

Abstract
Diabetic patients have increased susceptibility to infections, and urinary tract infections (UTI) are the most common type in women with diabetes mellitus. Knowledge of bacterial clearance effectiveness following UTI in diabetics is sparse. In this study, the effects of diabetes on bacterial clearance efficiency and components of the innate immune system in response to UTI in a murine model were investigated. Streptozotocin-induced diabetic and control female C57BL/6J mice were infected with uropathogenic Escherichia coli, and bacterial load, expression of chemokines, and neutrophil infiltration in the bladder over time were investigated. Expression levels of histone deacetylases were also measured to address a potential mechanism underlying the phenotype. Bacterial clearance during UTI was significantly prolonged in diabetic mice relative to controls. Neutrophil infiltration in bladder tissue and urine, and both mRNA and protein expression of chemokines MIP-2, KC, MCP-1 and IL-6 in bladder tissue were diminished at early time points after infection in diabetic mice relative to controls. In addition, mRNA levels of histone deacetylases 1–5 were increased in diabetic mice. This is the first study to show an association of impaired bacterial clearance in diabetic mice with suppression of UTI-induced chemokine expression and neutrophil infiltration in the bladder.
Keywords: type 1 fimbria, myeloperoxidase, quantitative real-time RT-PCR, ELISA
This study shows that the cytokine-driven neutrophil response to bacterial infection of the bladder is deficient in a mouse model of type 1 diabetes mellitus compared with healthy mice.
Graphical Abstract Figure.
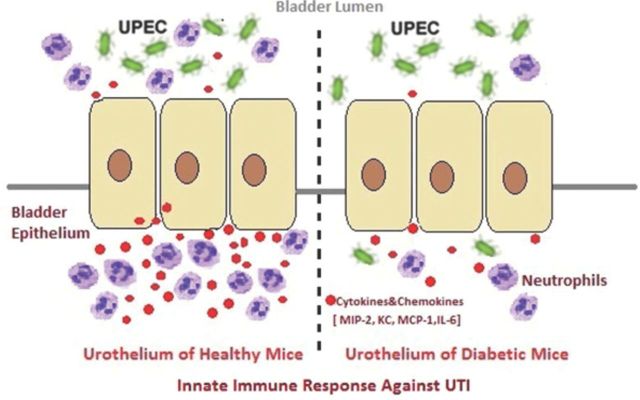
This study shows that the cytokine-driven neutrophil response to bacterial infection of the bladder is deficient in a mouse model of type 1 diabetes mellitus compared with healthy mice.
INTRODUCTION
Urinary tract infections (UTI) by uropathogenic bacteria are the most common urological disorders, and lower UTI involving bladder infections (or cystitis) specifically affect millions of individuals each year, carrying an annual cost of more than 3.5 billion dollars in the US (Griebling 2005a,b). Surveys have suggested that women are about four times more susceptible to UTI than men (Griebling 2007). Individuals with diabetes mellitus (DM) have a higher prevalence of several types of infection, including asymptomatic bacteriuria, lower UTI and acute pyelonephritis compared with people without DM (Scholes et al., 2005; Geerlings 2008). The documented increasing prevalence of DM in the US and throughout the world portends an increasing prevalence of lower UTI (Centers for Disease Control and Prevention 2012). The mechanistic basis of this association is still unclear; corollaries of DM that may be contributing factors include glucosuria, which can enhance growth of bacteria (Rosen, Hung and Kline 2008), alterations in the composition of the luminal urothelial surface that may enhance bacterial adherence and impaired innate immunity (Geerlings 2008).
Uropathogenic E. coli (UPEC) is the predominant uropathogen, responsible for approximately 80% of community-acquired UTIs (Ronald 2002). UPEC UTI is a complex process, and its fate in certain mouse strains is determined within the first 24 hours through dynamic host–pathogen interactions with various checkpoints and population bottlenecks (Hannan et al., 2012). UPEC adhere to the urothelium by means of adhesin-tipped fimbriae (pili), among which the type 1 fimbria is the predominant adhesion appendage on almost all cystitis-causing UPEC strains (Hultgren et al., 1985; Langermann et al., 1997). The FimH adhesin on type 1 fimbriae has been shown to mediate invasion and colonization of urothelial cells in vivo in murine models (Connell et al., 1996; Martinez et al., 2000), and clinical studies have provided strong circumstantial evidence for a critical role for type 1 fimbria in human UTI (Hannan et al., 2012). Host resistance to UTI is dependent on a strong innate immune response, initiated primarily by toll-like receptor 4 (TLR4) (Haraoka et al., 1999; Schilling et al., 2003), which can bind both lipopolysaccharide (LPS) and FimH on UPEC (Hoshino et al., 1999; Mossman et al., 2008). TLR4 signaling results in expulsion of UPEC via exocytosis (Song et al., 2007a) and activation of NF-κ B-dependent and -independent signaling pathways that lead to the production of cytokines and recruitment of neutrophils to the bladder (Song et al., 2007b). Although LPS-independent activation of TLR4 by FimH has been reported (Fischer et al., 2006; Mossman et al., 2008), both LPS and FimH may be required to yield the strongest cytokine and neutrophil response (Hedlund et al., 2001; Schilling et al., 2001).
Neutrophils and macrophages are the major phagocytic cells of the innate immune response against bacterial infections, and neutrophils are the major early responders recruited to the bladder during UTI (Haraoka et al., 1999). TLR4-defective C3H/HeJ mice (Poltorak et al., 1998) are deficient in both the neutrophil response to UPEC and in bacterial clearance compared with wild-type C3H/HeN mice, suggesting a role for neutrophils in clearance of UTI (Shahin et al., 1987). Further suggestive evidence for the importance of neutrophils was shown by impaired UPEC clearance from the urinary tract of C3H/HeN mice after pretreatment with an antibody that depletes peripheral neutrophils, as well as subsets of other leukocytes (Haraoka et al., 1999; Daley et al., 2008).
