Farid R, Berne B.J, Friesner R.A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008;130(9):2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rashin A.A, Bukatin M.A. Continuum based calculations of hydration entropies and the hydrophobic effect. J. Phys. Chem. 1991;95(8):2942–2944. doi: 10.1021/j100161a002. [DOI] [Google Scholar]
- 21.Michel J, Tirado-Rives J, Jorgensen W.L. Prediction of the water content in protein binding sites. J. Phys. Chem. B. 2009;113(40):13337–13346. doi: 10.1021/jp9047456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.WaterMap. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 23.Canada, H3A 2R7: Montreal, QC,; 2013. Molecular Operating Environment (MOE) St. West, Suite #910. [Google Scholar]
- 24.Jorgensen W.L, Tirado-Rives J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 2005;26(16):1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 25.SZMAP. OpenEye Scientific Software Inc.: Santa Fe, NM.
- 26.Protein Preparation Wizard. Maestro, Schrodinger LLC: [Google Scholar]
- 27.Repasky M.P, Murphy R.B, Banks J.L, Greenwood J.R, Tubert-Brohman I, Bhat S, Friesner R.A. Docking performance of the glide program as evaluated on the Astex and DUD datasets: a complete set of glide SP results and selected results for a new scoring function integrating WaterMap and glide. J. Comput. Aided Mol. Des. 2012;26(6):787–799. doi: 10.1007/s10822-012-9575-9. [DOI] [PubMed] [Google Scholar]
- 28.Corbeil C.R, Williams C.I, Labute P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012;26(6):775–786. doi: 10.1007/s10822-012-9570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.SiteMap, Schrodinger, LLC, 2013.
- 30.Ngan C.H, Bohnuud T, Mottarella S.E, Beglov D, Villar E.A, Hall D.R, Kozakov D, Vajda S. FTMAP: extended protein mapping with user-selected probe molecules. Nuc. Acids Res. 2012;40:W271–W275. doi: 10.1093/nar/gks441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidtke P, Bidon-Chanal A, Luque F.J, Barril X. MDpocket: open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics. 2011;27(23):3276–3285. doi: 10.1093/bioinformatics/btr550. [DOI] [PubMed] [Google Scholar]
- 33.Laurie A.T, Jackson R.M. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21(9):1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 34.Doppelt-Azeroual O, Delfaud F, Moriaud F, de Brevern A.G. Fast and automated functional classification with MED-SuMo: an application on purine-binding proteins. Protein Sci. 2010;19(4):847–867. doi: 10.1002/pro.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hernandez M, Ghersi D, Sanchez R. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009;37(Web Server issue):W413–W416. doi: 10.1093/nar/gkp281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seco J, Luque F.J, Barril X. Binding site detection and druggability index from first principles. J. Med. Chem. 2009;52(8):2363–2371. doi: 10.1021/jm801385d. [DOI] [PubMed] [Google Scholar]
- 37.Raman E.P, Yu W, Guvench O, Mackerell A.D. Reproducing crystal binding modes of ligand functional groups using Site-Identification by Ligand Competitive Saturation (SILCS) simulations. J. Chem. Inf. Model. 2011;51(4):877–896. doi: 10.1021/ci100462t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lexa K.W, Carlson H.A. Full protein flexibility is essential for proper hot-spot mapping. J. Am. Chem. Soc. 2011;133(2):200–202. doi: 10.1021/ja1079332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buch I, Giorgino T, De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2011;108(25):10184–10189. doi: 10.1073/pnas.1103547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herbert C, Schieborr U, Saxena K, Juraszek J, De Smet F, Alcouffe C, Bianciotto M, Saladino G, Sibrac D, Kudlinzki D, Sreeramulu S, Brown A, Rigon P, Herault J.P, Lassalle G, Blundell T.L, Rousseau F, Gils A, Schymkowitz J, Tompa P, Herbert J.M, Carmeliet P, Gervasio F.L, Schwalbe H, Bono F. