

Abstract
Mitochondria play a fundamental role in heart physiology, but are also key effectors of dysfunction and death. This dual role assumes a new meaning following recent advances on the nature and regulation of the permeability transition pore, an inner membrane channel whose opening requires matrix Ca2+ and is modulated by many effectors including reactive oxygen species, matrix cyclophilin D, Pi and matrix pH. The recent demonstration that the F-ATP synthase can reversibly undergo a Ca2+-dependent transition to form a channel that mediates the permeability transition opens new perspectives to the field. These findings demand a reassessment of the modifications of F-ATP synthase that take place in the heart under pathological conditions and of their potential role in determining the transition of F-ATP synthase from and energy-conserving into an energy-dissipating device.
Keywords: Mitochondria, heart pathophysiology, Ca2+, permeability transition pore, F-ATP synthase
Introduction
Besides their central role in ATP synthesis and oxidative metabolism of nutrients, mitochondria contribute to intracellular Ca2+ homeostasis and generation of reactive oxygen species (ROS), and are determinant for both cell survival and cell death. Important for any cell type, these functions are crucial for cardiac myocytes. Mitochondrial processes are highly compartmentalized due to the existence of 2 limiting membranes and to the quite selective localization of proteins, nucleotides and coenzymes in the intermembrane and matrix spaces.
The outer mitochondrial membrane (OMM) is the interface between mitochondria and other cellular components and represents a physical barrier preventing the release of mitochondrial proteins involved in apoptosis, such as cytochrome c. OMM proteins are involved in apoptosis1, intracellular signaling, tethering to the ER/SR,2 autophagy3,4 and mitophagy.5 OMM permeability to metabolites and small peptides (< 5kDa) is regulated by the voltage-dependent anion channel (VDAC) and other transporters including the translocator protein of 18 kDa (TSPO).6
The inner mitochondrial membrane (IMM), whose permeability to solutes is controlled by highly specific transporters and tightly regulated channels, is the site of coupling between substrate oxidation and ATP synthesis in the process of oxidative phosphorylation. Mitochondria operate a sequence of energy conversion processes through which the exergonic flow of electrons along the respiratory complexes supports the endergonic pumping of protons from the matrix to the intermembrane space. The resulting proton motive force (Δp) drives the rotation of the Fo sector of ATP synthase leading to the synthesis of ATP in the F1 sector. The two components of Δp, namely Δψm and ΔpH, are also utilized for the uptake of ADP and Pi, respectively, and for the release of ATP into the cytosol in exchange for ADP. In addition, Δp is also utilized by mitochondria for their biogenesis and for the maintenance of ion homeostasis, which is crucial for Ca2+.7,8 In particular, the mitochondrial Ca2+ uniporter (MCU)9,10 catalyzes Ca2+ uptake down the Ca2+ electrochemical gradient, which is largely driven by the inside-negative Δψm, whereas release from the matrix to the intermembrane space, which is catalyzed by the mitochondrial Na+/Ca2+ exchanger, ultimately depends on ΔpH. Indeed, the exchanger is functionally coupled with the mitochondrial Na+/H+ exchanger, so that mitochondrial Ca2+ release is paralleled by H+ reuptake.11
A rise in intramitochondrial [Ca2+] from a resting value of about 0.1 μM to ≤ 1 μM activates key enzymes of oxidative metabolism, such as pyruvic, isocitric, and oxoglutaric dehydrogenases.12 Therefore, the increase in ATP demand dictated by cytosolic Ca2+-activated processes, especially contractile activity, is matched by increased ATP synthesis.13 Higher values of matrix [Ca2+] could reflect the ability of mitochondria to buffer undesired elevation of cytosolic [Ca2+], but may also promote cell injury after opening of the permeability transition pore (PTP), an IMM channel, as discussed in detail in the following Sections. More controversial is the contribution of mitochondrial Ca2+ homeostasis to cytosolic Ca2+ oscillations related to myocardial contractile activity. Based upon kinetic analysis of mitochondrial Ca2+ uptake and release in vitro, beat-to-beat variations of matrix [Ca2+] should not occur because MCU exhibits high transport capacity but low affinity, so that even for values of cytosolic [Ca2+] measured at peak systole (≅2 μM) mitochondrial Ca2+ uptake should be negligible. However, due to the close proximity between mitochondria and SR, MCU might be exposed to local domains of [Ca2+] higher than those occurring in the bulk cytosolic space.14 As far as Ca2+ release is concerned, the Na+/Ca2+ exchanger may be too slow to cope with very fast cytosolic oscillations, such as those occurring in a beating mouse heart.7 Nevertheless, clear evidence of beat-to-beat changes of mitochondrial [Ca2+] in intact cardiomyocytes has been provided by different laboratories.15–17 This issue is still the matter of debate,18 and besides methodological questions a major point concerns the high energetic cost (or low convenience) that would be determined by beat-to-beat variations in mitochondrial [Ca2+]. An adequate pathway for Ca2+ release could be provided by transient openings of the PTP,19 which have been documented both in isolated mitochondria and in situ.20–23 The advantage of an unselective channel of high conductance would be to provide charge compensation within the channel itself, allowing mitochondrial Ca2+ release at zero potential, i.e. even for small Ca2+ gradients.7 Support for this idea comes from the finding that treatment of cardiomyocytes with CsA (which desensitizes the PTP) increased the uptake of 45 Ca2+ in mitochondria,24 and that mitochondrial [Ca2+] is elevated in hearts with genetic ablation of mitochondrial cyclophilin (CyP) D, the mitochondrial target of CsA.25 The reader is referred to a recent review for a general discussion of this hypothesis26 while the links between PTP and cardiac disease will be discussed further below.
Based upon a recent quantitative analysis, mitochondria do not appear to represent a significant dynamic buffer of cytosolic Ca2+ under physiological conditions while they might shape cellular [Ca2+] dynamics upon prolonged elevations of cytosolic [Ca2+].27 A limited contribution of mitochondrial Ca2+ homeostasis to cardiac function appears to be supported by the lack of a specific cardiac phenotype in MCU knockout mice.28 This experimental model seems also to rule out an involvement of mitochondrial Ca2+ overload in ischemia/reperfusion (I/R) injury.
Mitochondria are double-edged swords
Coupling between the proton gradient and the F-ATP synthase, which is essential for life, can become a life-threatening process under conditions of cell injury, especially when oxygen availability is curtailed. In the absence of oxygen electron flow stops, the mitochondrial membrane potential collapses and mitochondrial ATP synthesis can no longer occur. If cytosolic ATP is available it is taken up by mitochondria and hydrolyzed by F-ATP synthase, with regeneration of the proton gradient if the IMM permeability is low.29 If the IMM permeability increases following opening of the PTP, ATP hydrolysis is maximal and ATP depletion precipitates rigor contracture. This also explains the increased rate of glycolysis, the massive accumulation of lactate and the drop in intracellular pH determined by anoxia or any defect of the respiratory chain. These concepts support the inhibition of ATP hydrolysis as an effective strategy for myocardial protection against I/R injury that is elicited by both self-defense mechanisms and pharmacological treatments.30 Indeed, the rigor contracture was prevented by exposure to the F-ATP synthase inhibitor oligomycin, emphasizing the power of F-ATP synthase as an ATP consumer.31,32
Decreased flow of electrons along the respiratory chain promotes the partial reduction of oxygen, increasing ROS production. Complete reduction of dioxygen to H2O requires four electrons, a process occurring sequentially since oxygen accepts one electron at a time. Consequently, oxygen reduction inevitably implies the formation of partially reduced intermediates. The large majority of oxygen delivered to mitochondria is fully reduced to water at the level of complex IV. In this terminal step of the mitochondrial respiratory chain, oxygen is reduced sequentially, yet reaction intermediates remain bound to complex IV so that only the final product, i.e. water, is released. However, electrons flowing through the respiratory chain can be donated to oxygen at other sites where reduction is not complete resulting in ROS formation.33 Very significant amounts of ROS are also produced within mitochondria at sites other than the IMM such as monoamine oxidase in the OMM and p66Shc in the intermembrane space.34 In brain mitochondria the high rate of H2O2 formation from the respiratory chain elicited by the complex III inhibitor antimycin A, is still much lower than that originating from activity of monoamine oxidase.35,36
ROS generated within mitochondria may serve a function in signaling processes that are crucial for the optimal response to physiological and pathological stimuli.37–39 Most of the effects elicited by ROS can be explained by post-translational modifications of proteins, especially at the level of Cys residues40 that might impact also on protein (de)phosphorylation by modulating protein kinases41 and phosphatases.42 On the other hand, ROS can become detrimental, in a transition that has been dubbed mitohormesis.43,44 There is little doubt that formation of ROS can determine mitochondrial dysfunction, and accelerate evolution of cell injury towards necrosis and/or apoptosis;45,46 and that ROS formation is an essential element favoring onset of the permeability transition.47
The mitochondrial permeability transition pore
The PTP of mammalian mitochondria is an IMM channel with a radius of about 1.4 nm48 and a conductance ranging between 0.9 and 1.5 nS in symmetrical 150 mM KCl.49,50 It frequently enters a half-conductance substate that may be due to its dimeric composition,51–53 which will be further discussed below (see also a recent review by Szabò and Zoratti).54 Occurrence of mitochondrial permeabilization and its inhibition by adenine nucleotides had been appreciated since the early 1950s,55,56 but the potential relevance of this event to heart pathophysiology was first proposed by Haworth and Hunter,57–60 who coined the term permeability transition and provided the unique insight that it could be due to reversible opening of a proteinaceous pore of the IMM. The nature of the PTP has remained a mystery for 60 years.61 Theories about pore formation by the IMM adenine nucleotide translocator and Pi carrier, the OMM VDAC and TSPO and by a variety of additional proteins did not stand the test of genetic inactivation of each of these putative pore component(s) and will not be covered here, but for discussion the reader is referred to a recent review where the importance of the pore as a target for cardioprotection is also addressed.47
A turning point of research on the PTP was the discovery that the pore can be inhibited by the immunosuppressant drug CsA62,63 through its binding to CyPD,64 a matrix protein that like all CyPs has peptidyl-prolyl cis-trans isomerase activity that assists in protein folding.65,66 Through the use of CsA the role of the PTP in cardiac pathophysiology could be established in cellular models,67 in hearts subjected to ischemia-reperfusion injury68 and in a pilot study in infarcted patients.69 It should be stressed that CsA is not a blocker of the PTP, nor an inhibitor in the strict sense of the word. Indeed, at higher Ca2+ loads and/or higher concentrations of inducers the permeability transition can still occur in the presence of CsA,70 and a more appropriate term to describe the effects of CsA on the pore is desensitization.71 This key point was firmly established by the characterization of mitochondria from CyPD-null mice, which like CsA-treated wild-type mitochondria underwent PTP opening when the matrix Ca2+ load was increased72–75 and possessed a channel with electrophysiological features identical to those found in wild-type mice.76
Our recent identification of the F-ATP synthase complex as the likely channel-forming component of the PTP51 was made possible by two sets of critical observations. The first was that CyPD interacts with the F-ATP synthase, as shown by blue-native gel analysis and co-immunoprecipitation.77 Binding occurred at the lateral stalk of the complex, was favored by Pi (which promotes the permeability transition in mammalian mitochondria) and was counteracted by CsA with matching effects on the catalytic activity. Indeed, Pi-dependent CyPD binding decreased the catalytic activity of F-ATP synthase by about 30%, and the enzyme was fully reactivated by CsA-induced detachment of CyPD.77 It is of note that the interactions with the PTP are affected by posttranslational modifications (PTM) of CyPD including phosphorylation by GSK3β,78,79 acetylation80 and S-nitrosylation81.