Rosen et al. showed that UPEC infection of female C3H/HeN mice with streptozotocin (STZ)-induced type 1 DM yielded bladder titers 10 000 times higher than in non-diabetic C3H/HeN mice, whereas the difference in titers between UPEC-infected diabetic and non-diabetic C3H/HeJ mice was 100-fold, suggesting deficiencies in both TLR4-dependent and -independent responses to UPEC in diabetic mice (Rosen, Hung and Kline 2008). Numerous studies have shown functional defects in neutrophils from diabetic patients and animals (Alba-Loureiro et al., 2007), including impairments in chemotaxis (Pereira, Sannomiya and Leme 1987; Mowat and Baum 1971), phagocytosis (Bagdade, Nielson and Bulger 1972; Panneerselvam and Govindasamy 2003), and bactericidal activity (Tan et al., 1975; Yokoo, Kumamoto and Hirose 1994). However, while the initial recruitment of macrophages to the bladder after UPEC instillation has been reported to be higher in STZ-diabetic mice than in controls (Yokoo, Kumamoto and Hirose 1994), the neutrophil and related cytokine/chemokine responses to UTI in animal models of DM have not been studied.
Several cytokines with roles in neutrophil trafficking are expressed in response to UPEC in the bladder and in epithelial cell lines, and their expression can be induced by TLR4. These include interleukin (IL)-8 (CXCL8), two functional homologues of IL-8 that are also expressed in mice, namely macrophage inflammatory protein 2 (MIP-2, CXCL2) and keratinocyte chemoattractant (KC, CXCL1), as well as monocyte chemoattractant protein 1 (MCP-1, CCL2) and IL-6 (Boyd et al., 2006; Song et al., 2007b; Ao et al., 2009). In humans, IL-8 is the major attractant for neutrophils, and in mice, MIP-2 enabled neutrophils to traverse epithelial cells in UPEC-infected kidneys (Hang et al., 1999). Furthermore, both MIP-2 and KC have been shown to contribute to neutrophil infiltration of the kidney in a mouse model of renal inflammation (Roche et al., 2007). MCP-1, while primarily a monocyte chemoattractant, has been shown to promote neutrophil infiltration into injury sites in murine models of corneal and lung infection (Xue et al., 2007; Balamayooran et al., 2011). Incubation of type 1-fimbriated UPEC with human kidney carcinoma cells caused secretion and mRNA expression of MCP-1, KC and IL-8, but not LPS-induced CXC chemokine (LIX, CXCL5), a third functional homologue of IL-8 (Godaly et al., 2007). IL-6 is an important mediator of inflammation and has been shown to inhibit neutrophil apoptosis (Biffl et al., 1996; Gabay 2006). It is the most prominent cytokine detected in urine in response to UTI in humans and animals (de Man et al., 1989; Hedges et al., 1991) and is secreted by urothelial cells in response to FimH and LPS in a TLR4-dependent manner (Hedlund et al., 2001; Schilling et al., 2001).
Diabetic complications such as retinopathy and associated changes in gene expression persist even after good glycemic control is achieved, suggesting involvement of epigenetic modifications such as alterations in acetylation of histones (Intine and Sarras 2012). Consistent with that hypothesis, STZ-diabetic rats were found to have increased levels of three histone deacetylases (HDACs) and a reduced level of acetylated histone 3 in the retina, which persisted after restoration of good glycemic control (Zhong and Kowluru 2010).
In the current study, we aimed to evaluate the efficiency of neutrophil infiltration into the bladder during acute UTI in diabetic mice, with respect to the expression profiles of regulatory chemokines and HDACs, the latter as a potentially targetable mechanism underlying altered expression of the chemokines.
MATERIALS AND METHODS
Propagation and characterization of type 1-fimbriated E. coli
Growth of type 1-fimbriated UPEC and characterization of the fimbriae were conducted as described with minor modifications (Martinez et al., 2000). UPEC strain 53498 (American Type Culture Collection, Manassas, VA, USA) was freshly streaked from a frozen glycerol stock onto a Luria agar plate and grown at 37°C overnight. To stimulate expression of type 1 fimbriae, a single colony was inoculated into Luria-Bertani (LB) broth and incubated at 37°C overnight without shaking (static conditions), and then the bacterial suspension was subcultured at 1:1000 into 2 ml of fresh LB broth and incubated again at 37°C overnight without shaking. Bacteria were washed and concentrated to 1.6 × 109 cells ml−1 in 1 × PBS, and type 1 fimbria expression was confirmed by mannose-sensitive agglutination of a 2% solution of guinea pig erythrocytes. Bacteria were maintained on ice for a maximum of 2 h before installation into the bladder.
Type 1 DM induction in mice
C57BL/6J female mice (Jackson Laboratory, Bar Harbor, ME, USA) 8 weeks old received two intraperitoneal injections of high-dose STZ (150 mg kg−1 mouse) on consecutive days to induce DM, or 0.1 M sodium citrate vehicle, pH 4.5, as described (Rosen, Hung and Kline 2008). Blood glucose levels of mice were monitored weekly until UPEC inoculation 4 weeks after STZ or vehicle injection, and only STZ-injected mice that maintained glucose levels >300 mg dl−1 were used in experiments. Mice received food and water ad libitum. All protocols were pre-approved by the IACUC of Case Western Reserve University in compliance with the Public Health Service policy on humane care and use of laboratory animals.
Experimental UTI in mice and bacterial clearance assay
We followed the transurethral inoculation and clearance assay protocols of Mulvey, Schilling and Hultgren (2001) with some modifications. Twelve week-old female diabetic or control C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) were anesthetized with isoflurane. Prior to inoculation, the bladder was emptied by gentle abdominal massage, while collecting the urine for detection of pre-existing bacteriuria. Fifty microliters of UPEC suspension (1.6 × 109 CFU ml−1) in 1 × PBS were injected into the lumen via transurethral catheterization, using a 24 Ga. × 0.75 in. shielded I.V. catheter (BD AngiocathTM AutoguardTM, BD Medical Systems, Sandy, UT, USA). After 30 s, the catheter was removed. At the indicated times after UPEC inoculation, urine was collected aseptically by massaging the mouse abdomen over a sterile Eppendorf tube, and then mice were euthanized by cervical dislocation under anesthesia. Bladders were removed aseptically, weighed and homogenized in 1 ml of 0.025% Triton X-100 in 1 × PBS. Serial dilutions of urine samples or bladder homogenates were spread on LB agar plates and incubated at 37°C overnight. Bacteria numbers were counted and calculated as per ml of urine, or per whole bladder. Examination of urine prior to inoculation confirmed that none of the mice had pre-existing bacteriuria.
