

Abstract
Mitochondria are highly dynamic, except in adult cardiomyocytes. Yet, the fission and fusion-promoting proteins that mediate mitochondrial dynamism are highly expressed in, and essential to the normal functioning of, hearts. Here, we review accumulating evidence supporting important roles for mitochondrial fission and fusion in cardiac mitochondrial quality control, focusing on the PINK1-Parkin mitophagy pathway.Based in part on recent findings from in vivo mouse models in which mitofusin-mediated mitochondrial fusion or Drp1-mediated mitochondrial fission were conditionally interrupted in cardiac myocytes, we propose several new concepts that may provide insight into the cardiac mitochondrial dynamism-mitophagy interactome.
Keywords: mitochondria, autophagy, mitochondrial fusion, mitochondrial fission, Parkinson’s disease
Introduction to mitochondrial dynamics
Mitochondrial dynamics refers to organelle fission, fusion, and subcellular translocation. In many cell types mitochondria exist as interconnected reticular networks; mitochondrial dynamism is frequent, perhaps continuous, in such cells. In addition to structural remodeling of mitochondrial networks, mitochondrial fission and fusion are implicated in homeostatic maintenance of mitochondrial DNA stability and respiratory function, as well as preventing or propagating programmed cell death1. Recent results of in vivo cardiomyocyte-specific genetic manipulation have revealed that mitochondrial dynamics factors function in cardiomyocytes in other ways. This functional distinctiveness can be attributed in part to absence of mitochondrial networks and unusually slow mitochondrial turnover in hearts (at least compared to liver)2.Further, as irreplaceable cardiomyocytes do not normally undergo programmed cell death, mitochondrial dynamics proteins are not needed as part of apoptosis3. Nevertheless, the mitochondrial fusion factors mitofusins (Mfn) 1 and 2 and optic atrophy (Opa) 1, and the mitochondrial fission factor dynamin-related protein (Drp) 1, are highly expressed in mammalian hearts, wherein their genetic ablation provokes striking cardiac dysfunction4, 5. Here, we consider evidence supporting important roles performed byouter mitochondrial membrane (OMM) fusion factors, Mfn 1 and Mfn2, and fission factor Drp1, in cardiac mitochondrial quality control. Other roles for mitochondrial dynamics factors that may impact cardiac health and disease, such as physical tethering between cardiac mitochondria and sarcoplasmic reticulum that facilitates mitochondria calcium import6–8, Opa1-mediated regulation of mitochondrial cristae structure9,and Mfn- or Opa1-mediated regulation of cardiomyocyte differentiation5, 10, have been reviewed elsewhere.
In the first part of this manuscript we examine current concepts of how mitochondrial fission and fusion are accomplished through highly ordered processes necessitated by the dual membrane architecture of these essential, but potentially highly toxic, organelles. We introduce the paradigm of asymmetrical mitochondrial fission as a central event in mitochondrial quality control, followed by a discussion of the best understood molecular pathway by which defective mitochondria are culled from cells, PTEN-induced putative kinase 1 (PINK1)-Parkin mediated mitophagy. These notions were developed, and the molecular mechanisms elucidated, largely through in vitro studies using cell-types other than cardiomyocytes. Thus, the second part of the review discussesthe particular roles of Drp1, Mfn1, and Mfn2 in hearts, as revealed throughin vivo genetic manipulation. Finally, we explore points of controversy within the area of cardiacmitochondrial dynamism, focusing onunexpected molecular functions of mitochondrial dynamics and mitophagy factors.
Prior to examining the molecular mechanisms that mediate mitochondrial fission and fusion it may be helpful to consider why mitochondria remodel their structures through the seemingly complicated and energy intensive process of breaking apart (fission) and then re-forming (fusion) organelles. One reason may be cellular context. For example, complete dissolution of mitochondrialnetworks precedes mitosis. Such network disassembly conceivably not only facilitates cytokinesis, but helps to deliver roughly equal proportions of the parental cell mitochondrial pool to each daughter cell1; mitochondrial networks subsequently have to be re-established in each daughter cell via generalized organelle fusion.
Dismantling and then reconstituting the cellular mitochondria network through sequential organelle fission, distribution, and re-fusion may be the most efficient means of partitioning mitochondria in mitosis. Limited mitochondrial structural remodelingalso occurs between cell mitoses1, and one wonders about the biological advantages conferred by fusion-mediated incorporation of accessory organelles for focal network remodeling and growth, compared to (for example) organelle growth by enlargement and budding. We posit that the supramolecular structure ofthe mitochondrial respiratory apparatus lacks the plasticity necessary for existing organelles to morph into more complex structures. Thus, we discard the fluid-state model whereinmitochondrial respiratory chain complexes diffuse freely andinter-complex electron transfer occurs randomly11 in favor ofa solid state model in whichrespiratory complexes are organized into stable paracrystalline supercomplexes on the cristae12–14. Supporting this model, destabilizing respiratory supercomplexes(which promotes fluidity) diminishes mitochondrial respiratory function and increases oxidative damage15, 16. It seems that major structural modifications of respiratory supercomplexes on paracrystalline cristal membranes would first require destabilizing the membrane, then incorporating additional individual protein components, and finally re-constructing the original highly organized structure. This is complicated and potentially disruptive. It would beeasier to add pre-fabricated supercomplexes to pre-existing ones, as by fusingmitochondrial cristae.
In developing ideas about how fission and fusion of prefabricated mitochondria units is superior to fluidic mitochondrial remodeling we considered how military units are constituted and managed within an army’s hierarchical organization structure. Here, each soldier represents an individual respiratory complex protein. Grouped together, these proteins form a squad (analogous to a respiratory complex). Squads are arranged into platoons, and ~6 platoons comprise a functional unit, the company (like one complete respiratory chain). Approximately ~6 companies form a battalion (similar perhaps to one lamelliform crista), 2–6 battalions form a brigade (like one mitochondrion), and so on, to divisions and corps that collectively comprise an army; this is analogous to an entire cellular mitochondrial collective. Acknowledging obvious differences in stoichiometric relationships between military units and mitochondrial respiratory complexes,understandinghow the military manipulates its organization inspired thoughtsas to how the rigid mitochondrial structure could be modified:During major re-organizations an army minimizes disruption by adding units rather than individual soldiers. To increase the size of a company, platoons are added. To increase the size of a battalion, companies are added, etc. Individual soldiers (proteins) are incorporated at a slow and steady pace to replace soldiers lost through retirement or attrition (representing mitochondrial protein biogenesis), butnotto change the structure of the overall organization. By analogy, making a larger or different shaped mitochondrion through the wholesale incorporation of individual proteins (growing and budding) would interrupt ongoing organelle function. It would be better to add or subtract intact functional units in the form of respiratory supercomplexes, through mitochondrial fusion or fission.
Molecular mechanisms and cell biology of mitochondrial dynamism
Mitochondrial fission
As introduced above, mitochondria can undergo fission to remodel their physical structure. Additionally, they undergosymmetric fission to replicate and expand the cellular mitochondrial pool, and asymmetric fission as the initial step of a highly orchestrated process that maintains cell-wide mitochondrial fitness by sequestering and eliminating damaged organelle components. Replicative fission that facilitates distribution of mitochondria to daughter organelles has been reviewed elsewhere1, 5. Here we focus on asymmetric fission and targeted mitophagy as central steps in the overall mitochondrial quality control process.
The major protein effector of mitochondrial fission isDrp1. Structurally, Drp1 possesses an amino terminal GTPase domain, a middle domain, and a carboxyl terminal GTPase effector domain (Figure 1). Under normal cellular conditions, 97% of Drp1 is cytosolic17; it therefore must be recruited to the OMM to promote fission.Analogous to the actions of dynamin in endocytic vesicle fission18, OMM-localizedDrp1 self-assembles into spiralsand then constrictsin a GTP-dependent manner,ligating and separating the inner and outer mitochondrial membranes19, 20 (think of Drp1 as forming individual sausage links from a single long sausage casing; Figure 1). The particular factors that stimulate Drp1 translocation from the cytosol differ in apoptosis, mitosis, mitophagy, and mitochondrial remodeling. For example, during cell division Drp1 is phosphorylated by mitotic kinase cyclin B-CDK1 complex21 andthen recruited to the mitochondria through the actions of a phosphorylated small Ras-like GTPase called RALA and its effector RALBP122. On the other hand, during apoptosis Drp1 interacts with Bax and contributes to cytochrome c release23. Accordingly, genetic or chemical inhibition of Drp1 protect from some, but not all, forms of programmed cell death23–26.