The second insight was the identification of subunit OSCP as the binding partner of CyPD,51 and the observation by Glick and coworkers that OSCP is also the binding site of the F-ATP synthase inhibitor Bz-423.82 Following the demonstration that Bz-423 is a PTP inducer,51 we could show that purified dimers of F-ATP synthase form channels activated by Ca2+, Bz-423 and oxidative stress, and inhibited by Mg2+/ADP in preparations from bovine hearts,51 S. cerevisiae52 and D. melanogaster.53 Channel formation by F-ATP synthase was also observed by Alavian et al.,83 and a role for the enzyme is also supported by a study where translation of the c subunit was partially inhibited by siRNAs resulting in PTP inactivation.84
The mechanism through which the F-ATP synthase forms the PTP is the subject of intense investigation.47 We have proposed that the channel forms from dimers of F-ATP synthase after a conformational change that would follow replacement of Mg2+ with Ca2+ at the catalytic site, which is consistent with earlier literature on the F-ATP synthase. Indeed, when Ca2+ replaces Mg2+ ATP hydrolysis is not coupled to formation of a H+ gradient,85,86 suggesting that a conformational change takes place that leads to the apparent uncoupling of chemical catalysis from H+ transport. We suspect that the conformational change actually leads to PTP opening, and thus that the lack of measurable H+ translocation is due to H+ backflow through the channel. In mammalian mitochondria, binding of CyPD to OSCP would cause a conformational change affecting the accessibility of Me2+ to the catalytic binding sites, resulting in onset of the permeability transition if matrix [Ca2+] is sufficiently high. The conformational change would be independently favored by ROS-dependent thiol oxidation and counteracted by thiol reduction, making replacement of Mg2+ with Ca2+ possible also in mitochondria lacking CyPD. Once the conformational change has occurred, permeation would take place at the interface between dimers, consistent with the effect of genetic ablation of the e and g subunits in yeast.52 The F-ATP synthase would then return to its basal coupled state when the catalytic site is reoccupied by Mg2+, consistent with the full reversibility of the permeability transition in populations of isolated mitochondria87 as well as in individual organelles20,21 and in intact cells.22 If the opening is long-lasting, however, a set of events takes place (ATP depletion, loss of substrates and pyridine nucleotides, matrix swelling causing IMM remodeling and eventually OMM rupture, release of cytochrome c and other proapoptotic factors) that critically contributes to cell death.
Regulation of the PTP
Ca2+-dependent channel formation under condition of oxidative stress (thiol oxidation or cross-linking) and its inhibition by Mg2+, adenine nucleotides and acidic pH appear to be general features of F-ATP synthases, which have so far been detected in mitochondria from B. taurus, S. cerevisiae and D. melanogaster.51–53 On the other hand, marked differences exist in channel unit conductance, sensitivity to Pi, presence of a mitochondrial CyP and inhibition by CsA. In bilayer experiments with purified dimers of the F-ATP synthase, channel conductance was 53 pS in Drosophila, 300 pS in yeast and 500 pS in mammals, suggesting structural differences in the channel-forming subunit(s) of the enzyme.51–53 PTP opening is strongly favored by Pi in mammalian mitochondria (in spite of the Pi-dependent decrease of free matrix [Ca2+])88 while it is inhibited by Pi in yeast52,89,90 and Drosophila.53,91 Mammalian and S. cerevisiae mitochondria possess matrix CyPs (CyPD and CPR3, respectively) yet the PTP is inhibited by CsA only in mammalian mitochondria. On the other hand, Drosophila does not possess a mitochondrial CyP, yet expression of human CyPD in Drosophila S2R+ cells sensitizes the PTP to Ca2+ in a process that is insensitive to CsA.53 These studies suggest that regulatory interactions between CyPD and the F-ATP synthase only emerged in mammals. This interaction may also contribute to explain the unique inducing effects of Pi in mammalian mitochondria, which could (in part at least) be a consequence of increased binding of CyPD to the F-ATP synthase.77
Studies largely carried out in the 1990s defined key mechanisms of PTP modulation, and critical residues involved in its response to pathophysiological effectors. Pore opening requires the “permissive” presence of matrix Ca2+ which acts through a site that can bind other Me2+ ions like Mg2+, Sr2+ and Mn2+, resulting in PTP inhibition.92 A second, external binding site for Me2+ has been defined, whose occupancy results in PTP inhibition even when Ca2+ is bound.92 Pore opening is strongly promoted by oxidation of pyridine nucleotides and of matrix dithiols at discrete sites.93,94 These PTP-regulating dithiol-disulfide interconversions can be blocked by 1-chloro-2,4-dinitrobenzene and by monofunctional thiol reagents like N-ethylmaleimide and monobromobimane.95,96 A second class of regulatory thiols has been identified based on the effects of the impermeant oxidant copper-o-phenanthroline, which promotes PTP opening in a process that, unlike that mediated by matrix Cys residues, cannot be inhibited by monobromobimane.94,97 This site is located on the outer surface of the IMM, since mitoplasts displayed an identical inducing effect of copper-o-phenanthroline as intact mitochondria.98 The permeability transition is strongly affected by matrix pH. In deenergized mitochondria the pH optimum for PTP opening is 7.4, while the open probability decreases sharply below pH 7.4 through reversible protonation of His residues that can be prevented by diethylpyrocarbonate.99,100 The relevant His residues are not located on CyPD because PTP modulation by matrix pH is indistinguishable in mitochondria from CyPD-null and wild type animals.101 It should be kept in mind that in energized mitochondria an acidic pH can promote rather than inhibit PTP opening due to an increased rate of Pi uptake, an effect that could worsen PTP-dependent tissue damage in ischemic and postischemic acidosis.102 Finally, the PTP is affected by the transmembrane voltage, in the sense that depolarization increases the probability of pore opening95,103,104 in a process where Arg residues may play a critical role.105–107
Given that F-ATP synthase forms channels that are indistinguishable from the bona fide PTP, it may be useful to briefly present the structural features of the heart F-ATP synthase before discussing potential points of regulation, and residues where the effectors described above may exert their effects.
Structure of heart F-ATP synthase
The mitochondrial FOF1 ATP synthase (F-ATP synthase or complex V of the oxidative phosphorylation system) is a well conserved multi-subunit complex located in the IMM108,109 that is responsible for the majority (over 90%) of the aerobic ATP production in the heart. This large complex is a molecular motor and normally catalyzes the synthesis of ATP from ADP and Pi using the transmembrane proton-motive force generated by respiration. As mentioned in the Introduction, however, the complex is an ancestral ATPase that can act as an ATP-consuming device when the proton gradient is dissipated.110
Heart mitochondria of mammals contain high amounts of F-ATP synthase, which has been estimated to be 0.38–0.45 nmol/mg protein,111 and have been widely used as enzyme source for structural and functional studies. The complex is composed by a soluble catalytic F1 subcomplex protruding in the mitochondrial matrix and by a membrane-embedded FO subcomplex through which the protons flow (Figure 1, where the complex is shown in its physiological dimeric form). These subcomplexes are connected by central and peripheral stalks. The whole complex has a molecular mass that varies between 540 and 585 kDa depending on the source.112 Numerous atomic structures have revealed that F1 comprises three copies of each of the nucleotide-binding subunits α and β, which alternate around the central α-helical coiled coil of the γ subunit.113–117 Together with the F1 subunits δ and ε, subunit γ forms the central stalk, which is connected to the FO c-ring structure formed by 8 copies of subunit c (Figure 1) whose central region is probably occupied by phospholipids.118,119 The remaining FO part consists of the subunits abdefg(A6L)F6, two subunits of which - a and A6L - are encoded by mitochondrial DNA. The remaining FO and F1 subunits are encoded by nuclear genes, and complex assembly is a stepwise process assisted by several factors, which are still being actively investigated.120
Figure 1. Structure of bovine F-ATP synthase.