Neutrophil counts in urine
At different times after UPEC inoculation of mice, beginning 4 weeks after STZ or vehicle injection, urine was collected aseptically by massaging the mouse abdomen over a sterile Eppendorf tube. Urine was mixed 10:1 with Turk's stain (0.05 mg ml−1 crystal violet, 3% glacial acetic acid in distilled water), and neutrophils were counted with a Bürker chamber under a microscope. Examination of urine prior to inoculation of UPEC confirmed that none of the mice had pre-existing leukocyturia.
Neutrophil MPO assay
Urine was collected aseptically and bladders were harvested for ELISA of myeloperoxidase (MPO) at the times following UPEC inoculation indicated in Fig. 3 (beginning 4 weeks after injection of STZ or vehicle). Bladders were homogenized for 3 min on ice using PowerGen 125 homogenizer (Fisher Scientific, Waltham, MA, USA), and urine and bladder tissue neutrophils were quantified using a MPO ELISA kit from Hycult Biotech (Plymouth Meeting, PA) as described by Haraoka et al. (1999) and the manufacturer's protocol.
Figure 3.

Proinflammatory cytokine response to UPEC in the bladder. (a) qRT-PCR. Bladders were harvested from 4-week diabetic mice and age-matched controls at the indicated time points following UPEC inoculation (n = 5 per group). Total RNA was isolated, cDNA was synthesized, and MIP-2, KC, MCP-1 and IL-6 mRNA levels were quantified by qRT- PCR as described in the section ‘Materials and Methods’. Using the comparative CT method, chemokine threshold cycles (CT) were normalized to the corresponding β-actin CT values, and expression levels were calculated relative to the average normalized values in uninoculated (time 0), non-diabetic control mice (set at 1.0). Each bladder sample was assayed in triplicate. qRT- PCR data are expressed as means ± SEM. (b) ELISA. Bladders were harvested from 4-week diabetic mice and age-matched controls at the indicated time points following UPEC inoculation (n = 10 per group). Bladders were homogenized, and chemokine levels were quantified by ELISA. MIP-2, KC, MCP-1 and IL-6 MPO levels were calculated from the corresponding standard curves as pg/mg protein. Each bladder sample was assayed in quadruplicate. ELISA data are expressed as means ± 95% CI. Statistical analyses of qRT-PCR results and ELISA results in DM compared with control mice at each time point were performed by multiple Student's t tests with corrections for multiple comparisons by the Holm–Sidak method (‡P < 0.02, †P < 0.005, §P < 0.001 and *P < 0.0001).
Quantitative real-time RT-PCR (qRT-PCR)
Bladders were harvested for RNA isolation and qRT-PCR 4 weeks after injection of STZ or vehicle (before UPEC inoculation), and in the case of the chemokines, at the times following UPEC inoculation indicated in Fig. 3. Total RNA was isolated using Trizol (Life Technologies, Grand Island, NY, USA), and first-strand complementary DNA was synthesized from 1 μg of total RNA using a high-capacity cDNA synthesis kit (Life Technologies). cDNA was amplified in an ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), using SYBR Green PCR master mix with primer pairs for chemokines, HDACs and β-actin (Life Technologies). The primer sequences (Table 1) were designed using the online Universal Probe Library Assay Design Center (Roche, Mannheim, Germany). Chemokine and HDAC gene expression levels were normalized to the corresponding β-actin gene expression level and calculated relative to the mean level in healthy control mice before UPEC inoculation (set to 1.0) by the comparative CT method (Livak and Schmittgen 2001). The mean levels of β-actin mRNA did not differ significantly between the control and diabetic mice before or after inoculation.
Table 1.
Primer sequences used for qRT-PCR.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| MIP-2α | 5′-AAAATCATCCAAAAGATACTGAACAA | 5′-CTTTGGTTCTTCCGTTGAGG |
| KC | 5′-AGACTCCAGCCACACTCCAA | 5′-TGACAGCGCAGCTCATTG |
| MCP-1 | 5′-CATCCACGTGTTGGCTCA | 5′-GATCATCTTGCTGGTGAATGAGT |
| IL-6 | 5′-GCTACCAAACTGGATATAATCAGGA | 5′-CCAGGTAGCTATGGTACTCCAGAA |
| β-actin | 5′-GGTCATCACTATTGGCAACG | 5′-ACGGATGTCAACGTCACACT |
| HDAC1 | 5′-TGGTCTCTACCGAAAAATGGAG | 5′-TCATCACTGTGGTACTTGGTCA |
| HDAC2 | 5′-CTCCACGGGTGGTTCAGT | 5′-CCCAATTGACAGCCATATCA |
| HDAC3 | 5′-TTCAACGTGGGTGATGACTG | 5′-TTAGCTGTGTTGCTCCTTGC |
| HDAC4 | 5′-AATCCTGCCCGTGTGAAC | 5′-GTAGGGGCCACTTGCAGA |
| HDAC5 | 5′-GCATGAACTCTCCCAACGAG | 5′-TTCACCTCCACTGCCACAG |
| HDAC6 | 5′-GGCTGAGATTCGGAATGG | 5′-CCCATCCATAAGATTGTGCTG |
| HDAC7 | 5′-CGCCAGTTGGAAACAATGAT | 5′-GCTGAGAGCCTGGTGTGTCT |
| HDAC8 | 5′-GCAGCTGGCAACTCTGATT | 5′-GTCAAGTATGTCCAGCAACGAG |
| HDAC9 | 5′-CAAGAAGCGAGTGTTTGAGGT | 5′-GTTTGGTGAACTGGGACCTG |
| HDAC10 | 5′-ACCTTGCAGATGATGGGAGA | 5′-GCTCAGAAACCCTCCAGTTG |
| HDAC11 | 5′-TGCAGACATCACACTGGCTAT | 5′-GGTGGGCATCGAGATCAA |
Chemokine ELISA
Bladders were harvested for ELISA of chemokines at the times following UPEC inoculation indicated in Fig. 3 (beginning 4 weeks after injection of STZ or vehicle). Bladders were homogenized for 3 min on ice using PowerGen 125 homogenizer (Fisher Scientific), and levels of MIP-2, KC, MCP-1 and IL-6 (RayBiotech, Norcross, GA, USA) were quantified by ELISA as indicated in the manufacturer's protocols. Absorbances at 405 and 450 nm were measured using a FLUOstar OPTIMA Fluorescence plate reader (BMG LABTECH GmbH, Ortenberg, Hesse, Germany).