Figure 1. Sausage link model of mitochondrial fission by Drp1.

Top, schematic depiction of Drp1 molecular structure. The GTPase domain (green) is at the amino terminus and the GTPase-effector domain (GED; rust) is at the carboxyl terminus. The GED interacts with the middle domain of adjacent molecules (dashed lines) to promote head-to-toe oligomerization. GTP-mediated constriction of oligomeric ring structures constricts and severs the mitochondrion like forming sausage links (bottom). Sites for some post-translational modifications are represented on the uppermost structure; P is phosphorylation, NO is S-nitrosylation, SUMO is sumoylation.
Drp1 constriction sites are often marked by endoplasmic reticulum (ER); the ER-mitochondria contact is prior to, and independent of, Drp1 recruitment20. As a consequence of impaired mitochondrial fission, fibroblasts derived from Drp1-deficient mice are hyper-elongated and partially resistant to mitochondrial fragmentation induced by pharmacological uncoupling agents27. Nevertheless, the filamentous mitochondria of Drp1 null murine embryonic fibroblasts (MEFs) undergo fragmentation during mitosis, revealing the existence of one or more mechanisms capable of promoting mitochondrial fission independent of Drp127.
Because Drp1 lacks a clear hydrophobic transmembrane domain it is thought to bind to the OMM via resident receptor proteins. Fission 1 (Fis1) is a major Drp1 receptor protein in yeast, but Fis1 deficiency does not disrupt Drp1 association with mitochondriaor affect mitochondrial morphologyin colon carcinoma cells28. On the other hand, Mitochondrial fission factor (Mff) colocalizes with Drp1 on mammalian mitochondria, and RNAi-mediated Mff knockdown both reduces Drp1 association and induces mitochondrial elongation; Mff overexpression promotes mitochondrial fragmentation29.Other proteins that may help recruit Drp1 to OMM are mitochondrial dynamics protein of 49 and 51 kDa (MiD49 and MiD51)30, 31. Finally,there is intriguing evidence that Drp1 can bind to mitofusins and evoking a change in Mfn structure from a fusion-incompetent closed conformation to a fusion-competent open configuration32 (see Figure 2). This putative Drp1-Mfn interaction would have the effect of inverting the canonical fission-promoting function of Drp1 bypromoting Mfn-mediated fusion. Thus, Drp1 is functionally inert when sequestered in the cytosol. Drp1 provokes mitochondrial constriction and fission after binding to Mff and possibly other proteinsat sites of ER contact. And, mitochondria-localized but non-oligomerized Drp1 may promote mitochondrial fusion by binding to and dis-inhibiting mitofusins. How the various effects of Drp1 may be regulated through phosphorylation, S-nitrosylation, ubiquitination and SUMOylation (Figure 1) is under investigation33.
Figure 2. Turducken model of mitochondrial structure and fusion by Mfn1, Mfn2, and Opa1.
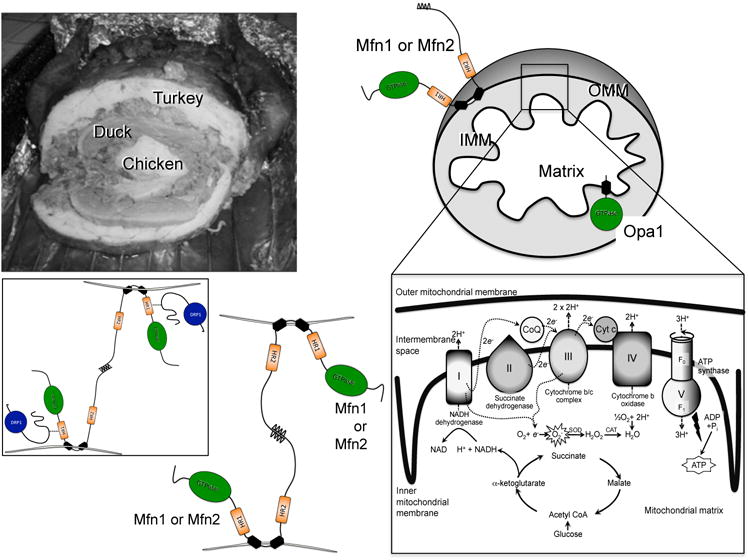
Top, multi-compartment structure of turducken (left) and cartoon mitochondrion (right). Outer mitochondrial membrane (OMM) fusion protein mitofusins (Mfn) 1 or 2 and inner mitochondrial membrane (IMM) fusion protein Opa1 are shown with GTPase domains in green. Exploded view of electron transport chain and associated pathways is bottom right. Bottom left, schematic depiction of Mfn-Mfn binding in trans, tethering two mitochondria; inset is authors’ conception of how pro-fission protein Drp1 (blue) may facilitate Mfn-mediated fusion.
Mitochondrial fusion
As an engineering problem, mitochondrial fission is straightforward: ligate and separate (like making sausage links). The double membrane/double space structure of mitochondria is no impediment to Drp1-mediated ligation. The sausage metaphor is imperfect, however, as mitochondria are constructed more like a turducken than a brautwurst.Turducken (popularized by the great NFL coach and sports announcer John Madden during Thanksgiving Day football broadcasts of the 1980s and 1990s34) is a chicken stuffed inside a duck stuffed inside a turkey. This creates concentric layers of poultry, resembling the central compartment ofmitochondrial matrix, enclosed by the crista/IMM, which is separated from the OMM by the inter-membrane space (Figure 2). This double membrane, double space architecture is critical for proper respiratory function andthe integrity of the compartments must be maintained throughout fusion. Thus (like making a large turducken from two turkeys, two ducks, and two chickens), mitochondrial fusion occurs layer by layer. The first step involves physical tethering of two mitochondria via trans interactions between the carboxyl terminal domains of OMM mitofusins of two organelles. Like Drp1, Mfn1 and Mfn2 are dynamin familyGTPases. Both Mfn isoforms have cytosolic N-terminal GTPase domains, two cytosolic hydrophobic heptad repeat coiled-coil domains (HR1 and HR2), and a small hydrophobic transmembrane domain(Figure 2). Consequently, mitofusins insert into the OMM much like a safety pin in cloth, with the vast majority of the protein being exposed to the cytosol and available to interact with cytosolic factors(Figure 2).Because Mfn1 and Mfn2 are similar, inter-molecular interactions between the cytosolic carboxyl-terminal HR2 domains of mitofusins located on different organelles can occur in either a homotypic (Mfn1-Mfn1 and Mfn2 –Mfn2) or heterotypic (Mfn1-Mfn2) manner35 with the heterotypic shown to be more efficient and to yield a higher rate of successful fusion events36.Additionally, Mfn2 (but not Mfn1) localizes to endo/sarcoplasmic (ER/SR) reticulum as well as mitochondria. For this reason, Mfn2 contributes to mitochondria-ER/SR calcium signaling, regulating acute metabolic demand andpathological ER stress37–39. Mitofusin-mediated physical tethering of mitochondria isGTP-independent and fully reversible, whereas GTP hydrolysis is essential to irreversible OMM fusion.
As noted, mitofusins are essential for the first two stages of mitochondrial fusion (tethering followed by OMM fusion). Accordingly, genetic deletion or RNAi-mediated suppression of mitofusins in cultured cells and Drosophila or mammalian cardiomyocytes produces abnormally small organelles, commonly referred to as “fragmented mitochondria” (although mechanistically they do not actively fragment, but are simply unable to undergo normal fusion)35, 40–43. The location of Mfn1 and Mfn2 on the OMM, which is the physical interface between mitochondria and cytosol, optimally positions them to participate in information exchange between the mitochondrion and its host cell. As discussed below, the pathophysiological implications of mitofusin expression, regulation, and dysfunction therefore extend far beyond simply modulating mitochondrial morphology.