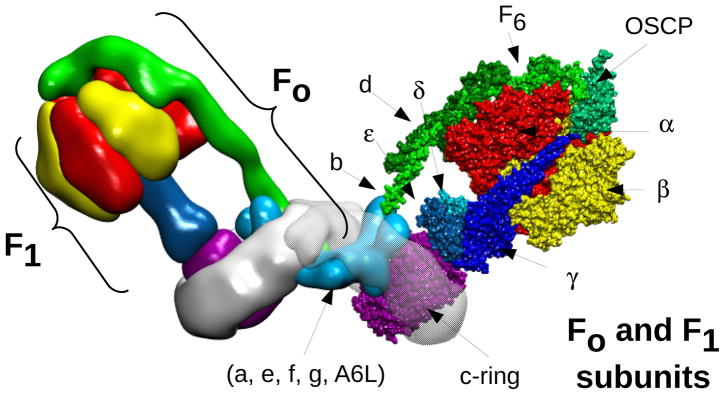
Left monomer, the F1 and Fo sectors are highlighted. Right monomer, the F1 and Fo subunits are shown. In the F1 sector the front α and β subunits have been removed to reveal the F1-rotor (central stalk). The F1 α and β subunits are colored in red and yellow, respectively. The F1-rotor γ, δ and ε subunits are colored in shades of blue, the peripheral stalk subunits b, d, F6 and OSCP in shades of green, and the c-ring in purple. The remaining Fo subunits a, e, f, g, A6L, whose structure has not been defined yet, are located in the Fo subcomplex colored in light blue. The image (lateral view) has been built starting from the yeast dimer molecular model133 (PDB id. 4b2q) and superimposing the cryo-electron microscopy map of bovine F-ATP synthase141 (EMD id. EMD-2091). The fit of molecular models to cryo-electron microscopy map was performed using the program ADP_EM198. The molecular model for bovine F-ATP synthase was obtained by superimposing the 3D structure of the bovine F1-c-ring complex (PDB id. 2xnd) onto each corresponding monomer of the yeast dimer. The superposition was performed using the swiss pdb viewer routine Iterative magic fit199. The lateral stalk was taken from the yeast dimer (PDB id. 4b2q) which contains the bovine subunits.
The structure and arrangement of the membrane subunits has not been defined yet except for subunit a, which associates with the c-ring peripherally, possibly forming an angle of approximately 70° relative to the c-ring helices, as recently resolved in the green alga Polytomella.121 Subunit a provides two half transmembrane water channels for H+ to access the conserved carboxylic residues present in each c subunit. Indeed, proton translocation into the mitochondrial matrix through these (half) channels is concurrent with the clockwise rotation of the FO c-ring (when viewed from the matrix side), which drives γ subunit rotation within the α3β3 F1 subcomplex at about 100 revolutions per second. Rotation of subunit γ takes each of the 3 catalytic sites through at least 3 major functional states denoted as βE, βDP and βTP, thereby supporting the synthesis of 3 ATP molecules for each 360° rotation.109,122,123 When the enzyme works in the direction of ATP hydrolysis, the counterclockwise rotation of the γ subunit and of the c-ring cause proton traslocation into the intermembrane space. This activity is selectively inhibited by reversible binding of the endogenous inhibitor factor 1 (IF1) to F1 with a 1:1 stoichiometry (Figure 2), resulting in full inhibition.124,125 Accordingly, the ability of IF1 to block the ATP-driven rotation of human F1 has been recently visualized126. The peripheral stalk, comprising the FO subunits oligomycin sensitivity conferring protein (OSCP), b, d and F6, is important to prevent the co-rotation of α and β subunits with γ and therefore the loss of enzyme efficiency.127 As already mentioned, this subcomplex is also the target of CyPD, which binds to the OSCP subunit (Figure 2), possibly sharing a common binding site with the F-ATP synthase inhibitor Bz-423.128,129 CyPD binding to OSCP is favored by Pi and counteracted by CsA;77 CyPD sensitizes the PTP to Ca2+, promoting the transition of the F-ATP synthase to a channel that may form at the interface between monomers51–53,61 (Figure 2). Interestingly, OSCP has also been recently recognized as the binding site of 17β-estradiol, whose binding promotes an intrinsically uncoupled state of ATP synthase,130 as well as of regulatory proteins, such as Sirtuin3,131 highlighting its ability to sense metabolic signals.
Figure 2. Interactors of F-ATP synthase and PTP formation.

Reversible binding of CyPD and IF1 to F-ATP synthase is shown, together with the putative region of channel formation (broken arrow). Factors promoting binding/release of interactors are also indicated.
Electron cryomicroscopy of mitochondria from different sources132 clearly revealed that F-ATP synthase is organized in dimers, which are essential to maintain a high local curvature of the IMM133 and normal cristae morphology,134 and form long rows of oligomers at the cristae edges of mitochondria. An additional proposed role of F-ATP synthase dimers/oligomers is higher catalytic efficiency.133,135 Biochemical and genetic studies provided evidence that, for dimer formation, preferential interactions occur between FO subunits a,136 b,137 e,138,139 and g,140 forcing the peripheral stalks (which are turned away from each other) and the two F1 heads at fixed angles of > 70°.133,141 In mammals the dimers have been reported to be further stabilized by interaction with IF1 favoring ATP synthesis in cells overexpressing IF1,142,143 but this proposal has been criticized based on biochemical studies in isolated ox heart membranes,144 as well as on the lack of effect on dimer stability of IF1 silencing in human osteosarcoma cells.145 Moreover, dimers seem to be stabilized by binding of the matrix metalloprotein Factor B (or subunit s of F-ATP synthase),146 which gets in contact with the e and g subunits as well as with the ADP/ATP carrier. The existence of a second interface (the oligomerization interface) through e/e and g/g interactions originally proposed in yeast is still debated because the distance between dimers appears variable in electron cryotomography of mitochondria from different sources, which would make direct protein contacts difficult.132 An interesting aspect of the dimeric/oligomeric structure is that the stabilizing contribution of the different subunits seems to be additive. Indeed, mutants lacking one or more of the above mentioned subunits, such as ρ0 cells (which lack mitochondrial DNA147 and therefore do not synthesize subunits a and A6L)136 and human cells totally or partially depleted of e and g subunits,148 still form lower amounts of F-ATP synthase dimers and oligomers as detected by native gel electrophoresis.136
Catalysis requires the binding of the nucleotide as a complex with Mg2+,149 which plays a key role in shaping the high affinity catalytic sites located in the β subunits, where Mg2+ is hexa-coordinated by βThr163 (bovine numbering), by the oxygen atoms βO2 and γO2 of ATP, and by 3 ordered water molecules (hydrogen-bonded to βArg189, βGlu192 and βAsp256)117 (Figure 3). Mg2+ can be replaced by other divalent cations, including Ca2+, and the ionic radius is the chief determinant of the ability to support catalysis.150 However, Ca2+ ions support γ subunit rotation and ATP hydrolysis, but not H+ translocation and ATP synthesis, as documented both in bacteria85 and in mammals,86 strongly suggesting that the catalytic site has a different conformation state when it is occupied by Ca2+.
Figure 3. Map of Mg2+/Ca2+ binding sites and Ca2+-dependent interactions in bovine F-ATP synthase.

Left monomer, the subunits involved in Ca2+-dependent interactions are highlighted, i.e. subunits α and β, which interact with the matrix protein S100A1154 (not shown in the picture), and the Fo region containing subunit e, which may interact with a hypothetical tropomyosin-like protein localized in the intermembrane space155. Ca2+-regulatory sites located in the c-ring are also shown156,157. Right monomer, the residues (T163, R189, E192, D256) of β subunit interacting with the catalytic metal ions are mapped onto the 3D structure of the bovine F1-c-ring complex.
F-ATP synthase and PTP formation in the heart
The mechanism of PTP formation from F-ATP synthase remains unsolved and is the matter of considerable debate47. It is clear, however, that all PTP effectors will eventually have to be assigned to specific sites of the F-ATP synthase. What follows is therefore a discussion of potential targets of well-characterized pore effectors in heart F-ATP synthase, and of PTM that may affect or cause the transition to the PTP.
The PT has been detected in ρ0 cells lacking subunit a and A6L,151 while it was markedly inhibited in yeast mutants lacking subunits e and/or g,52 suggesting a different contribution of the FO subunits in forming the PTP. Based on the observation that PTP has been conserved from yeast to Drosophila and mammals,152 it seems reasonable to suggest that the conserved subunits b, e and g of the Fo subcomplex, which mediate the dimer/oligomer formation, may also favor PTP formation in the presence of Ca2+ ions and thiol oxidants, possibly also through other subunits.
In the heart Ca2+ ions are also regulatory elements of F-ATP synthase activity, which must match ATP utilization for muscle contraction, whose rate and force can vary in vivo up to 5–10 fold.124 Indeed, it is now well established that F-ATP synthase does not respond simply to the bioenergetic parameters, i.e. to the levels of ATP, ADP, Pi and proton motive force, and reversible and rapid stimulation of F-ATP synthase by physiological intramitochondrial Ca2+ levels has been clearly demonstrated in isolated pig heart mitochondria14 and cultured rat cardiomyocytes.153 These changes were parallel to stimulation of the Ca2+-sensitive dehydrogenases of the Krebs cycle and rapid enough (100 ms) to potentially support steep changes in myocardial workload.14 Intriguingly, the ability to up-regulate the F-ATP synthase is lost in hypertension and hyperthyroidism,124 although its relationship to the genesis of these disease states is not clear. Although a direct allosteric mechanisms mediated by Ca2+ ions was ruled out already in the 1990s,124 how Ca2+ modulates the F-ATP synthase activity has not been established yet.
In cardiomyocytes a Ca2+-dependent interaction of F1 with the S100A1 protein, which is expressed predominantly in cardiac muscle, has been reported to lead to increased ATP production.154 The FO subunit e has been hypothesized to contain a putative Ca2+-dependent activating region exposed at the cytosolic site of FO sector, potentially ensuring the rapid decoding of increases of cytosolic [Ca2+].155 Indeed, residues 34–56 of e subunit are highly homologous to the Ca2+-dependent tropomyosin-binding region for troponin T, and their interaction with a specific antibody mediates an increase of F-ATP synthase activity. Due to exposure of the Ca2+-binding site to the mitochondrial intermembrane space, subunit e may be also involved in the Ca2+-dependent inhibition of PTP opening92. Subunit e is important for F-ATP synthase dimer formation,138,139 suggesting that Ca2+ inhibition could target the dimer structure and function (Figure 3).