Statistical analysis
Data are expressed as means ± 95% confidence intervals (95% CI) or SEM. Statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA). Urine (serial) measures were analyzed by calculating the area under the curve (AUC) for each mouse and comparing the AUC values in diabetic vs control groups by Student's t test. Comparisons of measures in bladder tissues were performed with the Student's t test for each time point, using the Holm–Sidak method to correct for multiple comparisons. Differences were considered significant for P < 0.05.
RESULTS
Characteristics of diabetic and control mice
At the time of euthanasia, the mean blood glucose level in diabetic mice was more than five times higher than in controls, while there were no statistically significant differences in body weights, bladder weights or bladder weight/body weight ratios between the two groups (Table 2).
Table 2.
General characteristics of DM and age-matched control mice.
| Group | Blood glucose (mg dl–1) | Body weight (g) | Bladder weight (mg) | Bladder weight/body weight (mg g–1) |
|---|---|---|---|---|
| Control | 106.4 ± 3.5 | 22.4 ± 0.75 | 21.5 ± 0.45 | 0.97 ± 0.02 |
| DM | 524.2 ± 9.7* | 21.6 ± 0.63 | 23.3 ± 1.36 | 1.10 ± 0.09 |
Values are means ± SEM, n = 10 mice per group.
*significantly different from controls by unpaired, two-tailed Student's t test, P < 0.0001.
Reduced bacterial clearance in diabetic mice
Lower UTI is an acute disease with a typical clearance pattern within 2 weeks in healthy C57BL/6 mice, but the process is determined within first 5 days post-infection, with a gradual decrease in bacterial load after a peak at about 24 h post-infection (Ingersoll et al., 2008; Rosen, Hung and Kline 2008). Here we show that, during the course of UTI from 3 h to 5 days post-infection, the overall bacterial count in urine was significantly higher in diabetic mice than in the control group (Fig. 1a, P < 0.0001). Control mice cleared the majority of UPEC from the bladder by day 5, whereas diabetic mice had significantly higher numbers of bacteria in the bladder at every time point (Fig. 1b). The data indicate less efficient bacterial clearance ability in these diabetic mice, resulting in prolonged infection.
Figure 1.

In vivo kinetics of UPEC infection and clearance in the bladder. Shown are UPEC colony forming units in urine and bladder tissue at the indicated times after inoculation of bacteria. Examination of urine prior to inoculation confirmed that none of the mice had pre-existing bacteriuria. Bacterial clearance assays were performed as described in the section ‘Materials and Methods’. (a) Bacterial counts per ml of urine were significantly higher in diabetic compared with control mice, determined by calculating area under the curve over the entire time course for each mouse and comparing the area values in diabetic vs control mice by a Student's t test (P < 0.0001). (b) Bacterial counts per whole bladder were significantly higher in diabetic compared with control mice at each time point, determined by multiple Student's t tests with corrections for multiple comparisons by the Holm–Sidak method. ‡P < 0.02, §P < 0.001, and *P < 0.0001. Data in (a) and (b) are expressed as means ± 95% CI.
Reduced neutrophil infiltration in response to UTI in diabetic mice
To evaluate the effect of DM on neutrophil infiltration into the bladder in response to UTI, we evaluated the neutrophil numbers in urine and bladder tissues of diabetic and control mice at the times after UPEC inoculation indicated in Fig. 2. In control mice, UPEC administration resulted in a strong increase in the number of neutrophils in urine, peaking at 6 h, followed by a sharp partial decrease by 12 h, and then a gradual reduction through 72 h. On the other hand, diabetic mice demonstrated a markedly attenuated influx of neutrophils in urine, peaking at 12 h and then gradually decreasing through 72 h (Fig. 2a, P < 0.0001 by Student's t test comparison of AUC values over the 3–12 h period in diabetic vs control mice). Neutrophil infiltration after UPEC administration was also assessed by measuring levels of the neutrophil marker MPO by ELISA. The time courses of MPO protein levels in urine and bladder paralleled the course of neutrophil counts in urine, with a robust peak at 6 h post-UPEC installation in controls that was markedly attenuated in diabetic mice (Fig. 2b, urine, P < 0.0001 by Student's t test comparison of AUC values over the 3–12 h period in diabetic vs control mice; Fig. 2c, bladder, P < 0.0001 comparing DM with control at 3 and 6 h).
Figure 2.