Membrane-by-membrane mitochondrial fusion maintains the structural integrity of the IMM and matrix, thus preservingoxidative phosphorylation and avoiding formation (or at least maintaining the physical sequestration)of cytotoxic oxidizing molecules generated upon interruption of the electron transport chain(Figure 2). Accordingly, after OMM fusion a related dynamin family GTPase, Opa1, promotes IMM fusion44,45. The consequences of Opa1 loss of function in cultured cells and in vivo models are both phenotypically and mechanistically complex. Mitochondrial tethering and OMM fusion still occur (through the actions of Mfn1 and Mfn2), but absence of Opa1-mediated IMM fusion produces mitochondria that are not only structurally heterogenous, but that exhibit generalizeddissipation of the normal IMM electrochemical potential andprofoundly impaired cellular respiration46. These results point to a major role for Opa1 in maintaining normal crista morphology that is essential for proper assembly and functioningof electron transport chain supercomplexes47, 48. The differing roles played by OMM and IMM fusion factors have been demonstrated in Drosophila heart tubes: interrupting Mfn-mediated OMM fusion evoked a cardiomyocyteER stress response that could be rescued by enhancing mitochondrial processing of unfolded proteins, whereas interrupting Opa1-mediated IMM fusion compromised mitochondrial function that was rescued by reactive oxygen species scavenging49.
Dys-dynamism; what happens when mitochondrial fission and fusion go wrong
The biological importance of mitochondrial fission and fusion is unambiguously revealed by embryonic lethality inhomozygousmouse knockouts of the genes encodingMfn1, Mfn2, Opa1 or Drp127, 40, 50.Loss of function genetic mutations linked to clinical disease underscorethe importance of intact fission and fusion in the human condition: Mfn2 mutations cause Charcot-Marie-Tooth syndrome type 2A, an autosomal dominant axonal peripheral neuropathy51,Opa1 mutations cause autosomal-dominant optic atrophy52, and a Drp1A395D mutation identified in an infant withmultisystem failure impairs Drp1 assembly and induced a mitochondrial fission defect53.
Altering the normal balance between mitochondrial fusion and fission in cells with interconnected mitochondrial networks has predictable effects on mitochondrial morphometry. A shift toward increased fusion and/or decreased fission produces elongated organelles with greater interconnectivity. Conversely, decreased fusion and/or increased fissionproduces shorter organelles havingpoorly connected networks, the so-called fragmentedmitochondria phenotype27, 40, 46, 54. Conventional wisdom holds that more connectivity (and by inference more fusion or less fission) is a healthier situation, and that less connectivity (or fragmentation) is deleterious. These pathophysiological inferences make sense given that mitochondrial networks disintegrate early in cells undergoing apoptosis55, and that fusion between healthy and damaged mitochondria has the potential to dilute out damaged components in the latter, referred to as repair via complementation56. Indeed, pharmacological inhibition of Drp1-induced mitochondrial fission is protective in a number of different tissue injury models57–61.However, the notion that small or fragmented mitochondria are necessarily bad (especially in cardiac myocytes wherein the mitochondria are already “fragmented” in comparison with most other cell types)is not supported by a number of observations: 1. Highly fused or interconnected mitochondria are more, not less, susceptible to staurosporine-stimulated apoptosis62; 2. Cells with defective mitochondrial fusion caused by Mfn2 deficiency are protected against programmed cell death63; 3. Forced Drp1 expression in Drosophila cardiomyocytes provokes mitochondrial fragmentation withoutany apparent adverse consequences49; and 4. Activation of nutrient oxidation by adrenergic stimulation of the brown adipose tissue is accompanied by mitochondrial fragmentation that is essential for increasing energy expenditure in this tissue64. Thus, as recently opined by Sun and Finkel65 it seems unreasonable to conclude that mitochondrial dysmorphology is the primary or only cause of organelle, cellular, or tissuedysfunction reported after genetic mitofusin ablation in vitro66 or in vivo67–69.Finally, a bidirectional exchange of mitochondrial components between healthy and damaged mitochondriahas the potential to harm the healthy organelle as well as to help the damaged one. We refer to fusion-mediated contamination of good mitochondria by bad as“mitochondrial contagion”, which occurs in mitophagically impaired Parkin-deficient Drosophila hearts andcan be prevented by suppressing mitofusins70. In the following sections we explore the inextricable interplay between mitophagy and mitochondrial dynamism.
How mitochondrial dynamism impacts mitochondrial quality control
The word “mitophagy” is a contraction of mitochondria and autophagy, and refers to the process by which cells eat their own mitochondria. Mitophagic engulfment of mitochondria by autophagosomes and subsequent transfer to degradative lysosomes can occur during generalized macroautophagy as during nutrient deprivation, or as a highly selective process that targets dysfunctional mitochondria71. Selective mitophagy as a means of mitochondrial quality control is the current focus.
There is a tendency to conceive of mitochondria as living linear lives. Mitochondria are “born” or assembled through biogenesis72, they function for a time, and after an inevitable age-related decline in function are either repaired (through fusion-mediated complementation)56, 73 or transported, via mitophagy, to the figurative mitochondrial junkyard. As comfortable as this anthropomorphic construct is, mitochondria are not people. Rather, the life cycle of mitochondria resembles that of the primordial bacteria from which they are descended74, 75 (Figure 3). Mitochondria replicate through symmetric fission: a healthy parent organelle produces two healthy daughters that grow by adding new components through mitochondrial biogenesis (i.e. induction of nuclear-encoded mitochondrial genes and incorporation of their protein products into pre-existing mitochondria) and by fusing with other healthy mitochondria. Over time or as a consequence of extrinsic stress a given mitochondrion will sustain damage too severe for correction through biogenic or fusion-mediated repair and is removed via mitophagy.
Figure 3. Replicative and asymmetric fission and intra-organelle partitioning of damaged components.

Left; diagram of symmetric replicative fission (top) and asymmetric fission leading to selective mitophagy of the impaired daughter organelle (bottom). Right; illustration with Lego blocks of how misfolded proteins (front right) might passively segregate from functioning respiratory supercomplexes (rear left).
To understand the importance of mitophagic quality control it is worth emphasizing that healthy mitochondria employ oxidative phosphorylation to produce energy in the form of ATP that drives virtually all biological processes, but damaged organelles transform into cytotoxic factories for reactive oxygen species (ROS). Thus to paraphrase Erasmus of Rotterdam: “Mitochondria – can’t live with them, nor without”. Mitochondrial ATP synthesis depends upon an electrochemical gradient generated across the inner mitochondrial membrane through a series of redox reactions (see Figure 2): the transfer of electrons between complexes of the electron transport chain is coupled to extrusion of protons (hydrogen ions) across the inner mitochondrial membrane and into the mitochondrial intermembrane space. Reversal of this proton flow powers the mitochondrial ATPsynthase, a molecular rotor76. The terminal electron acceptor is molecular oxygen, forming superoxide anion (O2−) that is normally reduced through the sequential actions of superoxide dismutase (to form hydrogen peroxide; O2− + 2H → H2O2) and catalase (to form water; 2 H2O2→ 2 H2O + O2). When electrons prematurely leak from complexes I or III of the electron transport chain77, 78, or when specific endogenous mitochondrial enzymes such as Romo1 (Reactive oxygen species modulator 1) are activated79, damaging superoxide radicals and/or hydrogen peroxide can accumulate within or escape from mitochondria and attack critical proteins, lipids, and mitochondrial or nuclear DNA. Because production of ROS in damaging amounts does not normally occur, substantial ROS production is a marker of mitochondrial dysfunction. As protection against injury from mitochondrial-derived ROS thatoverwhelm endogenous protective mechanisms (catalase and superoxide dismutase80) cells employ sophisticated surveillance and elimination mechanisms to identify and remove dysfunctional mitochondria.
The cellular decision to remove a dysfunctional mitochondrion is necessarily dichotomous; either the organelle is retained or it is targeted for autophagy. Thus, a continuum of mitochondrial fitness ranging from the perfectly healthy ATP generator at one extreme to the completely dysfunctional toxic ROS generator at the other has totriggera categorical decision regarding mitochondrial fate. A decision to reserve mitophagy for severely damaged mitochondria places the cell at risk from toxic effects of dysfunctional organelles that have not achieved the high threshold of dysfunction necessary to trigger their removal. If, however, the cell lowers its thresholdfor mitochondrial removal and targets less severely impaired organelles, it will inevitably eliminate mitochondria still functioning at some level, thus depriving itself of their ATP. Furthermore, the cell will have to expend resources to replace the recycled organelles. Nature has addressed this quandary by integrating mitochondrial dynamism with mitophagy in the form of asymmetric mitochondrialfission81.