Another conserved Ca2+ binding site has been described in the N-terminus of FO subunit c,156,157 which could also represent a candidate for PTP inhibition by Ca2+ in the intermembrane space. Ca2+ binding to subunit c purified from neuronal plasma membrane and reconstituted in artificial membranes was able to block monovalent cation currents157, and Ca2+ binding to subunit c inhibited H+ translocation in bacteria and chloroplasts,158 suggesting that Ca2+ could alter the c ring conformation. However, while the Ca2+-binding capacity of subunit c from bacteria and chloroplasts is well established, that of the mammalian subunit c is debated.158 These data emphasize the complexity of the Ca2+-dependent modulation of F-ATP synthase (Figure 3), and the need for further studies to clarify how different activating signals may be integrated in the normal heart and in the transition of F-ATP synthase to PTP formation.
We have already mentioned that when the proton motive force declines F-ATP synthase reverses to a proton-pumping ATPase, and that this event plays a major role in I/R injury of the heart. It is worth mentioning that this condition may also apply to mitochondrial diseases. 120,159 Interestingly, we observed that the sense of rotation of F-ATP synthase strongly affects the threshold of Ca2+ required for PTP opening in intact mitochondria.51,53 Because continuous ATP hydrolysis markedly reduces the Ca2+ sensitivity of PTP, the inverse rotation of F-ATP synthase during ischemia might contribute to maintain a closed pore. A tentative explanation is that rotation during ATP hydrolysis might destabilize the dimers by pulling the stators apart and vice versa, as proposed Buzhynskyy et al., who demonstrated the existence of two classes of dimers characterized by different stalk-to-stalk distance in native IMM.160
It is well established that heart mitochondria of both slow- and fast-beating species possess sufficiently high levels of IF1 relative to the F-ATP synthase to completely inhibit ATPase activity with an effect equivalent to the action of oligomycin.124 Consistently, both in vitro and in vivo experimental models demonstrated that during ischemia the ATPase activity is (at least partially) inhibited by the reversible binding of IF1, thus mitigating ATP depletion. Indeed, in ischemic hearts of dog,161 goat162 and rabbit163 the loss of ATPase activity correlates with an increased IF1 content. Down-regulation of F-ATP synthase activity is also observed in anoxic cardiomyocytes, although the IF1 contents were not measured.153 In HeLa and C2C12 cells transient IF1 overexpression preserves ATP and protects from cell death induced by oxygen and glucose deprivation, a finding that would enlist IF1 in prosurvival proteins.143 Nevertheless, the response to cell injury seems to depend on the cell type, as IF1 content varies between species, tissues, and even between cell types within a given tissue, such as in central nervous system where neurons contain much more IF1 than astrocytes.125 However, it is not yet clear how IF1 expression is modulated. Two modulators seem to be implicated, i.e. hypoxia inducible factor 1α and the immediate early response gene X-1, the latter targeting IF1 for degradation.164,165
IF1-F1 interactions are governed by the factors promoting IF1 release and rebinding, i.e. membrane potential and MgATP. The existence of a dynamic equilibrium, in which binding and release occur simultaneously involving the entire population of F-ATP synthase, has been demonstrated in energized ox heart mitochondria.124 In addition, IF1 binding is strongly favored by low pH when the inhibitory dimeric IF1 is formed166 so that in ischemia, when pH and membrane potential decrease, a new dynamic equilibrium is reached. In principle, the binding of IF1 might also contribute to decrease the open probability of PTP during ischemia (Figure 2). Indeed, the crystal structure of the IF1-F1 complex from ox heart has shown that the initial low-affinity binding of IF1 to the F1 surface is followed by the final inhibitory binding in response to ATP hydrolysis, so that two Mg2+-ADP molecules are contained in the F1 catalytic sites of βDP and βTP.167 Considering that Mg2+-ADP is a PTP inhibitor, it is conceivable that the final inhibited IF1-F1 complex may be unable to properly bind Ca2+, which is necessary for the switch of F-ATP synthase to the PTP. Intriguingly, the His reagent diethylpyrocarbonate, which restores the ability of mammalian mitochondria to undergo PTP opening at low matrix pH,100,168 also prevents IF1 binding at acidic pH.169 On the other hand, the inhibited IF1-F1 complex does not contain Pi,167 which is a PTP inhibitor at low concentrations, suggesting that IF1 binding might mask an inhibitory site favoring, rather than preventing, PTP opening.
Although direct modulation of PTP by IF1 remains an intriguing possibility that should be further explored, IF1 could also affect PTP formation as a consequence of its role in ischemic preconditioning. It is well established that massive PTP opening - which occurs during reperfusion when membrane potential is restored and Ca2+ overload occurs - is prevented by short ischemic periods before long-lasting ischemia and reperfusion, a phenomenon called ischemic preconditioning. During ischemic,162,170 as well pharmacological preconditioning,171,172 IF1 seems to mediate a peculiar, long-lasting ATPase inhibition in dog, goat and rat hearts (until 30–120 min of reperfusion depending on the species or the experimental model), although contradictory results have been published, which could be explained by the different analytic methods used173. As preconditioning results in sparing of ATP, lowering of membrane potential and Ca2+ accumulation, it may appear that IF1 favors conditions that inhibit PTP formation during reperfusion. Moreover, based on the observation that the lack of IF1 in Luft’s disease is associated with uncoupled mitochondria,174 long-lasting IF1 binding during ischemia might maintain tight coupling supporting oxidative phosphorylation during reperfusion. Clarification of the molecular events involving IF1 in preconditioning hold great promise for understanding preconditioning and for devising pharmacological strategies for cardioprotection.
Post-translational modifications of F-ATP synthase and PTP formation
Until recently, there were few reports regarding the PTMs of F-ATP synthase, which have been associated with specific biological function/processes in a very limited number of cases. Considerable information has been obtained about oxidative modifications of F-ATP synthase. Experiments with isolated mitochondria from bovine heart demonstrated that F-ATP synthase contains a set of thiols located in FO (Figure 4), whose environment senses membrane energization and whose oxidation results in complete and reversible mitochondrial uncoupling.175,176 Because uncoupling was not inhibited by oligomycin, the Authors proposed that the permeability pathway is located on the intermembrane side of the oligomycin binding site.175 This position would suggest involvement of the unique Cys residue of subunit c, which is located near the Glu58 residue essential for proton translocation.118 It is tempting to speculate that oxidation of this Cys residue may favor PTP formation in another part of FO. This does not exclude the involvement of species-specific Cys residues located in other subunits, such as subunit b in bovine and human heart (Figure 4), whose modification in the bovine enzyme also affects catalytic function.177 In animal F-ATP synthase Factor B contains two vicinal Cys residues, whose oxidation leads to mitochondrial uncoupling.178 However, Factor B is not present in yeast and Drosophila melanogaster, where the PTP is also modulated by SH reagents,52,53,91 thus suggesting that Factor B is probably not involved in PTP formation. We showed that isolated F1 from bovine heart F-ATP synthase is selectively inactivated by hydrogen peroxide through redox-active iron-protein adducts probably generating highly reactive oxygen species in proximity of the catalytic nucleotide binding sites.179 Due to their conservation it is tempting to hypothesize that also these sites may be candidates for ROS-mediated PTP modulation.
Figure 4. Map of Cys residues in bovine heart F-ATP synthase.

Position of Cys residues on the specified subunits (red dots) is mapped onto the 3D structure of the bovine F1-c-ring complex and of the bovine lateral stalk.
Information on oxidative modifications of F-ATP synthase subunits has widely increased with the development of mass spectrometry-based methods, which demonstrated that F-ATP synthase is susceptible to oxidative/nitrative stress associated with heart failure,180 CNS disorders,181,182 caloric restriction183 and aging.184,185 Specifically, the α subunit is S-nitrosylated in response to GSNO treatment of a mouse heart membrane fraction.186 Moreover, in the same subunit Tyr nitration was increased following I/R of mouse heart187 (Figure 5), suggesting that α subunit is actively involved in various oxidative modifications. Consistently, the formation of an inter-subunit disulfide bridge between αC251 and γC78 has been observed in canine dyssynchronous heart failure, which reverted following cardiac resynchronization therapy,188 and disulfide formation was proposed to be involved in PTP activation.95 However, these residues are quite distant in the assembled complex (Figure 4) suggesting that the disulfide may only form in a misfolded/aggregated enzyme. Interestingly, in mitochondria from aging cardiomyocytes, which are more prone to undergo the PT during reperfusion in spite of an impaired ability to accumulate Ca2+ ions during ischemia and reperfusion, quantitative proteomics revealed increased Cys oxidation at several subunits, including OSCP and d, which might potentially affect CyPD binding to the peripheral stalk and/or PTP formation.189 Moreover, a pharmaco-proteomic approach demonstrated that myocardial infarction in dogs caused several nitric oxide-related chemical modifications of d subunit, i.e., nitration of Y114 and hydroxylation of K71 (numbering according to PDB id 4b2q), followed by its myristoylation.190 These PTMs were abolished by the cardioprotective drug valsartan, which also induced phosphorylation of S29 (numbering according to PDB id 4b2q) of the same subunit (Figure 5). Intriguingly, δPKC, which plays key roles in I/R, is a modulator of F-ATP synthase in cardiomyocites through direct binding to subunit d, which requires cardiolipin191. Subunit d also contains three Trp residues, which have been classified as “hot spots” for oxidation in human heart being found in normoxic conditions192 (Figure 5). These data suggest subunit d as actively involved in various oxidative modifications, whose implication in PTP formation remains to be elucidated.
Figure 5. Posttranslational modifications of F-ATP synthase in the normal and failing heart.