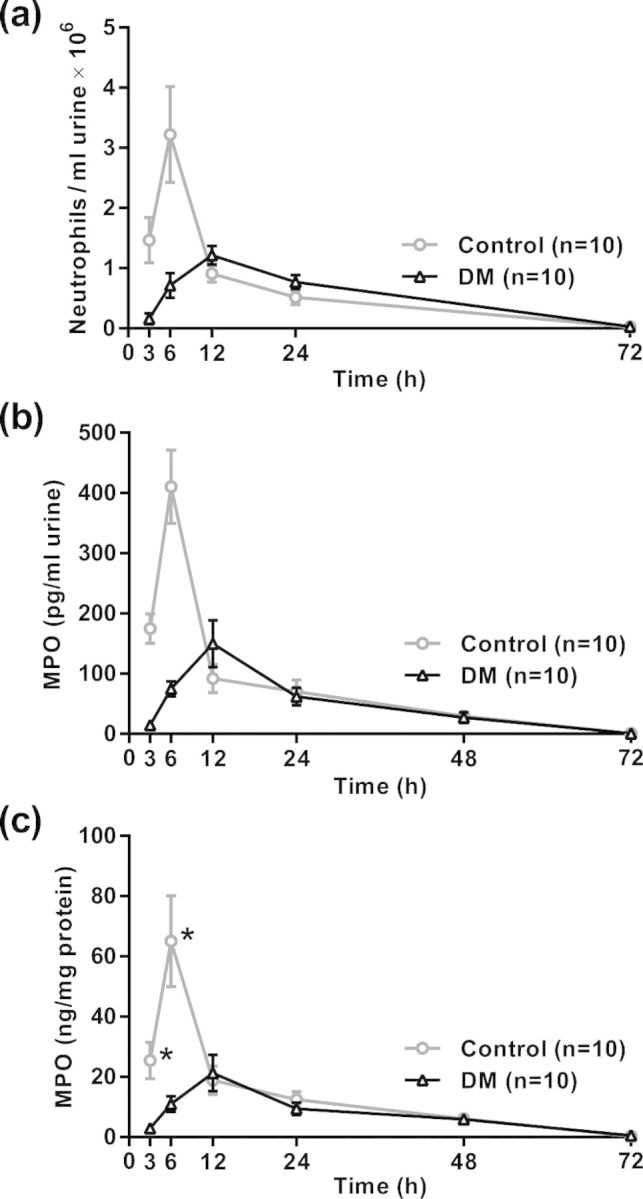
Influx of neutrophils into the urine, with confirmatory MPO kinetics in the urine and bladder. Urine was collected aseptically and bladders were harvested from diabetic and control mice at the indicated times after UPEC inoculation. (a) Urine was stained with Turk's solution and neutrophils were counted using a hemacytometer. The initial neutrophil response was markedly attenuated in diabetic mice compared with controls (P < 0.0001 by Student's t test comparison of AUC values over the 3–12 h period in diabetic vs control mice). (b) Results of ELISA quantification of the neutrophil marker MPO in urine, shown as ng MPO per ml of urine. P < 0.0001 by Student's t test comparison of AUC values over the 3–12 h period after UPEC challenge in diabetic vs control mice comparing DM with control at the indicated times. (c) ELISA analysis of MPO concentrations in bladder homogenates, shown as concentrations of MPO per mg total protein. *P < 0.0001 by multiple Student's t tests corrected for multiple comparisons by the Holm–Sidak method. Data in (a)–(c) are expressed as means ± 95% CI.
Attenuated chemokine expression in response to UTI in diabetic mice
Expression levels of MIP-2, KC, MCP-1 and IL-6 were measured at both the transcription and protein levels over time in response to UPEC installation (Fig. 3) Elevated chemokine mRNA levels were evident by 3 h post-infection, and peaked at 3 h (MCP-1), 6 h (MIP-2 and IL-6) or 12 h (KC) in both control and diabetic mice. However, the levels were significantly lower in the diabetic mice at multiple early time points (Fig. 3, left column). Chemokine protein levels followed the same pattern, except the MCP-1 protein level peaked later than its mRNA, at 6 h (Fig. 3, right column). The results show that early release of neutrophil-attractant chemokines upon infection with type 1-fimbriated UPEC is severely attenuated in female diabetic mice.
Altered HDAC gene expression in DM
Using qRT-PCR, we observed significantly increased expression of mRNAs for several proteins in the TLR4 signaling pathway to NF-κB activation, including MyD88, IRAK1 and IRAK4, in uninfected bladders of diabetic mice compared with controls, suggestive of sustained NF-κB activation and expression of proinflammatory cytokines (data not shown). Those results led us to investigate potential involvement of chromatin remodeling enzymes such as HDACs in the impaired cytokine response to UPEC infection in diabetic mice. We determined the effect of DM on gene expression of HDACs 1–11 in the bladder in uninfected mice. The mRNA levels of HDACs 1–5 were significantly higher in diabetic mice bladders compared with controls, suggesting a potential inhibitory effect on the expression of chemokines during DM (Fig. 4). On the other hand, the expression levels of HDACs 9 and 11 were significantly lower in the bladders of diabetic mice compared with non-diabetic control mice.
Figure 4.
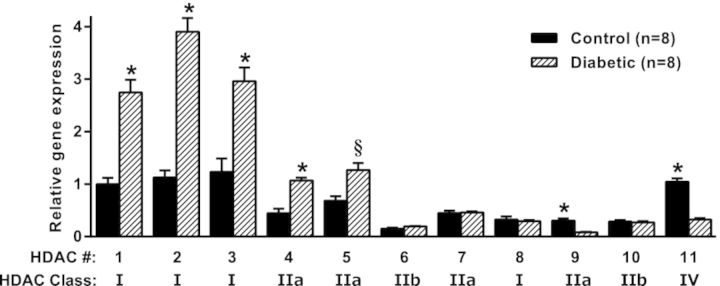
Relative HDAC mRNA levels in diabetic and control mice. Bladders were harvested from 4-week diabetic mice (n = 8) and age-matched controls (n = 8). Total RNA isolation, cDNA synthesis and qRT-PCR were performed as described in the section ‘Materials and Methods’. The results, normalized to β-actin expression, are expressed relative to the level of HDAC 1 in control mice, set at 1.0. *P < 0.0003, §P = 0.002 for DM vs control by multiple Student's t tests with corrections for multiple comparisons by the Holm–Sidak method.
DISCUSSION
This study demonstrated that clearance of UPEC strain 53498 is impaired in diabetic female C57BL/6 mice, characterized by diminished infiltration of neutrophils into the bladder and markedly attenuated induction of chemokine/cytokine expression in the bladder compared with non-diabetic controls. In contrast, the bladder chemokine response to experimental UTI in healthy mice was rapid and robust, and was accompanied by substantial accumulation of active neutrophils in the bladder and urine.
In the experimental UTI model, type 1 fimbriae promote colonization of the urothelium and initiate an early innate host response (Connell et al., 1996; Martinez et al., 2000; Hedlund et al., 2001; Schilling et al., 2001). An essential role for neutrophils in the innate immune response was suggested in a report demonstrating that pretreatment of female C3H/HeN mice with the neutrophil depleting antibody RB6–8C5 drastically impaired clearance of UPEC from the bladder and kidneys (Haraoka et al., 1999). However, subsequent work revealed that RB6–8C5 also depletes peripheral dendritic cells and a subset of macrophages in mice (Tvinnereim, Hamilton and Harty 2004; Daley et al., 2008). Although dendritic cells have been reported to be dispensable for UPEC clearance (Engel et al., 2006), the relative roles of neutrophils and macrophages remain to be clarified.