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell. 2013;23(4):489–501. doi: 10.1016/j.ccr.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 41.Shan Y, Kim E.T, Eastwood M.P, Dror R.O, Seeliger M.A, Shaw D.E. How does a drug molecule find its target binding site? J. Am. Chem. Soc. 2011;133(24):9181–9183. doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grove L.E, Hall D.R, Beglov D, Vajda S, Kozakov D. FTFlex: accounting for binding site flexibility to improve fragment-based identification of druggable hot spots. Bioinformatics. 2013;29(9):1218–1219. doi: 10.1093/bioinformatics/btt102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazaridis T. Inhomogeneous Fluid Approach to Solvation Thermodynamics. 2. Applications to Simple Fluids. J. Phys. Chem. B. 1998;102:3542–3550. doi: 10.1021/jp972358w. [DOI] [Google Scholar]
- 44.Lipinski C.A, Lombardo F, Dominy B.W, Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1-3):3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 45.Zhang M.Q, Wilkinson B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 2007;18(6):478–488. doi: 10.1016/j.copbio.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 46.Veber D.F, Johnson S.R, Cheng H.Y, Smith B.R, Ward K.W, Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 47.Congreve M, Carr R, Murray C, Jhoti H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today. 2003;8(19):876–877. doi: 10.1016/S1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 48.Köster H, Craan T, Brass S, Herhaus C, Zentgraf M, Neumann L, Heine A, Klebe G. A small nonrule of 3 compatible fragment library provides high hit rate of endothiapepsin crystal structures with various fragment chemotypes. J. Med. Chem. 2011;54(22):7784–7796. doi: 10.1021/jm200642w. [DOI] [PubMed] [Google Scholar]
- 49.Baell J, Congreve M, Leeson P, Abad-Zapatero C. Ask the experts: past, present and future of the rule of five. Future Med. Chem. 2013;5(7):745–752. doi: 10.4155/fmc.13.61. [DOI] [PubMed] [Google Scholar]
- 50.Yusof I, Segall M.D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today. 2013;18(13-14):659–666. doi: 10.1016/j.drudis.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Hughes J.D, Blagg J, Price D.A, Bailey S, Decrescenzo G.A, Devraj R.V, Ellsworth E, Fobian Y.M, Gibbs M.E, Gilles R.W, Greene N, Huang E, Krieger-Burke T, Loesel J, Wager T, Whiteley L, Zhang Y. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008;18(17):4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 52.Kerns E.H, Di L. Concepts, Structure Design and Methodsfrom ADME to Toxicity Optimization. 1. Academic Press; 2008. Drug-like Properties: . [Google Scholar]
- 53.Bickerton G.R, Paolini G.V, Besnard J, Muresan S, Hopkins A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012;4(2):90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuegg J, Cooper M.A. Drug-likeness and increased hydrophobicity of commercially available compound libraries for drug screening. Curr. Top. Med. Chem. 2012;12(14):1500–1513. doi: 10.2174/156802612802652466. [DOI] [PubMed] [Google Scholar]
- 55.Blagg J. Structure–Activity Relationships for In vitro and In vivo Toxicity. Annu. Rep. Med. Chem. 2006;41:353–368. doi: 10.1016/S0065-7743(06)41024-1. [DOI] [Google Scholar]
- 56.Baell J.B, Holloway G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 57.Metz J.T, Huth J.R, Hajduk P.J. Enhancement of chemical rules for predicting compound reactivity towards protein thiol groups. J. Comput. Aided Mol. Des. 2007;21(1-3):139–144. doi: 10.1007/s10822-007-9109-z. [DOI] [PubMed] [Google Scholar]
- 58.Athanasiadis E, Cournia Z, Spyrou G. ChemBioServer: a web-based pipeline for filtering, clustering and visualization of chemical compounds used in drug discovery. Bioinformatics. 2012;28(22):3002–3003. doi: 10.1093/bioinformatics/bts551. [DOI] [PubMed] [Google Scholar]