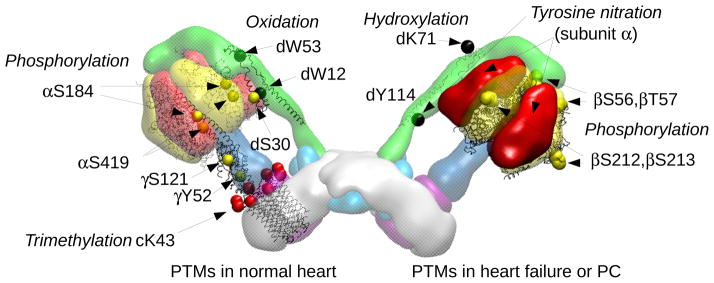
Left monomer, the residues involved in PTMs in normal heart are shown, i.e. complete trimethylation of cK43118; oxidation of W12, W53 of subunit d192; phosphorylation of αS184, αS419,194 γY52195 and γS121,196 dS30.193 Right monomer, residues involved in PTMs associated with preconditioning or heart failure are shown, i.e. phosphorylation of βS56, βT57, βT212, βS213197; nitration of d Y114 (numbering according to PDB id 4b2q) and hydroxylation of dK71190. Subunit α is also highlighted, being the target of tyrosine nitration187.
The most extensively characterized PTM concern Lys43 of the c subunit, which is completely trimethylated in all vertebrates and in the majority of invertebrates118,119 (Figure 5). These residues are located in the loops exposed to the matrix and possibly provide sites for the specific binding of cardiolipin. In the normal heart several phosphorylated residues have also been reported, and these are mainly located in the F1 sector193 where they may be functionally relevant. The modified residues include αS184, αS419,194 γY52195 and γS121196 (Figure 5). The β subunit was found to be modified by preconditioning of rabbit myocytes with adenosine, and contained at least 5 phosphorylated residues, 3 of which (βS56, βT57, and βT212 or βS213) are accessible and therefore potentially regulated197 (Figure 5).
Conclusions
Identification of the F-ATP synthase as the most likely channel-forming element of the PTP51–53,83 has been a turning point for our understanding of mitochondrial pathophysiology. Conservation of the channel-forming ability across species points to a conserved function, and species-specific differences in conductance and regulation suggest an evolutionary role that still needs to be addressed.51–53 We are aware that a coherent picture explaining the mechanisms through which the energy-conserving enzyme turns into an energy-dissipating device is not possible at present, but we hope that this review will help address the many open mechanistic issues and inspire the molecular analysis of appropriate F-ATP synthase mutants.
Supplementary Material
Acknowledgments
Sources of funding
Research in our laboratories is supported by AIRC, Telethon, the Italian Ministry of the University, NIH-PHS, the University of Padova and Fondazione Cassa di Risparmio di Padova e Rovigo.
Non-standard abbreviations and acronyms
- CsA
cyclosporin A
- CyP
cyclophilin
- IF1
inhibitor factor 1
- IMM
inner mitochondrial membrane
- I/R
ischemia/reperfusion
- MCU
mitochondrial Ca2+ uniporter
- OMM
outer mitochondrial membrane
- OSCP
oligomycin sensitivity conferring protein
- PTM
posttranslational modifications
- PTP
permeability transition pore
- ROS
reactive oxygen species
- TSPO
translocator protein of 18 kDa (TSPO)
- VDAC
voltage-dependent anion channel
Footnotes
Disclosures
None
In March 2015, the average time from submission to first decision for all original research papers submitted to Circulation Research was 12.68 days.
Contributor Information
Paolo Bernardi, Department of Biomedical Sciences, University of Padova, Italy.
Fabio Di Lisa, Department of Biomedical Sciences, University of Padova, Italy.
Federico Fogolari, Department of Medical and Biological Sciences, University of Udine, Italy.
Giovanna Lippe, Department of Food Science, University of Udine, Italy.
References
- 1.Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am J Physiol Cell Physiol. 2007;292:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- 2.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 3.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9:971–988. doi: 10.1038/nrd3295. [DOI] [PubMed] [Google Scholar]
- 7.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 8.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 9.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Stefani D, Raffaello A, Teardo E, Szabó I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crompton M, Heid I. The cycling of calcium, sodium, and protons across the inner membrane of cardiac mitochondria. Eur J Biochem. 1978;91:599–608. doi: 10.1111/j.1432-1033.1978.tb12713.x. [DOI] [PubMed] [Google Scholar]
- 12.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 13.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH and light scattering. J Biol Chem. 2001;276:2586–2599. doi: 10.1074/jbc.M002923200. [DOI] [PubMed] [Google Scholar]
- 15.Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20:4998–5007. doi: 10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kettlewell S, Cabrero P, Nicklin SA, Dow JA, Davies S, Smith GL. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J Mol Cell Cardiol. 2009;46:891–901. doi: 10.1016/j.yjmcc.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 17.Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca2+] in cardiac myocytes during sarcoplasmic reticulum Ca2+ release. Circ Res. 2013;112:424–431. doi: 10.1161/CIRCRESAHA.111.300501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46:767–774. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 20.Hüser J, Blatter LA. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J. 1999;343(Pt 2):311–317. [PMC free article] [PubMed] [Google Scholar]
- 21.Hüser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in single mitochondria. Biophys J. 1998;74:2129–2137. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem. 2001;276:12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- 24.Altschuld RA, Hohl CM, Castillo LC, Garleb AA, Starling RC, Brierley GP. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am J Physiol. 1992;262:H1699–704. doi: 10.1152/ajpheart.1992.262.6.H1699. [DOI] [PubMed] [Google Scholar]
- 25.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernardi P, von Stockum S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium. 2012;52:22–27. doi: 10.1016/j.ceca.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams GS, Boyman L, Chikando AC, Khairallah RJ, Lederer WJ. Mitochondrial calcium uptake. Proc Natl Acad Sci U S A. 2013;110:10479–10486. doi: 10.1073/pnas.1300410110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Lisa F, Bernardi P. Mitochondrial function as a determinant of recovery or death in cell response to injury. Mol Cell Biochem. 1998;184:379–391. [PubMed] [Google Scholar]
- 30.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 31.Rouslin W, Erickson JL, Solaro RJ. Effects of oligomycin and acidosis on rates of ATP depletion in ischemic heart muscle. Am J Physiol. 1986;250:H503–H508. doi: 10.1152/ajpheart.1986.250.3.H503. [DOI] [PubMed] [Google Scholar]
- 32.Jennings RB, Reimer KA. The cell biology of acute myocardial ischemia. Annu Rev Med. 1991;42:225–246. doi: 10.1146/annurev.me.42.020191.001301. [DOI] [PubMed] [Google Scholar]
- 33.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson EJ, Efird JT, Davies SW, O’Neal WT, Darden TM, Thayne KA, Katunga LA, Kindell LC, Ferguson TB, Anderson CA, Chitwood WR, Koutlas TC, Williams JM, Rodriguez E, Kypson AP. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc. 2014;3:e000713. doi: 10.1161/JAHA.113.000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 37.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balaban RS, Nemoto S, Finkel T. Mitochondria, Oxidants, and Aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finkel T. From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci Signal. 2012;5:e10. doi: 10.1126/scisignal.2002943. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Cole PA. Catalytic mechanisms and regulation of protein kinases. Methods Enzymol. 2014;548:1–21. doi: 10.1016/B978-0-12-397918-6.00001-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 43.Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med. 2014;20:709–711. doi: 10.1038/nm.3624. [DOI] [PubMed] [Google Scholar]
- 44.Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19:757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Di Lisa F, Kaludercic N, Carpi A, Menabò R, Giorgio M. Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66Shc and monoamine oxidase. Basic Res Cardiol. 2009;104:131–139. doi: 10.1007/s00395-009-0008-4. [DOI] [PubMed] [Google Scholar]
- 47.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massari S, Azzone GF. The equivalent pore radius of intact and damaged mitochondria and the mechanism of active shrinkage. Biochim Biophys Acta. 1972;283:23–29. doi: 10.1016/0005-2728(72)90094-1. [DOI] [PubMed] [Google Scholar]
- 49.Petronilli V, Szabó I, Zoratti M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett. 1989;259:137–143. doi: 10.1016/0014-5793(89)81513-3. [DOI] [PubMed] [Google Scholar]
- 50.Kinnally KW, Campo ML, Tedeschi H. Mitochondrial channel activity studied by patch-clamping mitoplasts. J Bioenerg Biomembr. 1989;21:497–506. doi: 10.1007/BF00762521. [DOI] [PubMed] [Google Scholar]
- 51.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carraro M, Giorgio V, Šileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabó I, Bernardi P. Channel Formation by Yeast F-ATP Synthase and the Role of Dimerization in the Mitochondrial Permeability Transition. J Biol Chem. 2014;289:15980–15985. doi: 10.1074/jbc.C114.559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.von Stockum S, Giorgio V, Trevisan E, Lippe G, Glick GD, Forte MA, Da-Rè C, Checchetto V, Mazzotta G, Costa R, Szabò I, Bernardi P. F-ATPase of D. melanogaster Forms 53 Picosiemen (53-pS) Channels Responsible for Mitochondrial Ca2+-induced Ca2+ Release. J Biol Chem. 2015;290:4537–4544. doi: 10.1074/jbc.C114.629766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szabó I, Zoratti M. Mitochondrial channels: ion fluxes and more. Physiol Rev. 2014;94:519–608. doi: 10.1152/physrev.00021.2013. [DOI] [PubMed] [Google Scholar]