Others have shown that UTI can trigger local mucosal expression of certain chemokines from the two main categories, CXC (MIP-2 and KC) and CC (MCP-1) (Hang et al., 1999; Mittal et al., 2004; Godaly et al., 2007; Duell et al., 2012). Treatment of mice with MIP-2 and KC neutralizing antibodies before E. coli Shiga toxin- and LPS-induced renal injury blocked neutrophil infiltration into the kidney in an additive manner (Roche et al., 2007). More recently, neutrophil influx into and bacterial clearance from the lungs of mice after intratracheal instillation of E. coli were markedly reduced by genetic disruption of MCP-1 (Balamayooran et al., 2011). Those results indicate important roles for MIP-2, KC and MCP-1 in neutrophil recruitment, and suggest that the impaired chemokine responses to UTI that we observed in diabetic mice are a likely cause of the diminished neutrophil infiltration into the bladder and impaired bacterial clearance.
In our time course, the levels of MIP-2, MCP-1 and IL-6 proteins in the bladder and neutrophils in the bladder and urine peaked at 6 h after UPEC instillation. A similar time course of MIP-2 induction accompanied by neutrophil infiltration was found in a mouse model of Pseudomonas aeruginosa-induced UTI (Mittal et al., 2004). Also, MIP-2 and KC levels in the lungs of mice peaked at 6 h after intratracheal installation of E. coli, although MCP-1 and neutrophil levels continued to increase through 24 h in that model (Balamayooran et al., 2011). In our model, KC mRNA and protein levels peaked at 12 h, and the mean MCP-1 mRNA level peaked at 3 h after UPEC instillation, suggesting different influences on the regulation of transcription of those chemokines relative to MIP-2 and IL-6. Our expression results are roughly consistent with Duell et al. (2012), who found increases of 67-, 22-, 28- and 133-fold in the mRNA levels of MIP-2, KC, MCP-1 and IL-6, respectively, 2 h after infection of mice with type 1-fimbriated UPEC strain CFT073.
Others have reported that expression of MIP-2, KC, MCP-1 and IL-6 can be induced by TLR4, likely through activation of the transcription factor NF-κB (Boyd et al., 2006; Song et al., 2007b; Ao et al., 2009). Our finding that the responses of all four of those cytokines to UPEC are impaired in diabetic mice, without altering the different individual time courses of expression, suggests that the impairment is due to a defect common to the regulation of the four cytokines, possibly at the level of TLR4 signaling to NF-κB, or epigenetic regulation of transcriptional activation by NF-κB through chromatin remodeling.
Although NF-κB is persistently activated in many tissues in DM, suggesting a sustained proinflammatory state (Patel and Santani 2009), its function is regulated at multiple levels. Our observation of increased expression of TLR4 signaling pathway genes in uninfected diabetic bladders (data not shown), which is consistent with sustained signaling to NF-κB, suggested possible involvement of epigenetic modifications such as protein deacetylation in the impaired cytokine response to UTI in diabetic mice. HDACs are important chromatin remodeling enzymes that can inhibit the transcription of genes by deacetylating lysine residues on histones, resulting in increased chromatin condensation. HDACs can also deacetylate and thereby regulate the activity of a variety of non-histone proteins, including transcription factors (Villagra, Sotomayor and Seto 2010). HDACs have been implicated in both pro- and antiinflammatory actions (Halili et al., 2009). HDACs 1, 2 and 3 have been shown to inhibit NF-κB activation by deacetylating or binding to NF-κB subunits, resulting in binding to IκB and export from the nucleus; on the other hand, HDACs have also been implicated in maintaining Iκ B kinase activity, leading to dissociation of IκB from and activation of NF-κB (Grabiec, Tak and Reedquist 2011). HDAC1 has also been shown to participate in the suppression of proinflammatory cytokine expression, including IL-6 and IL-8, in human breast cancer cells via chromatin modifications (Janzer et al., 2012). Thus, one or more of the three class I HDACs we found to be upregulated in diabetic bladders (HDACs 1, 2 and 3) could have contributed to the attenuation of the chemokine/cytokine response to UPEC at one or both of two levels: by promoting NF-κB binding to IκB, or by increasing chromatin condensation in the regions of the chemokine/cytokine gene promoters.
Our observation of increased expression of class I HDACs 1, 2 and 3 in the bladder in STZ-diabetic mice is consistent with a report that STZ-diabetic rats had increased expression of class I HDACs 1, 2 and 8 in the retina (Zhong and Kowluru 2010). We also observed upregulation of class IIa HDACs 4 and 5, consistent with a recent study showing increased expression of HDACs 2, 4 and 5 in both STZ-induced and db/db diabetic rat kidneys (Wang et al., 2014). On the other hand, class IIa HDAC 9 and class IV HDAC 11 were downregulated in diabetic bladders. Deletion studies have indicated that individual HDACs of both class I (widely expressed in mammalian tissues) and class II (limited expression patterns) can affect the expression of limited sets of genes (Witt et al., 2009). The novel class IV HDAC, HDAC11, has been shown to inhibit the expression of plasminogen activator inhibitor type-1, a promoter of macrophage migration, in a murine model of kidney ischemia and reperfusion injury (Kim et al., 2013). Such a role is consistent with our observation of lower HDAC11 expression in STZ-diabetic mice, a DM model previously reported to exhibit increased recruitment of macrophages to the bladder after UPEC instillation (Yokoo, Kumamoto and Hirose 1994). Further studies are required to clarify the roles of the specific HDACs up- or downregulated in the bladder in diabetic mice in the gene expression machinery of the immune response to UTI.
In conclusion, we showed that bacterial clearance is impaired in diabetic mice, likely due in part to attenuated expression of cytokines in response to UPEC challenge, resulting in lower recruitment of neutrophils to the bladder. Further characterization of the bacterial clearance defect in DM will include studies of upstream control of the expression of the related genes, as suggested by altered expression of HDACs.
Acknowledgments
We thank Michael Kavran for technical assistance with this research project and C. Thomas Powell for editorial assistance with this manuscript.
Footnotes
Co-first authors.
Present address: Department of Medical Biology, Harran University, Osmanbey Kampusu, 63200 Sanliurfa, Turkey.
Present address: Texas Institute of Biotechnology Education and Research, North American University, 10555 Stella Link Rd, Houston, TX 77025, USA.
Present address: Department of Medical Biochemistry, School of Medicine, Erciyes University, 38039 Melikgazi, Kayseri, Turkey.