- 59.Lagorce D, Sperandio O, Galons H, Miteva M.A, Villoutreix B.O. FAF-Drugs2: free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinformatics. 2008;9:396. doi: 10.1186/1471-2105-9-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lagorce D, Maupetit J, Baell J, Sperandio O, Tufféry P, Miteva M.A, Galons H, Villoutreix B.O. The FAF-Drugs2 server: a multistep engine to prepare electronic chemical compound collections. Bioinformatics. 2011;27(14):2018–2020. doi: 10.1093/bioinformatics/btr333. [DOI] [PubMed] [Google Scholar]
- 61.Kalliokoski T, Salo H.S, Lahtela-Kakkonen M, Poso A. The effect of ligand-based tautomer and protomer prediction on structure-based virtual screening. J. Chem. Inf. Model. 2009;49(12):2742–2748. doi: 10.1021/ci900364w. [DOI] [PubMed] [Google Scholar]
- 62.Sadowski J, Rudolph C, Gasteiger J. The generation of 3D-models of host-guest. Anal. Chim. Acta. 1992;265:233–241. doi: 10.1016/0003-2670(92)85029-6. [DOI] [Google Scholar]
- 63.Milletti F, Vulpetti A. Tautomer preference in PDB complexes and its impact on structure-based drug discovery. J. Chem. Inf. Model. 2010;50(6):1062–1074. doi: 10.1021/ci900501c. [DOI] [PubMed] [Google Scholar]
- 64.Morris G.M, Huey R, Lindstrom W, Sanner M.F, Belew R.K, Goodsell D.S, Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.LigPrep. New York, NY: Schrödinger, LLC; 2013. [Google Scholar]
- 66.MAPS, version 3.4. Paris, France: Scienomics SARL; 2014. [Google Scholar]
- 67.DISI http://wiki.uoft.bkslab.org/index.php/Preparing_the_ligand .
- 68.http://www.accelrys.com.
- 69.Hyperchem. HyperCube, Gainesville, FL.: [Google Scholar]
- 70.Grinter S.Z, Yan C, Huang S.Y, Jiang L, Zou X. Automated large-scale file preparation, docking, and scoring: evaluation of ITScore and STScore using the 2012 Community Structure-Activity Resource benchmark. J. Chem. Inf. Model. 2013;53(8):1905–1914. doi: 10.1021/ci400045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song C.M, Bernardo P.H, Chai C.L, Tong J.C. CLEVER: pipeline for designing in silico chemical libraries. J. Mol. Graph. Model. 2009;27(5):578–583. doi: 10.1016/j.jmgm.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 72.Douguet D. e-LEA3D: a computational-aided drug design web server. Nucleic Acids Res. 2010;38(Web Server issue):W615-21. doi: 10.1093/nar/gkq322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harris C.J, Hill R.D, Sheppard D.W, Slater M.J, Stouten P.F. The design and application of target-focused compound libraries. Comb. Chem. High Throughput Screen. 2011;14(6):521–531. doi: 10.2174/138620711795767802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lisurek M, Rupp B, Wichard J, Neuenschwander M, von Kries J.P, Frank R, Rademann J, Kühne R. Design of chemical libraries with potentially bioactive molecules applying a maximum common substructure concept. Mol. Divers. 2010;14(2):401–408. doi: 10.1007/s11030-009-9187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wirth M, Zoete V, Michielin O, Sauer W.H. SwissBioisostere: a database of molecular replacements for ligand design. Nucleic Acids Res. 2013;41(Database issue):D1137–D1143. doi: 10.1093/nar/gks1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weber J, Achenbach J, Moser D, Proschak E. VAMMPIRE: a matched molecular pairs database for structure-based drug design and optimization. J. Med. Chem. 2013;56(12):5203–5207. doi: 10.1021/jm400223y. [DOI] [PubMed] [Google Scholar]
- 77.Vainio M.J, Kogej T, Raubacher F. Automated recycling of chemistry for virtual screening and library design. J. Chem. Inf. Model. 2012;52(7):1777–1786. doi: 10.1021/ci300157m. [DOI] [PubMed] [Google Scholar]
- 78.Baell J.B. Broad coverage of commercially available lead-like screening space with fewer than 350,000 compounds. J. Chem. Inf. Model. 2013;53(1):39–55. doi: 10.1021/ci300461a. [DOI] [PubMed] [Google Scholar]
- 79.Xing L, McDonald J.J, Kolodziej S.A, Kurumbail R.G, Williams J.M, Warren C.J, O’Neal J.M, Skepner J.E, Roberds S.L. Discovery of potent inhibitors of soluble epoxide hydrolase by combinatorial library design and structure-based virtual screening. J. Med. Chem. 2011;54(5):1211–1222. doi: 10.1021/jm101382t. [DOI] [PubMed] [Google Scholar]