Pulse chase experiments show that in the absence of a general signal for the reduction in mitochondrial mass, mitochondria that are targeted for mitophagy have a relatively depolarized membrane potential before being targeted for autophagy81. This subpopulation of mitochondria also have reduced likelihood of being involved in fusion events as both mitofusions and OPA1 fusion proteins are either cleaved or degraded, a process induced by either inner membrane depolarization or reduced mitochondrial ATP production82. Thus, mitochondria targeted for autophagy are characterized by being relatively depolarized and remaining solitary. The time between mitochondrial depolarization and autophagosomal engulfment varies from less than 1 hour to ~3 hours, suggesting the existence of a transient population of pre-autophagic mitochondria81, 83. The existence of a pre-autophagic pool may explain the heterogeneity of mitochondrial membrane potential in most cell types84. The process that feeds mitochondria into the pre-autophagic pool is therefore expected to play a key role in determining the rate of removal of mitochondria. The development of a technology to label individual mitochondria and track their membrane potential using membrane potential dyes allowed for the identification of the event at which depolarized mitochondria are produced85. A photo-activatable form of green fluorescent protein (PA-GFP) has been targeted to the matrix of mitochondria. Laser mediated photo-conversion of the PA-GFP in one mitochondrion allowed its tagging and tracking, and staining with positively charged dyes such as TMRE provided information on membrane potential. This kind of analysis showed that fission events produce functionally dissimilar daughter mitochondria; in other words, asymmetric mitochondrial fission. While one daughter leaves the fission event with membrane potential that is similar or higher compared to the mother mitochondrion, the other daughter may have depolarized membrane potential that may recover or persist. The latter daughter has reduced chance of being involved in a fusion event and if membrane potential is not recovered, it is becoming part of the pre-autophagic pool. Evidence supports that asymmetric fission is the result of uneven distribution of dysfunctional mitochondrial components including oxidized or older proteins81, 86. This model explains the role of fusion in mitochondria quality control as it may allow for the redistribution of damaged components while fission and mitophagy are responsible for the elimination. Since fusion and mitophagy are two competing fates of the post fission daughter mitochondria, giving preference to the daughter with the higher membrane potential generates a sorting mechanism where damaged material is segregated before being eliminated. The selective nature of fusion is therefore key to the efficiency of the quality control mechanism. The selectivity is dependent on transitioning of dysfunctional daughter mitochondria from being fusion competent to permanently solitary mitochondria, a process that may be mediated by coupling the degradation or post-translational modification of fusion proteins to the respective bioenergetic functioning of the healthy and impaired daughter mitochondria.
The mechanism by which sub-mitochondrial partitioning of damaged from healthy components is achieved prior to asymmetric fission is not known, but similar asymmetrical fission quality control processes are observed in budding yeast87–89. The paracrystalline structure of IMM mitochondrial respiratory supercomplexes suggests an intriguing possibility12–14: because misfolded and damaged proteins are physically incompatible with the supermolecular structure of IMM respiratory complexes they are passively excluded. To illustrate this point, consider (instead of soldiers and army units) respiratory proteins of the IMM to be Lego blocks, each fitting with the next to form respiratory complexes that fit together to form supermolecules, whichin turn fit together to form supercomplexes (Figure 3). Normal Lego proteins can be readily inserted (like biogenesis) or entire units added (like fusion). But misfolded proteins, represented here by Legos melted in the microwave (likely by some toddling future cardiovascular scientist), cannot be integrated into the highly ordered structure. Because they do not fit they segregate and accumulate (Figure 3). The same process may apply to proteins of the IMM. After partitioning, physically separating normal from abnormal Lego proteins (as by mitochondrial fission), placing the normal Legos back in the box with other functional Legos for future use (as with mitochondrial fusion), and disposing of damaged components in the trash (as by mitophagy) follows naturally.
Mitochondrial dynamism-mitophagy cross talk in the heart
If the concept of asymmetric mitochondrial fission in mitophagy is correct, then interruption of mitochondrial fission or fusion should impact not only mitochondrial shape, but mitochondrial quality. Several recent reports suggest that this is, in fact, the case.
The first suggestion that genetic perturbation of cardiomyocyte mitochondrial dynamics factors would compromise mitochondrial quality control mechanisms was the observation that unusually small, degenerated mitochondria accumulated in adult mouse hearts after conditional combined ablation of the mitochondrial fusion factors Mfn1 and Mfn243. In that study an increase in physically abnormal mitochondria was associated with impaired cardiomyocyte respiration, but not with measurable alterations in substrate-stimulated oxygen consumption by isolated cardiac mitochondria. This internal inconsistency was subsequently resolved when it was discovered that standard isolation procedures did not efficiently capture the fragmented mitochondria produced by interrupting mitochondrial fusion. After the procedure was appropriately modified not only was it clear that Mfn1/Mfn2 double deficient mitochondria are functionally impaired, but the smallest mitochondria exhibit the greatest dysfunction54. So, why weren’t these impaired mitochondria eliminated by the mitophagy quality control apparatus? Simply interrupting fusion should not directly interrupt mitophagy (see Figure 3). Indeed, individual cardiac deletion of Mfn1 had little effect on cardiomyocyte mitochondria, whereas deletion of Mfn2 provoked a chronically progressive cardiomyopathy associated with accumulation of enlarged (not fragmented) mitochondria90. This result suggested a role for Mfn2, but not Mfn1, in mitochondrial quality control, prompting the discovery that PINK1-phosphorylated Mfn2 is a mitochondrial binding partner for Parkin. Studies of Mfn2-deficient hearts alsoled to detection of a Parkin-independent, ROS-dependent alternate pathwayfor mitochondrial quality control91. Thus, the mitochondrial fusion protein Mfn2 is essential to Parkin-mediated mitophagy, at least for mouse cardiomyocytes and neurons90, 92.
Three recent papers have also implicated the mitochondrial fission factor Drp1 in cardiac mitophagy. Cardiac-specific ablation of Drp1 in the early postnatal period54, 93 or in adult mice54, 94 induced cardiomyopathies and perturbed cardiac macroautophagy or mitochondrial autophagy; the exact nature of the abnormality in mitophagy/autophagy attributed to Drp1 deficiency differed between studies. It is worth considering that if asymmetric mitochondrial fission normally precedes mitophagy as depicted in Figure 3, then chronic suppression of fission by ablating Drp1 would be predicted to have different consequences on mitophagy, depending upon whether mitophagy is assayed early or late after Drp1 deletion4. Shortly after loss of Drp1-mediated fission, mitophagy should be suppressed because the mechanism by which damaged and dysfunctional components are segregated into the depolarized daughter organelle is lost. With time, however, as the overall health of the cellular mitochondrial population deteriorates due to interruption of the asymmetric fission/mitophagy process, the parent organelles will themselves become sufficiently dysfunctional to trigger mitophagy; when this occurs cell-wide, mitophagy will be seen to be increased. Indeed, if mitochondrial biogenesis is inadequate to replace mitochondria in sufficient numbers, then accelerated mitophagy late after Drp1 ablation will consume a significant portion of the mitochondrial pool, resulting in mitochondrial insufficiency. The aggregate data to date support this time-dependent evolution in phenotype4, 54, 94, but more definitive studies using better genetic models are necessary to concretely establish a role for Parkin-mediated mitophagy in the cardiomyopathy that develops after cardiac Drp1 ablation. In the following section we examine our current understanding of the molecular events underlying mitophagy, and explore the implications of functional cross-talk between mitophagy and mitochondrial dynamism mediated by the mitochondrial fusion factor, Mfn2.
Mfn2 and PINK1-Parkin mitophagy signaling
The most thoroughly explored mechanism for homeostatic mitochondrial quality control is PINK1-Parkin mediated mitophagy. PINK1 (PTEN-induced putative kinase) and Parkin are prototypical Parkinson’s disease factors; mutations in their genes were the first identified causative events linked to hereditary (autosomal recessive) early onset Parkinson’s disease95, 96. The foundation for our current understanding of mitophagy was discoveringhow the mitochondrial localized kinase PINK1 interacts functionally with the cytosolic E3 ubiquitin ligase Parkin, which was definedin Drosophila97, 98 and extended to mammalian systems99. A detailed discussion of the individual fruit fly studies that linked Parkin and PINK1 to mitochondrial fitness, and eventually proved that PINK1 is upstream of Parkin in a single mitochondrial quality control pathway, is beyond the scope of this review, but it is a fascinating tale of how genetic manipulation in flies provided fundamental biological insights into the human condition100.