FUNDING
This study was supported by NIH grant R01 DK083733 to F. Daneshgari. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- Alba-Loureiro TC, Munhoz CD, Martins JO, et al. Neutrophil function and metabolism in individuals with diabetes mellitus. Braz J Med Biol Res. 2007;40:1037–44. doi: 10.1590/s0100-879x2006005000143. [DOI] [PubMed] [Google Scholar]
- Ao L, Zou N, Cleveland JC, et al. Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: mediating KC and MCP-1 expression induced by extracellular HSC70. Am J Physiol-Heart C. 2009;297:H21–8. doi: 10.1152/ajpheart.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdade JD, Nielson KL, Bulger RJ. Reversible abnormalities in phagocytic function in poorly controlled diabetic patients. Am J Med Sci. 1972;263:451–6. doi: 10.1097/00000441-197206000-00005. [DOI] [PubMed] [Google Scholar]
- Balamayooran G, Batra S, Balamayooran T, et al. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect Immun. 2011;79:2567–77. doi: 10.1128/IAI.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffl WL, Moore EE, Moore FA, et al. Interleukin-6 delays neutrophil apoptosis. Arch Surg. 1996;131:24–9. doi: 10.1001/archsurg.1996.01430130026005. [DOI] [PubMed] [Google Scholar]
- Boyd JH, Mathur S, Wang Y, et al. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93. doi: 10.1016/j.cardiores.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention . Diabetes Report Card 2012. Atlanta, GA: US Department of Health and Human Services; 2012. [Google Scholar]
- Connell I, Agace W, Klemm P, et al. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. P Natl Acad Sci USA. 1996;93:9827–32. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, et al. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukocyte Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- de Man P, van Kooten C, Aarden L, et al. Interleukin-6 induced at mucosal surfaces by gram-negative bacterial infection. Infect Immun. 1989;57:3383–8. doi: 10.1128/iai.57.11.3383-3388.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duell BL, Carey AJ, Tan CK, et al. Innate transcriptional networks activated in bladder in response to uropathogenic Escherichia coli drive diverse biological pathways and rapid synthesis of IL-10 for defense against bacterial urinary tract infection. J Immunol. 2012;188:781–92. doi: 10.4049/jimmunol.1101231. [DOI] [PubMed] [Google Scholar]
- Engel D, Dobrindt U, Tittel A, et al. Tumor necrosis factor alpha- and inducible nitric oxide synthase-producing dendritic cells are rapidly recruited to the bladder in urinary tract infection but are dispensable for bacterial clearance. Infect Immun. 2006;74:6100–7. doi: 10.1128/IAI.00881-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Yamamoto M, Akira S, et al. Mechanism of pathogen-specific TLR4 activation in the mucosa: fimbriae, recognition receptors and adaptor protein selection. Eur J Immunol. 2006;36:267–77. doi: 10.1002/eji.200535149. [DOI] [PubMed] [Google Scholar]
- Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerlings SE. Urinary tract infections in patients with diabetes mellitus: epidemiology, pathogenesis and treatment. Int J Antimicrob Ag. 2008;31(Suppl 1):S54–7. doi: 10.1016/j.ijantimicag.2007.07.042. [DOI] [PubMed] [Google Scholar]
- Godaly G, Otto G, Burdick MD, et al. Fimbrial lectins influence the chemokine repertoire in the urinary tract mucosa. Kidney Int. 2007;71:778–86. doi: 10.1038/sj.ki.5002076. [DOI] [PubMed] [Google Scholar]
- Grabiec AM, Tak PP, Reedquist KA. Function of histone deacetylase inhibitors in inflammation. Crit Rev Immunol. 2011;31:233–63. doi: 10.1615/critrevimmunol.v31.i3.40. [DOI] [PubMed] [Google Scholar]
- Griebling T. Urinary tract infection in women. In: Litwin M. S., Saigal C. S., editors. Urologic Diseases in America. Washington, DC: US Government Printing Office, NIH Pub. No. 07-5512; 2007. pp. 587–620. [Google Scholar]
- Griebling TL. Urologic diseases in America project: trends in resource use for urinary tract infections in women. J Urol. 2005a;173:1281–7. doi: 10.1097/01.ju.0000155596.98780.82. [DOI] [PubMed] [Google Scholar]
- Griebling TL. Urologic diseases in America project: trends in resource use for urinary tract infections in men. J Urol. 2005b;173:1288–94. doi: 10.1097/01.ju.0000155595.98120.8e. [DOI] [PubMed] [Google Scholar]
- Halili MA, Andrews MR, Sweet MJ, et al. Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem. 2009;9:309–19. doi: 10.2174/156802609788085250. [DOI] [PubMed] [Google Scholar]
- Hang L, Haraoka M, Agace WW, et al. Macrophage inflammatory protein-2 is required for neutrophil passage across the epithelial barrier of the infected urinary tract. J Immunol. 1999;162:3037–44. [PubMed] [Google Scholar]
- Hannan TJ, Totsika M, Mansfield KJ, et al. Host-pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol Rev. 2012;36:616–48. doi: 10.1111/j.1574-6976.2012.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraoka M, Hang L, Frendeus B, et al. Neutrophil recruitment and resistance to urinary tract infection. J Infect Dis. 1999;180:1220–9. doi: 10.1086/315006. [DOI] [PubMed] [Google Scholar]
- Hedges S, Anderson P, Lidin-Janson G, et al. Interleukin-6 response to deliberate colonization of the human urinary tract with gram-negative bacteria. Infect Immun. 1991;59:421–7. doi: 10.1128/iai.59.1.421-427.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund M, Frendeus B, Wachtler C, et al. Type 1 fimbriae deliver an LPS- and TLR4-dependent activation signal to CD14-negative cells. Mol Microbiol. 2001;39:542–52. doi: 10.1046/j.1365-2958.2001.02205.x. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–52. [PubMed] [Google Scholar]