- 80.Ewing T.J, Makino S, Skillman A.G, Kuntz I.D. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001;15(5):411–428. doi: 10.1023/A:1011115820450. [DOI] [PubMed] [Google Scholar]
- 81.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261(3):470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 82.Friesner R.A, Banks J.L, Murphy R.B, Halgren T.A, Klicic J.J, Mainz D.T, Repasky M.P, Knoll E.H, Shelley M, Perry J.K, Shaw D.E, Francis P, Shenkin P.S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 83.Jones G, Willett P, Glen R.C, Leach A.R, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 84.Jain A.N. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003;46(4):499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 85.Abagyan R, Totrov M, Kuznetsov D. ICM: a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994;15:488–506. doi: 10.1002/jcc.540150503. [DOI] [Google Scholar]
- 86.Venkatachalam C.M, Jiang X, Oldfield T, Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003;21(4):289–307. doi: 10.1016/S1093-3263(02)00164-X. [DOI] [PubMed] [Google Scholar]
- 87.Vlachakis D, Tsagrasoulis D, Megalooikonomou V, Kossida S. Introducing Drugster: a comprehensive and fully integrated drug design, lead and structure optimization toolkit. Bioinformatics. 2013;29(1):126–128. doi: 10.1093/bioinformatics/bts637. [DOI] [PubMed] [Google Scholar]
- 88.Zsoldos Z, Szabo I, Szabo Z. A.P.J. Software tools for structure based rational drug design. J. Mol. Struct. THEOCHEM. 2003;666-667:659–665. doi: 10.1016/j.theochem.2003.08.105. [DOI] [Google Scholar]
- 89.Guido R.V, Oliva G, Andricopulo A.D. Virtual screening and its integration with modern drug design technologies. Curr. Med. Chem. 2008;15(1):37–46. doi: 10.2174/092986708783330683. [DOI] [PubMed] [Google Scholar]
- 90.Cross J.B, Thompson D.C, Rai B.K, Baber J.C, Fan K.Y, Hu Y, Humblet C. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2009;49(6):1455–1474. doi: 10.1021/ci900056c. [DOI] [PubMed] [Google Scholar]
- 91.McGann M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012;26(8):897–906. doi: 10.1007/s10822-012-9584-8. [DOI] [PubMed] [Google Scholar]
- 92.Liebeschuetz J.W, Cole J.C, Korb O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided Mol. Des. 2012;26(6):737–748. doi: 10.1007/s10822-012-9551-4. [DOI] [PubMed] [Google Scholar]
- 93.Spitzer R, Jain A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012;26(6):687–699. doi: 10.1007/s10822-011-9533-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Neves M.A, Totrov M, Abagyan R. Docking and scoring with ICM: the benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012;26(6):675–686. doi: 10.1007/s10822-012-9547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gkeka P, Eleftheratos S, Kolocouris A, Cournia Z. Free Energy Calculations Reveal the Origin of Binding Preference for Aminoadamantane Blockers of Influenza A/M2TM Pore. J. Chem. Theory Comput. 2013;9(2):1272–1281. doi: 10.1021/ct300899n. [DOI] [PubMed] [Google Scholar]
- 96.Baggett A.W, Cournia Z, Han M.S, Patargias G, Glass A.C, Liu S.Y, Nolen B.J. Structural characterization and computer-aided optimization of a small-molecule inhibitor of the Arp2/3 complex, a key regulator of the actin cytoskeleton. ChemMedChem. 2012;7(7):1286–1294. doi: 10.1002/cmdc.201200104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zilian D, Sotriffer C.A. SFCscore(RF): a random forest-based scoring function for improved affinity prediction of protein-ligand complexes. J. Chem. Inf. Model. 2013;53(8):1923–1933. doi: 10.1021/ci400120b. [DOI] [PubMed] [Google Scholar]
- 98.Schneider N, Hindle S, Lange G, Klein R, Albrecht J, Briem H, Beyer K, Claußen H, Gastreich M, Lemmen C, Rarey M. Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J. Comput. Aided Mol. Des. 2012;26(6):701–723. doi: 10.1007/s10822-011-9531-0. [DOI] [PubMed] [Google Scholar]