PINK1, the ignition switch for Parkin-mediated mitophagy
If mitochondrial depolarization, ROS, or protein misfolding are the input variables that activate mitophagy101,102, 103, then PINK1 senses these inputs and initiates the appropriate response. Mitochondrial damage stimulates PINK1 kinase activity, which turns on Parkin-mediated mitophagy. The molecular mechanisms by which this happens were elucidated by Youle and colleagues101. For those interested in a detailed history of these discoveries and the neurosciences perspective, Youle recently authored a detailed overview of the fundamentals of PINK1 – Parkin biology in the context of Parkinson’s disease104.
The revelation that PINK1 is central to mitochondrial quality control was unexpected because PINK1 protein levels are so low in normal mitochondria as to be virtually undetectable101; normal mitochondria are therefore PINK1-deficient. The key to unlocking mitophagy signaling was the observation that PINK1 is readily detected in damaged mitochondria, appearing after mitochondrial depolarization101. This area of investigation has seen rapid advances over the past few years; here and in Figure 4 we summarizecurrent concepts. PINK1 kinase is encoded by the PINK1 (previously called Park6) gene, which like all but the 13 proteins encoded by mitochondrial DNA is part of the nuclear genome. PINK1 protein is therefore translated by the ribosomal apparatus and imported into mitochondria. In normal mitochondria, as fast as PINK1 is imported it is proteolytically degraded. Consequently, normal mitochondria have very little PINK1 protein or PINK1 kinase activity101. Upon mitochondrial depolarization however, this import-and-immediately-degrade PINK1 process is interrupted, causing PINK1 to accumulate and phosphorylate its substrate proteins. Conceptually, this has been likened to a mitochodrial “dead-man switch”5, in which healthy mitochondria expend effort to actively degrade PINK1, thus staying alive by avoiding mitophagic destruction. In damaged mitochondria that no longer support PINK1 degradation, passive accumulation of PINK1 triggers the organelle’s mitophagic destruction.
Figure 4. Molecular events leading to selective mitophagy of dysfunctional mitochondria.
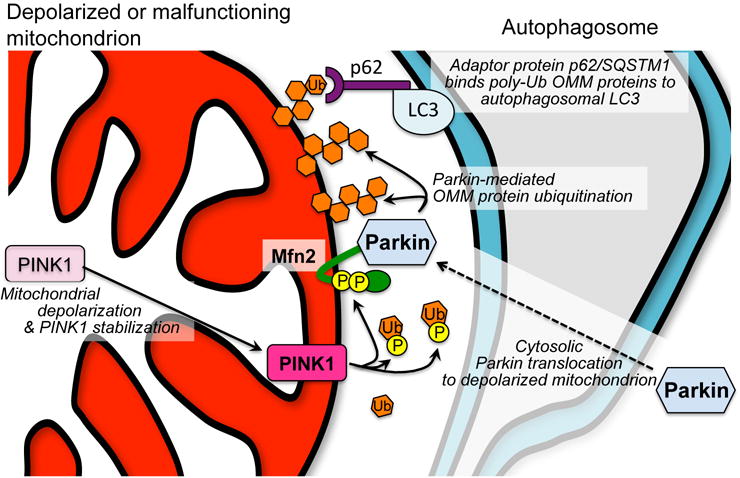
Left; mitochondrial depolarization or other impairment interrupts normal proteolytic processing of PINK1 kinase. PINK1 accumulates on the OMM and phosphorylates Mfn2, promoting its recruitment of Parkin, and ubiquitin (Ub), enabling its utilization by Parkin. Poly-ubiquitinated OMM proteins bind p62 linked to autophagosomal LC3, thus targeting the mitochondrion for mitophagy.
The specifics of mitochondrial PINK1 import and degradation are inextricably linked to Parkin-mediated mitophagy. (How mitochondria generally import nuclear-encoded proteins via membrane translocases for the OMM and IMM [called TOM and TIM, respectively] has been recently reviewed105.) Unprocessed 63 kDa PINK1 protein is transported across OMM by the TOM complex, delivered to the IMM translocase, TIM, and proteolytically processed in a manner typical for imported mitochondrial matrix proteins106. In healthy mitochondria, processed PINK1 is rapidly cleaved by PARL (presenilin-associated rhomboid-like protein)107, 108, generating a 52 kDa PINK1 fragment that escapes into the cytosol and undergoes proteasomal degradation directed by an N-Degron109. Thus,mitochondrial PINK1 levels are suppressed. Mitochondrial dysfunction impairs TIM-mediated PINK1 translocation across the IMM,protectingit from degradation by PARL and maintaining its physical association with TOM on the OMM110. From here PINK1 phosphorylates available substrates111.
Mfn2 is a phosphorylation substrate of PINK1 and a receptor for Parkin
To promote mitophagy, PINK1 that accumulates on damaged mitochondria must induce cytosolic Parkin to translocate to, and ubiquitinate OMM proteins on, the organelle. Simultaneously, to prevent contamination of the healthy mitochondrial pool by the damaged organelle, PINK1 inhibits fusion of the damaged mitochondrion. There are differing views regarding the biochemical events that induce mitochondrial Parkin localization and that interrupt mitochondrial fusion. Indeed, it is almost certain that signaling redundancy is built into this important mechanism of mitochondrial quality control and that multiple pathways involved.
PINK1 phosphorylates Parkin on Ser65112–116, and Parkin poly-ubiquitinates mitofusins82, 117–120. The former observation implicates PINK1-mediated phosphorylation in Parkin recruitmentto damaged mitochondria, whereas the latter observation suggests that selective Mfn degradation can suppress mitochondrial fusion in organelles targeted for mitophagic destruction121–124. But as Sherlock Homes declared (in Sir Arthur Conan Doyle’s The Bascombe Valley Mystery) “There is nothing more deceptive than an obvious fact”. Indeed, although PINK1 does phosphorylate Parkin (on Ser65 within the Parkin ubiquitin-like domain), recent studies from multiple laboratories have revealed that it is PINK1-phosphorylated ubiquitin, and not PINK1-mediated phosphorylation of Parkin itself, which is essential to enable Parkin as an E3 ubiquitin ligase125–128 (Figure 4).
The observation that PINK1 can phosphorylate ubiquitin complexes on OMM proteins128, and not just free ubiquitin125, 126, has suggested that non-specific anchoring of Parkin to phospho-ubiquitinated OMM proteins is a mechanism for its PINK1-mediated recruitment to mitochondria104, 129. This is consistent with the idea that Parkin binding to phospho-ubiquitin can accelerate Parkin-mediated OMM protein ubiquitination as a positive feedback amplification loop128. But this is a loop after all, and it seems inefficient to have the primary event of Parkin binding, which is a prerequisite for OMM protein ubiquitination, to rely upon pre-existing ubiquitinated OMM proteins. Chen and Dorn reported that PINK1-mediated phosphorylation of Mfn2 on Thr111 and Ser442 conferred Parkin binding activity to Mfn290. Functional ablation of these phosphorylation sites by their mutational substitution with Ala abrogated Mfn2-Parkin binding, whereas mimicking Mfn2 phosphorylation by mutational substitution with Glu conferred constitutive Parkin binding. According to this mechanism, stabilized PINK1 located on the OMM phosphorylates Mfn2, transforming it into a receptor to which Parkin can bind, thereby bringing it into physical proximity of its many mitochondrial ubiquitination substrates90. PINK1-mediated phosphorylation of free ubiquitin activates Parkin’s E3 ubiquitin ligase activity125, 126, and its phosphorylation of ubiquitinated OMM proteins amplifies mitophagy signaling128 (Figure 4).