- Hultgren SJ, Porter TN, Schaeffer AJ, et al. Role of type 1 pili and effects of phase variation on lower urinary tract infections produced by Escherichia coli. Infect Immun. 1985;50:370–7. doi: 10.1128/iai.50.2.370-377.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingersoll MA, Kline KA, Nielsen HV, et al. G-CSF induction early in uropathogenic Escherichia coli infection of the urinary tract modulates host immunity. Cell Microbiol. 2008;10:2568–78. doi: 10.1111/j.1462-5822.2008.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intine RV, Sarras MP. Metabolic memory and chronic diabetes complications: potential role for epigenetic mechanisms. Curr Diabetes Rep. 2012;12:551–9. doi: 10.1007/s11892-012-0302-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzer A, Lim S, Fronhoffs F, et al. Lysine-specific demethylase 1 (LSD1) and histone deacetylase 1 (HDAC1) synergistically repress proinflammatory cytokines and classical complement pathway components. Biochem Bioph Res Co. 2012;421:665–70. doi: 10.1016/j.bbrc.2012.04.057. [DOI] [PubMed] [Google Scholar]
- Kim JI, Jung KJ, Jang HS, et al. Gender-specific role of HDAC11 in kidney ischemia- and reperfusion-induced PAI-1 expression and injury. Am J Physiol-Renal. 2013;305:F61–70. doi: 10.1152/ajprenal.00015.2013. [DOI] [PubMed] [Google Scholar]
- Langermann S, Palaszynski S, Barnhart M, et al. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science. 1997;276:607–11. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martinez JJ, Mulvey MA, Schilling JD, et al. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000;19:2803–12. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal R, Chhibber S, Sharma S, et al. Macrophage inflammatory protein-2, neutrophil recruitment and bacterial persistence in an experimental mouse model of urinary tract infection. Microbes Infect/Institut Pasteur. 2004;6:1326–32. doi: 10.1016/j.micinf.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Mossman KL, Mian MF, Lauzon NM, et al. Cutting edge: FimH adhesin of type 1 fimbriae is a novel TLR4 ligand. J Immunol. 2008;181:6702–6. doi: 10.4049/jimmunol.181.10.6702. [DOI] [PubMed] [Google Scholar]
- Mowat A, Baum J. Chemotaxis of polymorphonuclear leukocytes from patients with diabetes mellitus. New Engl J Med. 1971;284:621–7. doi: 10.1056/NEJM197103252841201. [DOI] [PubMed] [Google Scholar]
- Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun. 2001;69:4572–9. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneerselvam S, Govindasamy S. Sodium molybdate improves the phagocytic function in alloxan-induced diabetic rats. Chem Biol Interact. 2003;145:159–63. doi: 10.1016/s0009-2797(02)00254-5. [DOI] [PubMed] [Google Scholar]
- Patel S, Santani D. Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol Rep. 2009;61:595–603. doi: 10.1016/s1734-1140(09)70111-2. [DOI] [PubMed] [Google Scholar]
- Pereira MA, Sannomiya P, Leme JG. Inhibition of leukocyte chemotaxis by factor in alloxan-induced diabetic rat plasma. Diabetes. 1987;36:1307–14. doi: 10.2337/diab.36.11.1307. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Roche JK, Keepers TR, Gross LK, et al. CXCL1/KC and CXCL2/MIP-2 are critical effectors and potential targets for therapy of Escherichia coli O157: H7-associated renal inflammation. Am J Pathol. 2007;170:526–37. doi: 10.2353/ajpath.2007.060366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald A. The etiology of urinary tract infection: traditional and emerging pathogens. Am J Med. 2002;113(Suppl 1A):14S–19S. doi: 10.1016/s0002-9343(02)01055-0. [DOI] [PubMed] [Google Scholar]
- Rosen DA, Hung CS, Kline KA, et al. Streptozocin-induced diabetic mouse model of urinary tract infection. Infect Immun. 2008;76:4290–8. doi: 10.1128/IAI.00255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling JD, Martin SM, Hung CS, et al. Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. P Natl Acad Sci USA. 2003;100:4203–8. doi: 10.1073/pnas.0736473100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling JD, Mulvey MA, Vincent CD, et al. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J Immunol. 2001;166:1148–55. doi: 10.4049/jimmunol.166.2.1148. [DOI] [PubMed] [Google Scholar]
- Scholes D, Hooton TM, Roberts PL, et al. Risk factors associated with acute pyelonephritis in healthy women. Ann Intern Med. 2005;142:20–7. doi: 10.7326/0003-4819-142-1-200501040-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin RD, Engberg I, Hagberg L, et al. Neutrophil recruitment and bacterial clearance correlated with LPS responsiveness in local gram-negative infection. J Immunol. 1987;138:3475–80. [PubMed] [Google Scholar]
- Song J, Bishop BL, Li G, et al. TLR4-initiated and cAMP-mediated abrogation of bacterial invasion of the bladder. Cell Host Microbe. 2007a;1:287–98. doi: 10.1016/j.chom.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Duncan MJ, Li G, et al. A novel TLR4-mediated signaling pathway leading to IL-6 responses in human bladder epithelial cells. PLoS Path. 2007b;3:e60. doi: 10.1371/journal.ppat.0030060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JS, Anderson JL, Watanakunakorn C, et al. Neutrophil dysfunction in diabetes mellitus. J Lab Clin Med. 1975;85:26–33. [PubMed] [Google Scholar]
- Tvinnereim AR, Hamilton SE, Harty JT. Neutrophil involvement in cross-priming CD8+ T cell responses to bacterial antigens. J Immunol. 2004;173:1994–2002. doi: 10.4049/jimmunol.173.3.1994. [DOI] [PubMed] [Google Scholar]
- Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene. 2010;29:157–73. doi: 10.1038/onc.2009.334. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu J, Zhen J, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014;86:712–25. doi: 10.1038/ki.2014.111. [DOI] [PubMed] [Google Scholar]
- Witt O, Deubzer HE, Milde T, et al. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Xue ML, Thakur A, Cole N, et al. A critical role for CCL2 and CCL3 chemokines in the regulation of polymorphonuclear neutrophils recruitment during corneal infection in mice. Immunol Cell Biol. 2007;85:525–31. doi: 10.1038/sj.icb.7100082. [DOI] [PubMed] [Google Scholar]
- Yokoo A, Kumamoto Y, Hirose T. Study on local immune response in diabetic mice, in which bactericidal capacity of the neutrophils had been damaged—Escherichia coli induced experimental urinary tract infection. Kansenshōgaku Zasshi: J Jpn Ass Infect Dis. 1994;68:861–71. doi: 10.11150/kansenshogakuzasshi1970.68.861. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J Cell Biochem. 2010;110:1306–13. doi: 10.1002/jcb.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]