- 99.Ravindranathan K, Tirado-Rives J, Jorgensen W.L, Guimarães C.R. Improving MM-GB/SA Scoring through the Application of the Variable Dielectric Model. J. Chem. Theory Comput. 2011;7(12):3859–3865. doi: 10.1021/ct200565u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korb O, Olsson T.S, Bowden S.J, Hall R.J, Verdonk M.L, Liebeschuetz J.W, Cole J.C. Potential and limitations of ensemble docking. J. Chem. Inf. Model. 2012;52(5):1262–1274. doi: 10.1021/ci2005934. [DOI] [PubMed] [Google Scholar]
- 101.Craig I.R, Essex J.W, Spiegel K. Ensemble docking into multiple crystallographically derived protein structures: an evaluation based on the statistical analysis of enrichments. J. Chem. Inf. Model. 2010;50(4):511–524. doi: 10.1021/ci900407c. [DOI] [PubMed] [Google Scholar]
- 102.Rueda M, Bottegoni G, Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 2010;50(1):186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Osguthorpe D.J, Sherman W, Hagler A.T. Generation of receptor structural ensembles for virtual screening using binding site shape analysis and clustering. Chem. Biol. Drug Des. 2012;80(2):182–193. doi: 10.1111/j.1747-0285.2012.01396.x. [DOI] [PubMed] [Google Scholar]
- 104.Kalid O, Toledo Warshaviak D, Shechter S, Sherman W, Shacham S. Consensus Induced Fit Docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J. Comput. Aided Mol. Des. 2012;26(11):1217–1228. doi: 10.1007/s10822-012-9611-9. [DOI] [PubMed] [Google Scholar]
- 105.Houston D.R, Walkinshaw M.D. Consensus docking: improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013;53(2):384–390. doi: 10.1021/ci300399w. [DOI] [PubMed] [Google Scholar]
- 106.Waszkowycz B. Towards improving compound selection in structure-based virtual screening. Drug Discov. Today. 2008;13(5-6):219–226. doi: 10.1016/j.drudis.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 107.Malmstrom R.D, Watowich S.J. Using free energy of binding calculations to improve the accuracy of virtual screening predictions. J. Chem. Inf. Model. 2011;51(7):1648–1655. doi: 10.1021/ci200126v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ding B, Wang J, Li N, Wang W. Characterization of small molecule binding. I. Accurate identification of strong inhibitors in virtual screening. J. Chem. Inf. Model. 2013;53(1):114–122. doi: 10.1021/ci300508m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ben Nasr N, Guillemain H, Lagarde N, Zagury J.F, Montes M. Multiple structures for virtual ligand screening: defining binding site properties-based criteria to optimize the selection of the query. J. Chem. Inf. Model. 2013;53(2):293–311. doi: 10.1021/ci3004557. [DOI] [PubMed] [Google Scholar]
- 110.Planesas J.M, Claramunt R.M, Teixidó J, Borrell J.I, Pérez-Nueno V.I. Improving VEGFR-2 docking-based screening by pharmacophore postfiltering and similarity search postprocessing. J. Chem. Inf. Model. 2011;51(4):777–787. doi: 10.1021/ci1002763. [DOI] [PubMed] [Google Scholar]
- 111.Kumar N.V, Eblen S.T, Weber M.J. Identifying specific kinase substrates through engineered kinases and ATP analogs. Methods. 2004;32(4):389–397. doi: 10.1016/j.ymeth.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 112.Kumar N.V, Eblen S.T, Weber M.J. Identifying specific kinase substrates through engineered kinases and ATP analogs. Methods. 2004;32(4):389–397. doi: 10.1016/j.ymeth.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 113.Islam K, Chen Y, Wu H, Bothwell I.R, Blum G.J, Zeng H, Dong A, Zheng W, Min J, Deng H, Luo M. Defining efficient enzyme-cofactor pairs for bioorthogonal profiling of protein methylation. Proc. Natl. Acad. Sci. USA. 2013;110(42):16778–16783. doi: 10.1073/pnas.1216365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhao S, Kumar R, Sakai A, Vetting M.W, Wood B.M, Brown S, Bonanno J.B, Hillerich B.S, Seidel R.D, Babbitt P.C, Almo S.C, Sweedler J.V, Gerlt J.A, Cronan J.E, Jacobson M.P. Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature. 2013;502(7473):698–702. doi: 10.1038/nature12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stumpfe D, Bill A, Novak N, Loch G, Blockus H, Geppert H, Becker T, Schmitz A, Hoch M, Kolanus W, Famulok M, Bajorath J. Targeting multifunctional proteins by virtual screening: structurally diverse cytohesin inhibitors with differentiated biological functions. ACS Chem. Biol. 2010;5(9):839–849. doi: 10.1021/cb100171c. [DOI] [PubMed] [Google Scholar]