The proposed role for PINK-phosphorylated Mfn2 as a Parkin receptor90 is viewed as controversial by some. The most common concern is reflected in comments by Pickrell and Youle104 that “Parkin translocates constitutively to mitochondria in Mfn1/Mfn2-KO cells, arguing that Mfn2 is not involved in translocation.” This may over-interpret the results of experiments performed using fibroblasts derived from germ-line knockout mice. While Parkin can indeed translocate to mitochondria of Mfn2-deficient murine embryonic fibroblasts (MEFs), it should be noted that mitophagy (measured as lysosomal incorporation of mitochondria after pharmacological uncoupling of respiration from ATP synthase) takes place in MEFs lacking any of the three postulated mitophagy effectors, PINK1, Mfn2, and Parkin (G Dorn, unpublished results). Results such as these130 do not demonstrate that Mfn2 (or PINK or Parkin) are unimportant in mitophagy. Rather, they support the existence of secondary compensatory mitophagy mechanisms131, 132,91 that are induced when primary pathway are interrupted. Indeed, both cardiomyocyte-specific and neuronal-specific deletion of Mfn2 in mice induce distinctive defects in Parkin localization to depolarized mitochondria90, 92. Absence of Parkin translocation in Mfn2 deficient neurons suggests thatanother concern, that Mfn2 may act as a Parkin receptor only in hearts133, is alsooverstated.
A mechanistic link between PINK1, Mfn2, and Parkin is attractive because it can explain how the PINK1-Parkin pathway simultaneously initiates mitophagy and shuts down fusion, as required to preclude mitochondrial contagion70. It is possible that PINK1-mediated phosphorylation may instantaneously convert Mfn2 from a fusion protein to a Parkin binding protein. This mechanism of modulating mitochondrial fusion in mitophagy would have the advantage of being more rapid and direct than Parkin-mediated Mfn2 ubiquitination, extraction, and proteasomal degradation82, 117–120. Indeed, the importance of Parkin-mediated ubiquitination of specific proteins, leading to their selective elimination from soon-to-be autophagocytized organelles,is unclear. Parkin-mediated ubiquitination of Mfn1 and Mfn2 might deplete these fusion promoting proteins by targeting them for proteasomal degradation, thus interrupting mitochondrial fusion and placing the organelle in quarantine until it can be mitophagically eliminated121, 122. Parkin does not selectively ubiquitinate pro-fusion proteins; it also ubiquitinates the pro-fission protein, Drp1134, which would move the fission/fusion equilibrium in the opposite way toward mitochondrial fusion. Indeed, Parkin promiscuously ubiquitinates a hundred or more OMM proteins135, 136, essentially painting the organelle with a coat of ubiquitin that attracts autophagosomes137. Thus, during mitophagy at least, Parkin-mediated OMM ubiquitination does not seem to fine tune OMM protein expression of organelles that, in any case, are shortly headed to the graveyard. (A more surgical role for Parkin in mitochondrial protein turnover via vesicular export is described below.) Furthermore, there is little delay between Parkin localization to mitochondria and their engulfment by autophagosomes. Live cell microscopy of cultured cells revealed that Parkin localization, focal protein ubiquitination, and regional mitochondrial fragmentation with autophagosomal engulfment all occur within minutes of mitochondrial injury83. This provides little time for selective extraction and proteasomal degradation of mitofusins.
As noted, this area of investigation is rapidly advancing. Uncertainties about the mechanism for Parkin translocation (are there chaperones?) and the identity of putative Parkin receptors require additional experimentation. For example, one can conceive of studies using Mfn2PINK1 phosphorylation site mutants having constitutive, or that completely lack, Parkin binding activity to interrogate PINK1-Parkin signaling in vivo.
In vivo studies of PINK1-Parkin signaling
The roles of PINK1 in Parkin-mediated mitophagy, as a proximal effector that accumulates only in depolarized mitochondria, as an inducer (via phosphorylation of Mfn2) of Parkin translocation to mitochondria, and as a Parkin activator through phosphorylation of ubiquitin, support its central role in this form of mitochondrial quality control. Given the nearly unassailable evidence that loss-of-function mutations in the PINK1 gene cause hereditary Parkinson’s disease in humans96, 138, 139, one might anticipate that PINK1 gene deletions engineered into mice would recapitulate the human neuropathology. That has not been the case. Indeed, multiple efforts to ablate PINK1 or Parkin in mice by genetically deleting exons encoding critical protein domains have produced a “negligible neurodegenerative pattern in the (dopaminergic neuron-rich) substantia nigra pars compacta, a region clearly affected in Parkinson’s disease patients”140. Even combined deletion of the mouse genes for PINK1, Parkin, and DJ-1 (another gene in which loss-of-function mutations have been causally linked to early Parkinsons disease) has failed to recapitulate the hallmark loss of dopaminergic neurons in the brain141. Perhaps not surprisingly, baseline cardiac phenotypes in PINK1 and Parkin knockout mice are also modest, although interruption of PINK1-Parkin mediated mitophagy increases susceptibility to myocardial damage in conditions such as advancing age and ischemic injury wherein chronic mitochondrial impairment likely requires aggressive mitophagic culling142–146. The failure of germ-line PINK1 or Parkin gene deletion in mice to recapitulate human disease phenotypes may be attributable to developmental plasticity that promotes induction of compensatory pathways. Favoring this notion is transcriptional upregulation of other E3 ubiquitin ligases in germ-line Parkin-knockout mouse hearts70.
What the future may hold…
One of the questions raised by apparent compensatory induction of alternate mitochondrial quality control pathways in mice (and possibly humans147) having genetic loss of PINK1 or Parkin function is “What is the nature of these other pathways?”. This area of research is active and our understanding is both incomplete and evolving. Conventional wisdom has been that mitochondrial depolarization or a downstream reduction in ATP synthesis may constitute central stimuli for mitophagy. However, depolarization may be a late and final result of a decompensating organelle. In the sections above we considered how it could be detrimental for cells to wait until mitochondria are completely depolarized before triggering their sequestration and removal. New work has identified other, potentially earlier stimuli for mitophagy. In one example, induction of the mitochondrial unfolded protein response prompted PINK1 accumulation and Parkin recruitment; these mitochondria were subsequently eliminated despite maintaining a healthy polarization status102. Likewise, increased mitochondrial release of ROS can stimulate alternate mechanisms of mitochondrial quality control in hearts having a primary impairment of Parkin recruitment91. It seems that different triggers exist for mitophagy that provoke a quality control reaction proportional to organelle damage. Here, we describe a variation on this theme, PINK1-Parkin involvement in preserving mitochondrial fitness via export of damaged proteins in mitochondrial-derived vesicles.
It may be helpful to consider PINK1-Parkin dependent mitophagy and mitochondria vesicle formation in the broader context of mitochondrial quality control mechanisms by introducing a new metaphor, your car. A new car (like a healthy mitochondrion) will, over time or because of untoward circumstances, run down and/or sustain damage. In this circumstance mitophagy is analogous to waiting until the car no longer runs, and then trading it in on a new model. Although necessary every few years, this is inconvenient and expensive. It is preferable to delay purchasing a new car by properly maintaining it and performing necessary repairs; our cells seem to take a similar approach to their mitochondria.
Functionality of cars (and mitochondria)can be optimized through preventative maintenance. Routine car maintenance includes exchanging used engine oil and filter, replacing worn tires and belts, and restoring any parts that unexpectedly fail; individual components are removed and replaced as needed. Mitochondria may accomplish the same thing, protein by protein, using endogenous proteases and the ubiquitin/proteasome pathway (Figure 5). Thus, unfolded or oxidized internal mitochondrial proteins (i.e. those that are located within the matrix or intermembrane space) are proteolytically degraded and replaced148. Mitochondrial outer membrane proteins that are not readily accessible to these proteases are subject to degradation via the standard ubiquitin/proteasome pathway after ubiquitination in a Parkin-independent manner, as by Mule/ARF-Bp1 mediated ubiquitination and removal of the anti-apoptotic factor Mcl-1149. Both of these mechanisms promote turnover of damaged or undesirable mitochondrial proteins in a manner that maintainsbasal function.
Figure 5. Mechanisms of mitochondrial quality improvement, from macro to micro.