- 55.Raaflaub J. Die schwellung isolierter leberzell mitochondrien und ihre physikalisch beeinfluβarkeit. Helv Physiol Pharmacol Acta. 1953;11:142–156. [PubMed] [Google Scholar]
- 56.Brenner-Holzach O, Raaflaub J. Die korrelation zwischen der schwellung isolierter mitochondrien und dem abbau intramitochondrialen adenosinnucleotide (ATP, ADP, AMP, CoA) Helv Physiol Pharmacol Acta. 1954;12:242–252. [PubMed] [Google Scholar]
- 57.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- 58.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys. 1979;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- 59.Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- 60.Haworth RA, Hunter DR. The Ca2+-induced membrane transition of rat liver mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 61.Bernardi P. The mitochondrial permeability transition pore: A mystery solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 63.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- 64.Halestrap AP, Davidson AM. Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tropschug M, Barthelmess IB, Neupert W. Sensitivity to cyclosporin A is mediated by cyclophilin in Neurospora crassa and Saccharomyces cerevisiae. Nature. 1989;342:953–955. doi: 10.1038/342953a0. [DOI] [PubMed] [Google Scholar]
- 66.Matouschek A, Rospert S, Schmid K, Glick BS, Schatz G. Cyclophilin catalyzes protein folding in yeast mitochondria. Proc Natl Acad Sci U S A. 1995;92:6319–6323. doi: 10.1073/pnas.92.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc Res. 1993;27:1790–1794. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- 68.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 69.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier J-P, Derumeaux G, Ovize M. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. New Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 70.Broekemeier KM, Pfeiffer DR. Cyclosporin A-sensitive and insensitive mechanisms produce the permeability transition in mitochondria. Biochem Biophys Res Commun. 1989;163:561–566. doi: 10.1016/0006-291x(89)92174-8. [DOI] [PubMed] [Google Scholar]
- 71.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 72.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 73.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 74.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 75.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Marchi U, Basso E, Szabó I, Zoratti M. Electrophysiological characterization of the Cyclophilin D-deleted mitochondrial permeability transition pore. Mol Membr Biol. 2006;23:521–530. doi: 10.1080/09687860600907644. [DOI] [PubMed] [Google Scholar]
- 77.Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci U S A. 2010;107:726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J Cell Sci. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson KM, Chen X, Boitano A, Swenson L, Opipari AW, Jr, Glick GD. Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem Biol. 2005;12:485–496. doi: 10.1016/j.chembiol.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 83.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nathanson L, Gromet-Elhanan Z. Mutations in the beta-subunit Thr159 and Glu184 of the Rhodospirillum rubrum F0F1 ATP synthase reveal differences in ligands for the coupled Mg2+- and decoupled Ca2+-dependent F0F1 activities. J Biol Chem. 2000;275:901–905. doi: 10.1074/jbc.275.2.901. [DOI] [PubMed] [Google Scholar]
- 86.Papageorgiou S, Melandri AB, Solaini G. Relevance of divalent cations to ATP-driven proton pumping in beef heart mitochondrial F0F1-ATPase. J Bioenerg Biomembr. 1998;30:533–541. doi: 10.1023/a:1020528432609. [DOI] [PubMed] [Google Scholar]
- 87.Petronilli V, Nicolli A, Costantini P, Colonna R, Bernardi P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim Biophys Acta. 1994;1187:255–259. doi: 10.1016/0005-2728(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 88.Zoccarato F, Nicholls DG. The role of phosphate in the regulation of the independent calcium-efflux pathway of liver mitochondria. Eur J Biochem. 1982;127:333–338. doi: 10.1111/j.1432-1033.1982.tb06875.x. [DOI] [PubMed] [Google Scholar]
- 89.Jung DW, Bradshaw PC, Pfeiffer DR. Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J Biol Chem. 1997;272:21104–21112. doi: 10.1074/jbc.272.34.21104. [DOI] [PubMed] [Google Scholar]
- 90.Yamada A, Yamamoto T, Yoshimura Y, Gouda S, Kawashima S, Yamazaki N, Yamashita K, Kataoka M, Nagata T, Terada H, Pfeiffer DR, Shinohara Y. Ca2+-induced permeability transition can be observed even in yeast mitochondria under optimized experimental conditions. Biochim Biophys Acta. 2009;1787:1486–1491. doi: 10.1016/j.bbabio.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 91.von Stockum S, Basso E, Petronilli V, Sabatelli P, Forte MA, Bernardi P. Properties of Ca2+ Transport in Mitochondria of Drosophila melanogaster. J Biol Chem. 2011;286:41163–41170. doi: 10.1074/jbc.M111.268375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bernardi P, Veronese P, Petronilli V. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J Biol Chem. 1993;268:1005–1010. [PubMed] [Google Scholar]
- 93.Costantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J Biol Chem. 1996;271:6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 94.Costantini P, Colonna R, Bernardi P. Induction of the mitochondrial permeability transition pore by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochim Biophys Acta. 1998;1365:385–392. doi: 10.1016/s0005-2728(98)00090-5. [DOI] [PubMed] [Google Scholar]
- 95.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 96.Costantini P, Chernyak BV, Petronilli V, Bernardi P. Selective inhibition of the mitochondrial permeability transition pore at the oxidation-reduction sensitive dithiol by monobromobimane. FEBS Lett. 1995;362:239–242. doi: 10.1016/0014-5793(95)00256-9. [DOI] [PubMed] [Google Scholar]
- 97.Costantini P, Colonna R, Bernardi P. Induction of the mitochondrial permeability transition by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochim Biophys Acta. 1998;1365:385–392. doi: 10.1016/s0005-2728(98)00090-5. [DOI] [PubMed] [Google Scholar]
- 98.Sileikyte J, Petronilli V, Zulian A, Dabbeni-Sala F, Tognon G, Nikolov P, Bernardi P, Ricchelli F. Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor) J Biol Chem. 2011;286:1046–1053. doi: 10.1074/jbc.M110.172486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem. 1992;267:8834–8839. [PubMed] [Google Scholar]
- 100.Nicolli A, Petronilli V, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry. 1993;32:4461–4465. doi: 10.1021/bi00067a039. [DOI] [PubMed] [Google Scholar]
- 101.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 102.Kristian T, Bernardi P, Siesjö BK. Acidosis promotes the permeability transition in energized mitochondria: implications for reperfusion injury. J Neurotrauma. 2001;18:1059–1074. doi: 10.1089/08977150152693755. [DOI] [PubMed] [Google Scholar]
- 103.Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem. 1992;267:8834–8839. [PubMed] [Google Scholar]
- 104.Petronilli V, Cola C, Massari S, Colonna R, Bernardi P. Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J Biol Chem. 1993;268:21939–21945. [PubMed] [Google Scholar]
- 105.Eriksson O, Fontaine E, Bernardi P. Chemical modification of arginines by 2,3-butanedione and phenylglyoxal causes closure of the mitochondrial permeability transition pore. J Biol Chem. 1998;273:12669–12674. doi: 10.1074/jbc.273.20.12669. [DOI] [PubMed] [Google Scholar]
- 106.Linder MD, Morkunaite-Haimi S, Kinnunen PKJ, Bernardi P, Eriksson O. Ligand-selective modulation of the permeability transition pore by arginine modification. Opposing effects of p-hydroxyphenylglyoxal and phenylglyoxal. J Biol Chem. 2002;277:937–942. doi: 10.1074/jbc.M107610200. [DOI] [PubMed] [Google Scholar]
- 107.Johans M, Milanesi E, Franck M, Johans C, Liobikas J, Panagiotaki M, Greci L, Principato G, Kinnunen PKJ, Bernardi P, Costantini P, Eriksson O. Modification of permeability transition pore arginine(s) by phenylglyoxal derivatives in isolated mitochondria and mammalian cells: Structure-function relationship of arginine ligands. J Biol Chem. 2005;280:12130–12136. doi: 10.1074/jbc.M413454200. [DOI] [PubMed] [Google Scholar]
- 108.Boyer PD. The ATP synthase--a splendid molecular machine. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 109.Futai M, Nakanishi-Matsui M, Okamoto H, Sekiya M, Nakamoto RK. Rotational catalysis in proton pumping ATPases: From E. coli F-ATPase to mammalian V-ATPase. Biochim Biophys Acta. 2012;1817:1711–1721. doi: 10.1016/j.bbabio.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 110.Cross RL, Muller V. The evolution of A-, F-, and V-type ATP synthases and ATPases: reversals in function and changes in the H+/ATP coupling ratio. FEBS Lett. 2004;576:1–4. doi: 10.1016/j.febslet.2004.08.065. [DOI] [PubMed] [Google Scholar]
- 111.Schägger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J Biol Chem. 2001;276:37861–37867. doi: 10.1074/jbc.M106474200. [DOI] [PubMed] [Google Scholar]
- 112.Wittig I, Schägger H. Structural organization of mitochondrial ATP synthase. Biochim Biophys Acta. 2008;1777:592–598. doi: 10.1016/j.bbabio.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 113.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2. 8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 114.Menz RI, Walker JE, Leslie AG. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell. 2001;106:331–341. doi: 10.1016/s0092-8674(01)00452-4. [DOI] [PubMed] [Google Scholar]