- 116.Mandelker D, Gabelli S.B, Schmidt-Kittler O, Zhu J, Cheong I, Huang C.H, Kinzler K.W, Vogelstein B, Amzel L.M. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc. Natl. Acad. Sci. USA. 2009;106(40):16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sali A, Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 118.Phillips J.C, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R.D, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen W.L. Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J. Med. Chem. 2009;52(2):416–424. doi: 10.1021/jm801100v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gkeka P, Athanasiadis E, Spyrou G, Cournia Z. 2012. [DOI] [PubMed] [Google Scholar]
- 121.Crowe D.L, Chandraratna R.A. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-alpha is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res. 2004;6(5):R546–R555. doi: 10.1186/bcr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tanaka T, De Luca L.M. Therapeutic potential of “rexinoids” in cancer prevention and treatment. Cancer Res. 2009;69(12):4945–4947. doi: 10.1158/0008-5472.CAN-08-4407. [DOI] [PubMed] [Google Scholar]
- 123.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark A.E, Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 124.QikProp, version 3.8. New York, NY: Schrödinger, LLC; 2013. [Google Scholar]
- 125.Maestro, version 9.4, New York, NY: Schrödinger, LLC; 2013. [Google Scholar]
- 126.Schnecke V, Swanson C.A, Getzoff E.D, Tainer J.A, Kuhn L.A. Screening a peptidyl database for potential ligands to proteins with side-chain flexibility. Proteins. 1998;33(1):74–87. doi: 10.1002/(SICI)1097-0134(19981001)33:1<74::AID-PROT7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 127.Chaudhury S, Gray J.J. Conformer selection and induced fit in flexible backbone protein-protein docking using computational and NMR ensembles. J. Mol. Biol. 2008;381(4):1068–1087. doi: 10.1016/j.jmb.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ouyang X, Zhou S, Su C.T, Ge Z, Li R, Kwoh C.K. CovalentDock: automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J. Comput. Chem. 2013;34(4):326–336. doi: 10.1002/jcc.23136. [DOI] [PubMed] [Google Scholar]
- 129.Lill M.A, Danielson M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011;25(1):13–19. doi: 10.1007/s10822-010-9395-8. [DOI] [PubMed] [Google Scholar]
- 130.Truchon J.F, Bayly C.I. GLARE: A free open source software for combinatorial library design. J. Chem. Inf. Model. 2006;46:1536–1548. doi: 10.1021/ci0504871. [DOI] [PubMed] [Google Scholar]
- 131.Truszkowski A, Jayaseelan K.V, Neumann S, Willighagen E.L, Zielesny A, Steinbeck C. New developments on the cheminformatics open workflow environment CDK-Taverna. J. Cheminform. 2011;3:54. doi: 10.1186/1758-2946-3-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schüller A, Hähnke V, Schneider G. SmiLib v2.0: A Java-based tool for rapid combinatorial library enumeration. QSAR Comb. Sci. 2007;26(3):407–410. doi: 10.1002/qsar.200630101. [DOI] [Google Scholar]
- 133.Library synthesizer, Tripod,http://tripod.nih.gov/?p=370.
- 134.Reactor http://www.chemaxon.com.
- 135.OELib library enumeration http://www.eyesopen.com.
- 136.CombiLibMaker and Legion http://www.tripos.com.
- 137.QuaSAR-CombiGen http://www.chemcomp.com.
- 138.ChemOffice,CombiChem http://http://www.cambridgesoft.com.
- 139.ICM-Chemist http://www.molsoft.com.
- 140.LUCIA Transform-based virtual library generation http://www.eidogensertanty.com.