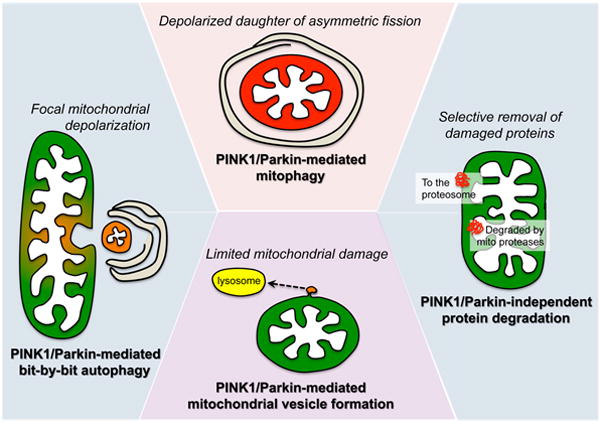
At the top is PINK1/Parkin-mediated mitophagy of an intact organelle, as with the depolarized daughter of an asymmetric fission event (see Figure 3). To the left is bit-by-bit autophagy of localized mitochondrial damage, which is excised and engulfed by an autophagosome via Parkin activity. At the bottom is PINK1/Parkin-mediated formation of mitochondria-derived vesicle, incorporating damaged components into a vesicle that transports them to lysosomes. On the right is shown PINK1-Parkin independent proteasomal removal of outer mitochondrial membrane proteins, and protease-dependent degradation of internal mitochondrial proteins, the most selective of the quality improvement processes.
Sometimes a car requires more than routine maintenance, but is not a total loss, such as needing new brakes or a water pump. In mitochondria this seems to be accomplished throughPINK1 and Parkin-mediated formation of mitochondrial vesicles and their export to lysosomes150. These mitochondrial vesicles contain oxidized proteins generated during times of oxidant stress151, 152. Heidi McBridehas proposed that focal oxidative damage to mitochondrial proteins provokes local accumulation of PINK1 kinase and limited recruitment of Parkin, promoting incorporation of a “patch” of damaged proteins into a newly-generated vesicle that separates from the mitochondrion and travels to degradatory lysosomes153. The specific mechanisms for cargo segregation, vesicle formation, and lysosomal targeting are poorly understood, but a role for Parkin in this process is consistent with previous evidence for its involvement in endocytic vesicle formation at cell membranes154, 155.
Finally, there are circumstances in which major damage has occurred but the car is still useful after major repairs, such as replacing the engine or transmission or some other integrated sub-system. We envision that this is the function of Parkin-mediated bit-by-bit autophagy83 (Figure 5). Here, substantial but still localized damage is excised from the parent mitochondrion via spatially restricted fragmentation, and the damaged fragment is removed via the usual mitophagy apparatus, thus both repairing and sparing the parent. It should be noted that categorization of different forms of PINK1-Parkin mediated mitochondrial protein turnover mechanisms on the basis of scale, i.e. vesicles, bit-by-bit, and mitophagy, may be artificial. Given the mechanistic commonalities, the labels may be misapplied to what is actually a continuum of response that is variably invoked according to need.
Summary
Mitophagy and mitochondrial dynamism are highly integrated at the functional level. Molecular crosstalk between the primary effectors of each pathway evokes complex regulatory and counter-regulatory mechanisms that exclusively eliminate damaged mitochondria, thus preserving fitness of the overall cellular mitochondrial collective. The newly described role of Mfn2 as mitochondrial fusion factor when not acted upon by PINK1, and Parkin receptor when it is, suggests that mitochondrial fusion and mitophagy are contextual and mutually exclusive, which protects healthy mitochondria from fusion-mediated contamination by dysfunctional organelles. Likewise, parallel involvement in PINK1 and Parkin in multiple mitochondrial quality control mechanisms points to functional redundancies in this crucial activity, revealing how deterioration in mitochondrial fitness has become an emerging theme in chronic degenerative disease, and uncovering multiple opportunities for therapeutic intervention.
Supplementary Material
Acknowledgments
We gratefully acknowledge thought-provoking discussions with Dr. Roberta Gottlieb on the paracrystalline nature of mitochondrial respiratory supercomplexes, which prompted the Lego model described herein.
Sources of funding
Supported by NIH HL59888 and HL128071 (GWD) and an American Heart Association pre-doctoral fellowship award (MS).
Abbreviations
- Drp1
Dynamin-related protein 1
- Fis1
Fission 1
- HR
heptad repeat
- IMM
Inner mitochondrial membrane
- MEF
murine embryonic fibroblast
- Mff
Mitochondrial fission factor
- Mfn
Mitofusin
- Opa1
Optic atrophy 1
- OMM
Outer mitochondrial membrane
- PA-GFP
Photo-activatable green fluorescent protein
- PARL
presenilin-associated rhomboid-like protein
- PINK1
PTEN-induced putative kinase 1
- ROS
Reactive oxygen species
- TIM
Translocase of the inner mitochondrial membrane
- TOM
Translocase of the outer mitochondrial membrane
Footnotes
Disclosures – None
References
- 1.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15:634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lotz C, Lin AJ, Black CM, et al. Characterization, design, and function of the mitochondrial proteome: from organs to organisms. J Proteome Res. 2014;13:433–446. doi: 10.1021/pr400539j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Berlo JH, Molkentin JD. An emerging consensus on cardiac regeneration. Nat Med. 2014;20:1386–1393. doi: 10.1038/nm.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorn GW., 2nd Gone fission…: diverse consequences of cardiac drp1 deficiency. Circ Res. 2015;116:225–228. doi: 10.1161/CIRCRESAHA.114.305672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorn GW, 2nd, Kitsis RN. The mitochondrial dynamism-mitophagy-cell death interactome: Multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res. 2015;116:167–182. doi: 10.1161/CIRCRESAHA.116.303554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorn GW, 2nd, Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ Res. 2010;107:689–699. doi: 10.1161/CIRCRESAHA.110.225714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorn GW, 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol. 2015;78:123–128. doi: 10.1016/j.yjmcc.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song M, Dorn GW., 2nd Mitoconfusion: Noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015;21:195–205. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellegrini L, Scorrano L. A cut short to death: Parl and Opa1 in the regulation of mitochondrial morphology and apoptosis. Cell Death Differ. 2007;14:1275–1284. doi: 10.1038/sj.cdd.4402145. [DOI] [PubMed] [Google Scholar]
- 10.Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Hackenbrock CR, Chazotte B, Gupte SS. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J Bioenerg Biomembr. 1986;18:331–368. doi: 10.1007/BF00743010. [DOI] [PubMed] [Google Scholar]
- 12.Green DE, Ji S. Transductional and structural principles of the mitochondrial transducing unit. Proc Natl Acad Sci U S A. 1973;70:904–908. doi: 10.1073/pnas.70.3.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathaniel DR. Paracrystalline arrays in atypical cristae and mitochondrial division. J Cell Sci. 1980;42:23–32. doi: 10.1242/jcs.42.1.23. [DOI] [PubMed] [Google Scholar]
- 14.Dudkina NV, Kouril R, Peters K, Braun HP, Boekema EJ. Structure and function of mitochondrial supercomplexes. Biochim Biophys Acta. 2010;1797:664–670. doi: 10.1016/j.bbabio.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 16.Chen YC, Taylor EB, Dephoure N, Heo JM, Tonhato A, Papandreou I, Nath N, Denko NC, Gygi SP, Rutter J. Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 2012;15:348–360. doi: 10.1016/j.cmet.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid SL, Frolov VA. Dynamin: functional design of a membrane fission catalyst. Annu Rev Cell Dev Biol. 2011;27:79–105. doi: 10.1146/annurev-cellbio-100109-104016. [DOI] [PubMed] [Google Scholar]
- 19.Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 22.Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108–1115. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 24.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- 27.Ishihara N, Nomura M, Jofuku A, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 28.Tieu Q, Nunnari J. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol. 2000;151:353–366. doi: 10.1083/jcb.151.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang P, Galloway CA, Yoon Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS One. 2011;6:e20655. doi: 10.1371/journal.pone.0020655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta. 2013;1833:150–161. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Madden J. John Madden celebrates Thanksgiving with his own set of traditions. Chicago Tribune. 2001 [Google Scholar]
- 35.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 36.Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41:150–160. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW, 2nd, Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res. 2012;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–1328. doi: 10.1128/MCB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012;111:1012–1026. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dorn GW, 2nd, Clark CF, Eschenbacher WH, Kang MY, Engelhard JT, Warner SJ, Matkovich SJ, Jowdy CC. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res. 2011;108:12–17. doi: 10.1161/CIRCRESAHA.110.236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 45.Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cogliati S, Frezza C, Soriano ME, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 49.Bhandari P, Song M, Dorn GW., 2nd Dissociation of mitochondrial from sarcoplasmic reticular stress in Drosophila cardiomyopathy induced by molecularly distinct mitochondrial fusion defects. J Mol Cell Cardiol. 2014;80C:71–80. doi: 10.1016/j.yjmcc.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davies VJ, Hollins AJ, Piechota MJ, Yip W, Davies JR, White KE, Nicols PP, Boulton ME, Votruba M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet. 2007;16:1307–1318. doi: 10.1093/hmg/ddm079. [DOI] [PubMed] [Google Scholar]
- 51.Verhoeven K, Claeys KG, Zuchner S, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006;129:2093–2102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