- 115.Bowler MW, Montgomery MG, Leslie AG, Walker JE. Ground state structure of F1-ATPase from bovine heart mitochondria at 1. 9 Å resolution. J Biol Chem. 2007;282:14238–14242. doi: 10.1074/jbc.M700203200. [DOI] [PubMed] [Google Scholar]
- 116.Bason JV, Runswick MJ, Fearnley IM, Walker JE. Binding of the inhibitor protein IF1 to bovine F1-ATPase. J Mol Biol. 2011;406:443–453. doi: 10.1016/j.jmb.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rees DM, Montgomery MG, Leslie AG, Walker JE. Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2012;109:11139–11143. doi: 10.1073/pnas.1207587109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Watt IN, Montgomery MG, Runswick MJ, Leslie AG, Walker JE. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci U S A. 2010;107:16823–16827. doi: 10.1073/pnas.1011099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Walpole TB, Palmer DN, Jiang H, Ding S, Fearnley IM, Walker JE. Conservation of complete trimethylation of lysine 43 in the rotor ring of c-subunits of metazoan ATP synthase. Mol Cell Proteomics. 2015 doi: 10.1074/mcp.M114.047456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hejzlarová, Mrácek T, Vrbacky M, Kaplanová V, Karbanová V, Nusková H, Pecina P, Houštek J. Nuclear genetic defects of mitochondrial ATP synthase. Physiol Res. 2014;63(Suppl 1):S57–S71. doi: 10.33549/physiolres.932643. [DOI] [PubMed] [Google Scholar]
- 121.Allegretti M, Klusch N, Mills DJ, Vonck J, Kühlbrandt W, Davies KM. Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature. 2015 doi: 10.1038/nature14185. [DOI] [PubMed] [Google Scholar]
- 122.Noji H, Yasuda R, Yoshida M, Kinosita K., Jr Direct observation of the rotation of F1-ATPase. Nature. 1997;386:299–302. doi: 10.1038/386299a0. [DOI] [PubMed] [Google Scholar]
- 123.Nam K, Pu J, Karplus M. Trapping the ATP binding state leads to a detailed understanding of the F1-ATPase mechanism. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1419486111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Harris DA, Das AM. Control of mitochondrial ATP synthesis in the heart. Biochem J. 1991;280(Pt 3):561–573. doi: 10.1042/bj2800561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF1: setting the pace of the F1Fo-ATP synthase. Trends Biochem Sci. 2009;34:343–350. doi: 10.1016/j.tibs.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 126.Suzuki T, Tanaka K, Wakabayashi C, Saita E, Yoshida M. Chemomechanical coupling of human mitochondrial F1-ATPase motor. Nat Chem Biol. 2014;10:930–936. doi: 10.1038/nchembio.1635. [DOI] [PubMed] [Google Scholar]
- 127.Rees DM, Leslie AG, Walker JE. The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci U S A. 2009;106:21597–21601. doi: 10.1073/pnas.0910365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cleary J, Johnson KM, Opipari AW, Jr, Glick GD. Inhibition of the mitochondrial F1F0-ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg Med Chem Lett. 2007;17:1667–1670. doi: 10.1016/j.bmcl.2006.12.102. [DOI] [PubMed] [Google Scholar]
- 129.Stelzer AC, Frazee RW, Van Huis C, Cleary J, Opipari AW, Jr, Glick GD, Al-Hashimi HM. NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F1F0-ATPase. Biopolymers. 2010;93:85–92. doi: 10.1002/bip.21306. [DOI] [PubMed] [Google Scholar]
- 130.Moreno AJ, Moreira PI, Custodio JB, Santos MS. Mechanism of inhibition of mitochondrial ATP synthase by 17β-estradiol. J Bioenerg Biomembr. 2013;45:261–270. doi: 10.1007/s10863-012-9497-1. [DOI] [PubMed] [Google Scholar]
- 131.Vassilopoulos A, Pennington JD, Andresson T, Rees DM, Bosley AD, Fearnley IM, Ham A, Flynn CR, Hill S, Rose KL, Kim HS, Deng CX, Walker JE, Gius D. SIRT3 deacetylates ATP synthase F1 complex proteins in response to nutrient- and exercise-induced stress. Antioxid Redox Signal. 2014;21:551–564. doi: 10.1089/ars.2013.5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kühlbrandt W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci U S A. 2011;108:14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Davies KM, Anselmi C, Wittig I, Faraldo-Gomez JD, Kühlbrandt W. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci U S A. 2012;109:13602–13607. doi: 10.1073/pnas.1204593109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Habersetzer J, Ziani W, Larrieu I, Stines-Chaumeil C, Giraud MF, Brèthes D, Dautant A, Paumard P. ATP synthase oligomerization: from the enzyme models to the mitochondrial morphology. Int J Biochem Cell Biol. 2013;45:99–105. doi: 10.1016/j.biocel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 135.Bisetto E, Di Pancrazio F, Simula MP, Mavelli I, Lippe G. Mammalian ATPsynthase monomer versus dimer profiled by blue native PAGE and activity stain. Electrophoresis. 2007;28:3178–3185. doi: 10.1002/elps.200700066. [DOI] [PubMed] [Google Scholar]
- 136.Wittig I, Meyer B, Heide H, Steger M, Bleier L, Wumaier Z, Karas M, Schägger H. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim Biophys Acta. 2010;1797:1004–1011. doi: 10.1016/j.bbabio.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 137.Spannagel C, Vaillier J, Arselin G, Graves PV, Grandier-Vazeille X, Velours J. Evidence of a subunit 4 (subunit b) dimer in favor of the proximity of ATP synthase complexes in yeast inner mitochondrial membrane. Biochim Biophys Acta. 1998;1414:260–264. doi: 10.1016/s0005-2736(98)00174-6. [DOI] [PubMed] [Google Scholar]
- 138.Everard-Gigot V, Dunn CD, Dolan BM, Brunner S, Jensen RE, Stuart RA. Functional analysis of subunit e of the F1Fo-ATP synthase of the yeast Saccharomyces cerevisiae: importance of the N-terminal membrane anchor region. Eukaryot Cell. 2005;4:346–355. doi: 10.1128/EC.4.2.346-355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bisetto E, Picotti P, Giorgio V, Alverdi V, Mavelli I, Lippe G. Functional and stoichiometric analysis of subunit e in bovine heart mitochondrial F0F1ATP synthase. J Bioenerg Biomembr. 2008;40:257–267. doi: 10.1007/s10863-008-9183-5. [DOI] [PubMed] [Google Scholar]
- 140.Bustos DM, Velours J. The modification of the conserved GXXXG motif of the membrane-spanning segment of subunit g destabilizes the supramolecular species of yeast ATP synthase. J Biol Chem. 2005;280:29004–29010. doi: 10.1074/jbc.M502140200. [DOI] [PubMed] [Google Scholar]
- 141.Baker LA, Watt IN, Runswick MJ, Walker JE, Rubinstein JL. Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proc Natl Acad Sci U S A. 2012;109:11675–11680. doi: 10.1073/pnas.1204935109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Garcia JJ, Morales-Rios E, Cortés-Hernandez P, Rodriguez-Zavala JS. The inhibitor protein IF1 promotes dimerization of the mitochondrial F1F0-ATP synthase. Biochemistry. 2006;45:12695–12703. doi: 10.1021/bi060339j. [DOI] [PubMed] [Google Scholar]
- 143.Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 144.Tomasetig L, Di Pancrazio F, Harris DA, Mavelli I, Lippe G. Dimerization of F0F1ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim Biophys Acta. 2002;1556:133–141. doi: 10.1016/s0005-2728(02)00344-4. [DOI] [PubMed] [Google Scholar]
- 145.Barbato S, Sgarbi G, Gorini G, Baracca A, Solaini G. The Inhibitor Protein (IF1) of the F1F0-ATPase Modulates Human Osteosarcoma Cell Bioenergetics. J Biol Chem. 2015;290:6338–6348. doi: 10.1074/jbc.M114.631788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lee JK, Belogrudov GI, Stroud RM. Crystal structure of bovine mitochondrial factor B at 0. 96-A resolution. Proc Natl Acad Sci U S A. 2008;105:13379–13384. doi: 10.1073/pnas.0805689105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 148.Habersetzer J, Larrieu I, Priault M, Salin B, Rossignol R, Brèthes D, Paumard P. Human F1F0 ATP synthase, mitochondrial ultrastructure and OXPHOS impairment: a (super-)complex matter? PLoS One. 2013;8:e75429. doi: 10.1371/journal.pone.0075429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Frasch WD. The participation of metals in the mechanism of the F1-ATPase. Biochim Biophys Acta. 2000;1458:310–325. doi: 10.1016/s0005-2728(00)00083-9. [DOI] [PubMed] [Google Scholar]
- 150.Selwyn MJ. Model reaction for mitochondrial adenosine triphosphatase. Nature. 1968;219:490–493. doi: 10.1038/219490a0. [DOI] [PubMed] [Google Scholar]
- 151.Masgras I, Rasola A, Bernardi P. Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochimica et Biophysica Acta. 2012;1817:1860–1866. doi: 10.1016/j.bbabio.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 152.Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Lett. 2010;584:2504–2509. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Das AM, Harris DA. Reversible modulation of the mitochondrial ATP synthase with energy demand in cultured rat cardiomyocytes. FEBS Lett. 1989;256:97–100. doi: 10.1016/0014-5793(89)81725-9. [DOI] [PubMed] [Google Scholar]
- 154.Boerries M, Most P, Gledhill JR, Walker JE, Katus HA, Koch WJ, Aebi U, Schoenenberger CA. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol. 2007;27:4365–4373. doi: 10.1128/MCB.02045-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Arakaki N, Ueyama Y, Hirose M, Himeda T, Shibata H, Futaki S, Kitagawa K, Higuti T. Stoichiometry of subunit e in rat liver mitochondrial H+-ATP synthase and membrane topology of its putative Ca2+-dependent regulatory region. Biochim Biophys Acta. 2001;1504:220–228. doi: 10.1016/s0005-2728(00)00248-6. [DOI] [PubMed] [Google Scholar]
- 156.Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynelä J, Saris N-EL. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium. 2014;55:69–77. doi: 10.1016/j.ceca.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 157.McGeoch JE, McGeoch MW, Mao R, Guidotti G. Opposing actions of cGMP and calcium on the conductance of the F0 subunit c pore. Biochem Biophys Res Commun. 2000;274:835–840. doi: 10.1006/bbrc.2000.3231. [DOI] [PubMed] [Google Scholar]