- 52.Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 53.Chang CR, Manlandro CM, Arnoult D, Stadler J, Posey AE, Hill RB, Blackstone C. A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem. 2010;285:32494–32503. doi: 10.1074/jbc.M110.142430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–285. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest. 2013;123:5371–5388. doi: 10.1172/JCI70911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 60.Lackner LL, Nunnari J. Small molecule inhibitors of mitochondrial division: tools that translate basic biological research into medicine. Chem Biol. 2010;17:578–583. doi: 10.1016/j.chembiol.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 63.Whelan RS, Konstantinidis K, Wei AC, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wikstrom JD, Mahdaviani K, Liesa M, et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014;33:418–436. doi: 10.1002/embj.201385014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun N, Finkel T. Cardiac mitochondria: A surprise about size. J Mol Cell Cardiol. 2015 doi: 10.1016/j.yjmcc.2015.01.009. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 67.Pham AH, Meng S, Chu QN, Chan DC. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum Mol Genet. 2012;21:4817–4826. doi: 10.1093/hmg/dds311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 69.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW., 2nd Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res. 2014;114:257–265. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. 2013;1833:205–212. doi: 10.1016/j.bbamcr.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 72.Rabinowitz M, Swift H. Mitochondrial nucleic acids and their relation to the biogenesis of mitochondria. Physiol Rev. 1970;50:376–427. doi: 10.1152/physrev.1970.50.3.376. [DOI] [PubMed] [Google Scholar]
- 73.Schon EA, Gilkerson RW. Functional complementation of mitochondrial DNAs: mobilizing mitochondrial genetics against dysfunction. Biochim Biophys Acta. 2010;1800:245–249. doi: 10.1016/j.bbagen.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 74.Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. Mitochondrial origins. Proc Natl Acad Sci U S A. 1985;82:4443–4447. doi: 10.1073/pnas.82.13.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 76.Nakamoto RK, Baylis Scanlon JA, Al-Shawi MK. The rotary mechanism of the ATP synthase. Arch Biochem Biophys. 2008;476:43–50. doi: 10.1016/j.abb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chung YM, Kim JS, Yoo YD. A novel protein, Romo1, induces ROS production in the mitochondria. Biochem Biophys Res Commun. 2006;347:649–655. doi: 10.1016/j.bbrc.2006.06.140. [DOI] [PubMed] [Google Scholar]
- 80.Wanagat J, Dai DF, Rabinovitch P. Mitochondrial oxidative stress and mammalian healthspan. Mech Ageing Dev. 2010;131:527–535. doi: 10.1016/j.mad.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang JY, Yang WY. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Commun. 2013;4:2428. doi: 10.1038/ncomms3428. [DOI] [PubMed] [Google Scholar]
- 84.Wikstrom JD, Twig G, Shirihai OS. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol. 2009;41:1914–1927. doi: 10.1016/j.biocel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 85.Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M, Zurgil N, Reynolds N, Shirihai OS. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Cell Physiol. 2006;291:C176–184. doi: 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- 86.Abeliovich H, Zarei M, Rigbolt KT, Youle RJ, Dengjel J. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat Commun. 2013;4:2789. doi: 10.1038/ncomms3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barker MG, Walmsley RM. Replicative ageing in the fission yeast Schizosaccharomyces pombe. Yeast. 1999;15:1511–1518. doi: 10.1002/(sici)1097-0061(199910)15:14<1511::aid-yea482>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- 88.Erjavec N, Cvijovic M, Klipp E, Nystrom T. Selective benefits of damage partitioning in unicellular systems and its effects on aging. Proc Natl Acad Sci U S A. 2008;105:18764–18769. doi: 10.1073/pnas.0804550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Coelho M, Dereli A, Haese A, Kuhn S, Malinovska L, DeSantis ME, Shorter J, Alberti S, Gross T, Tolic-Norrelykke IM. Fission yeast does not age under favorable conditions, but does so after stress. Curr Biol. 2013;23:1844–1852. doi: 10.1016/j.cub.2013.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW., 2nd Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res. 2014;115:348–353. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet. 2012;21:4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- 93.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798–2813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 95.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 96.Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91:1161–1218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- 97.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 98.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 99.Exner N, Treske B, Paquet D, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guo M. Drosophila as a model to study mitochondrial dysfunction in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy. 2013;9:1750–1757. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pickrell AM, Youle RJ. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron. 2015;85:257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014;19:357–372. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 106.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem. 2011;117:856–867. doi: 10.1111/j.1471-4159.2011.07253.x. [DOI] [PubMed] [Google Scholar]
- 109.Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9:1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem. 2013;288:36372–36384. doi: 10.1074/jbc.M113.509653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377:975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- 113.Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kazlauskaite A, Kelly V, Johnson C, Baillie C, Hastie CJ, Peggie M, Macartney T, Woodroof HI, Alessi DR, Pedrioli PG, Muqit MM. Phosphorylation of Parkin at Serine65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 2014;4:130213. doi: 10.1098/rsob.130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, Knebel A, Alessi DR, Muqit MM. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP, Lohmann K, Klein C. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One. 2011;6:e16746. doi: 10.1371/journal.pone.0016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ziviani E, Whitworth AJ. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy. 2010;6:660–662. doi: 10.4161/auto.6.5.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol. 2011;23:476–482. doi: 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: a mitochondrial quality control system? J Bioenerg Biomembr. 2009;41:499–503. doi: 10.1007/s10863-009-9253-3. [DOI] [PubMed] [Google Scholar]
- 124.Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koyano F, Okatsu K, Kosako H, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 126.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stolz A, Dikic I. PINK1-PARKIN interplay: down to ubiquitin phosphorylation. Mol Cell. 2014;56:341–342. doi: 10.1016/j.molcel.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 130.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA, Dreier L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem. 2012;287:40652–40660. doi: 10.1074/jbc.M112.419721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bertolin G, Ferrando-Miguel R, Jacoupy M, et al. The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy. 2013;9:1801–1817. doi: 10.4161/auto.25884. [DOI] [PubMed] [Google Scholar]
- 133.Pallanck L. Mitophagy: mitofusin recruits a mitochondrial killer. Curr Biol. 2013;23:R570–572. doi: 10.1016/j.cub.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 134.Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 138.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 139.Kawajiri S, Saiki S, Sato S, Hattori N. Genetic mutations and functions of PINK1. Trends Pharmacol Sci. 2011;32:573–580. doi: 10.1016/j.tips.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 140.Oliveras-Salva M, Van Rompuy AS, Heeman B, Van den Haute C, Baekelandt V. Loss-of-function rodent models for parkin and PINK1. J Parkinsons Dis. 2011;1:229–251. doi: 10.3233/JPD-2011-11041. [DOI] [PubMed] [Google Scholar]
- 141.Kitada T, Tong Y, Gautier CA, Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem. 2009;111:696–702. doi: 10.1111/j.1471-4159.2009.06350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol. 2013;6:e24511. doi: 10.4161/cib.24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Siddall HK, Yellon DM, Ong SB, Mukherjee UA, Burke N, Hall AR, Angelova PR, Ludtmann MH, Deas E, Davidson SM, Mocanu MM, Hausenloy DJ. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS One. 2013;8:e62400. doi: 10.1371/journal.pone.0062400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 146.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bezard E, Gross CE, Brotchie JM. Presymptomatic compensation in Parkinson’s disease is not dopamine-mediated. Trends Neurosci. 2003;26:215–221. doi: 10.1016/S0166-2236(03)00038-9. [DOI] [PubMed] [Google Scholar]
- 148.Tatsuta T, Langer T. AAA proteases in mitochondria: diverse functions of membrane-bound proteolytic machines. Res Microbiol. 2009;160:711–717. doi: 10.1016/j.resmic.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 149.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 150.McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014;33:282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 152.Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One. 2012;7:e52830. doi: 10.1371/journal.pone.0052830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, Brice A, van Bergen En Henegouwen PM, Fon EA. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- 155.Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, Gehring K, Fon EA. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell. 2009;36:1034–1047. doi: 10.1016/j.molcel.2009.11.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.