- 158.Van Walraven HS, Scholts MJ, Zakharov SD, Kraayenhof R, Dilley RA. pH-dependent Ca2+ binding to the F0 c-subunit affects proton translocation of the ATP synthase from Synechocystis 6803. J Bioenerg Biomembr. 2002;34:455–464. doi: 10.1023/a:1022518109371. [DOI] [PubMed] [Google Scholar]
- 159.Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. 2012;35:211–225. doi: 10.1007/s10545-011-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Buzhynskyy N, Sens P, Prima V, Sturgis JN, Scheuring S. Rows of ATP synthase dimers in native mitochondrial inner membranes. Biophys J. 2007;93:2870–2876. doi: 10.1529/biophysj.107.109728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rouslin W, Pullman ME. Protonic inhibition of the mitochondrial adenosine 5′-triphosphatase in ischemic cardiac muscle. Reversible binding of the ATPase inhibitor protein to the mitochondrial ATPase during ischemia. J Mol Cell Cardiol. 1987;19:661–668. doi: 10.1016/s0022-2828(87)80374-7. [DOI] [PubMed] [Google Scholar]
- 162.Di Pancrazio F, Mavelli I, Isola M, Losano G, Pagliaro P, Harris DA, Lippe G. In vitro and in vivo studies of F0F1ATP synthase regulation by inhibitor protein IF1 in goat heart. Biochim Biophys Acta. 2004;1659:52–62. doi: 10.1016/j.bbabio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 163.Rouslin W. The mitochondrial adenosine 5′-triphosphatase in slow and fast heart rate hearts. Am J Physiol. 1987;252:H622–H627. doi: 10.1152/ajpheart.1987.252.3.H622. [DOI] [PubMed] [Google Scholar]
- 164.Huang LJ, Chuang IC, Dong HP, Yang RC. Hypoxia-inducible factor 1α regulates the expression of the mitochondrial ATPase inhibitor protein (IF1) in rat liver. Shock. 2011;36:90–96. doi: 10.1097/SHK.0b013e318219ff2a. [DOI] [PubMed] [Google Scholar]
- 165.Shen L, Zhi L, Hu W, Wu MX. IEX-1 targets mitochondrial F1Fo-ATPase inhibitor for degradation. Cell Death Differ. 2009;16:603–612. doi: 10.1038/cdd.2008.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cabezon E, Arechaga I, Jonathan P, Butler G, Walker JE. Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1. J Biol Chem. 2000;275:28353–28355. doi: 10.1074/jbc.C000427200. [DOI] [PubMed] [Google Scholar]
- 167.Bason JV, Montgomery MG, Leslie AG, Walker JE. Pathway of binding of the intrinsically disordered mitochondrial inhibitor protein to F1-ATPase. Proc Natl Acad Sci U S A. 2014;111:11305–11310. doi: 10.1073/pnas.1411560111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- 169.Vinogradov AD, Gyurova ZS, Fitin AF. Effect of SH-reagents on the mitochondrial ATPase and induction of respiratory control in EDTA particles. FEBS Lett. 1975;54:230–233. doi: 10.1016/0014-5793(75)80080-9. [DOI] [PubMed] [Google Scholar]
- 170.Ylitalo K, Ala-Rämi A, Vuorinen K, Peuhkurinen K, Lepojarvi M, Kaukoranta P, Kiviluoma K, Hassinen I. Reversible ischemic inhibition of F1F0-ATPase in rat and human myocardium. Biochim Biophys Acta. 2001;1504:329–339. doi: 10.1016/s0005-2728(00)00261-9. [DOI] [PubMed] [Google Scholar]
- 171.Contessi S, Metelli G, Mavelli I, Lippe G. Diazoxide affects the IF1 inhibitor protein binding to F1 sector of beef heart F0F1ATPsynthase. Biochem Pharmacol. 2004;67:1843–1851. doi: 10.1016/j.bcp.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 172.Comelli M, Metelli G, Mavelli I. Downmodulation of mitochondrial F0F1 ATP synthase by diazoxide in cardiac myoblasts: a dual effect of the drug. Am J Physiol Heart Circ Physiol. 2007;292:H820–H829. doi: 10.1152/ajpheart.00366.2006. [DOI] [PubMed] [Google Scholar]
- 173.Bosetti F, Yu G, Zucchi R, Ronca-Testoni S, Solaini G. Myocardial ischemic preconditioning and mitochondrial F1F0-ATPase activity. Mol Cell Biochem. 2000;215:31–37. doi: 10.1023/a:1026558922596. [DOI] [PubMed] [Google Scholar]
- 174.Yamada EW, Huzel NJ. Distribution of the ATPase inhibitor proteins of mitochondria in mammalian tissues including fibroblasts from a patient with Luft’s disease. Biochim Biophys Acta. 1992;1139:143–147. doi: 10.1016/0925-4439(92)90093-3. [DOI] [PubMed] [Google Scholar]
- 175.Yagi T, Hatefi Y. Thiols in oxidative phosphorylation: inhibition and energy-potentiated uncoupling by monothiol and dithiol modifiers. Biochemistry. 1984;23:2449–2455. doi: 10.1021/bi00306a020. [DOI] [PubMed] [Google Scholar]
- 176.Yagi T, Hatefi Y. Thiols in oxidative phosphorylation: thiols in the F0 of ATP synthase essential for ATPase activity. Arch Biochem Biophys. 1987;254:102–109. doi: 10.1016/0003-9861(87)90085-3. [DOI] [PubMed] [Google Scholar]
- 177.Lippe G, Dabbeni-Sala F, Sorgato MC. ATP synthase complex from beef heart mitochondria. Role of the thiol group of the 25-kDa subunit of Fo in the coupling mechanism between Fo and F1. J Biol Chem. 1988;263:18627–18634. [PubMed] [Google Scholar]
- 178.Joshi S, Hughes JB. Inhibition of coupling factor B activity by cadmium ion, arsenite-2,3-dimercaptopropanol, and phenylarsine oxide, and preferential reactivation by dithiols. J Biol Chem. 1981;256:11112–11116. [PubMed] [Google Scholar]
- 179.Lippe G, Comelli M, Mazzilis D, Sala FD, Mavelli I. The inactivation of mitochondrial F1 ATPase by H2O2 is mediated by iron ions not tightly bound in the protein. Biochem Biophys Res Commun. 1991;181:764–770. doi: 10.1016/0006-291x(91)91256-c. [DOI] [PubMed] [Google Scholar]
- 180.Kane LA, Van Eyk JE. Post-translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr. 2009;41:145–150. doi: 10.1007/s10863-009-9218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Poon HF, Calabrese V, Calvani M, Butterfield DA. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid Redox Signal. 2006;8:381–394. doi: 10.1089/ars.2006.8.381. [DOI] [PubMed] [Google Scholar]
- 182.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 183.Poon HF, Shepherd HM, Reed TT, Calabrese V, Stella AM, Pennisi G, Cai J, Pierce WM, Klein JB, Butterfield DA. Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: Mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiol Aging. 2006;27:1020–1034. doi: 10.1016/j.neurobiolaging.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 184.Haynes V, Traaseth NJ, Elfering S, Fujisawa Y, Giulivi C. Nitration of specific tyrosines in FoF1 ATP synthase and activity loss in aging. Am J Physiol Endocrinol Metab. 2010;298:E978–E987. doi: 10.1152/ajpendo.00739.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Groebe K, Krause F, Kunstmann B, Unterluggauer H, Reifschneider NH, Scheckhuber CQ, Sastri C, Stegmann W, Wozny W, Schwall GP, Poznanovic S, Dencher NA, Jansen-Durr P, Osiewacz HD, Schrattenholz A. Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Exp Gerontol. 2007;42:887–898. doi: 10.1016/j.exger.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 186.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 187.Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta. 2009;1794:476–485. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang SB, Foster DB, Rucker J, O’Rourke B, Kass DA, Van Eyk JE. Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ Res. 2011;109:750–757. doi: 10.1161/CIRCRESAHA.111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fernandez-Sanz C, Ruiz-Meana M, Castellano J, Miro-Casas E, Nunez E, Inserte J, Vazquez J, Garcia-Dorado D. Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost. 2015;113:441–451. doi: 10.1160/TH14-10-0901. [DOI] [PubMed] [Google Scholar]
- 190.Sawicki G, Jugdutt BI. Valsartan reverses post-translational modifications of the δ-subunit of ATP synthase during in vivo canine reperfused myocardial infarction. Proteomics. 2007;7:2100–2110. doi: 10.1002/pmic.200601022. [DOI] [PubMed] [Google Scholar]
- 191.Nguyen T, Ogbi M, Johnson JA. Delta protein kinase C interacts with the d subunit of the F1F0 ATPase in neonatal cardiac myocytes exposed to hypoxia or phorbol ester. Implications for F1F0 ATPase regulation. J Biol Chem. 2008;283:29831–29840. doi: 10.1074/jbc.M801642200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J Biol Chem. 2003;278:19587–19590. doi: 10.1074/jbc.C300135200. [DOI] [PubMed] [Google Scholar]
- 193.Covian R, Balaban RS. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am J Physiol Heart Circ Physiol. 2012;303:H940–H966. doi: 10.1152/ajpheart.00077.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Di Pancrazio F, Bisetto E, Alverdi V, Mavelli I, Esposito G, Lippe G. Differential steady-state tyrosine phosphorylation of two oligomeric forms of mitochondrial F0F1 ATPsynthase: a structural proteomic analysis. Proteomics. 2006;6:921–926. doi: 10.1002/pmic.200500077. [DOI] [PubMed] [Google Scholar]
- 196.Boja ES, Phillips D, French SA, Harris RA, Balaban RS. Quantitative mitochondrial phosphoproteomics using iTRAQ on an LTQ-Orbitrap with high energy collision dissociation. J Proteome Res. 2009;8:4665–4675. doi: 10.1021/pr900387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, Robinson J, Murata M, Murphy AM, Marban E, Van Eyk JE. Proteomic analysis of pharmacological preconditioning: novel protein targets converge to mitochondrial metabolism pathways. Circ Res. 2006;99:706–714. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 198.Garzón JI, Kovacs J, Abagyan R, Chacón P. ADP_EM: fast exhaustive multi-resolution docking for high-throughput coverage. Bioinformatics. 2007;23:427–433. doi: 10.1093/bioinformatics/btl625. [DOI] [PubMed] [Google Scholar]
- 199.Guex N, Diemand A, Peitsch MC. Protein modelling for all. Trends Biochem Sci. 1999;24:364–367. doi: 10.1016/s0968-0004(99)01427-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.