

Abstract
Microsystems designed for cell-based studies or applications inherently require fluid handling. Flows within such systems inevitably generate fluid shear stress (FSS) that may adversely affect cell health. Simple assays of cell viability, morphology or growth are typically reported to indicate any gross disturbances to cell physiology. However, no straightforward metric exists to specifically evaluate physiological implications of FSS within microfluidic devices, or among competing microfluidic technologies. This paper presents the first genetically encoded cell sensors that fluoresce in a quantitative fashion upon FSS pathway activation. We picked a widely used cell line (NIH3T3s) and created a transcriptional cell-sensor where fluorescence turns on when transcription of a relevant FSS-induced protein is initiated. Specifically, we chose Early Growth Factor-1 (a mechanosensitive protein) upregulation as the node for FSS detection. We verified our sensor pathway specificity and functionality by noting induced fluorescence in response to chemical induction of the FSS pathway, seen both through microscopy and flow cytometry. Importantly, we found our cell sensors to be inducible by a range of FSS intensities and durations, with a limit of detection of 2 dynes/cm2 when applied for 30 minutes. Additionally, our cell-sensors proved their versatility by showing induction sensitivity when made to flow through an inertial microfluidic device environment with typical flow conditions. We anticipate these cell sensors to have wide application in the microsystems community, allowing the device designer to engineer systems with acceptable FSS, and enabling the end-user to evaluate the impact of FSS upon their assay of interest.
Introduction
Fluid flow is an essential feature of every microsystem involving cell handling, culture or sorting. The particular application determines the relevant flow rates used in a device1. One way to characterize microfluidic technologies is by virtue of their operational flow rate and experimental duration. There is a whole gamut of devices which operate at high flow fluid rates for short durations, such as high-throughput cell sorters2, inertial-force devices3 and droplet-based microsystems4. Such devices commonly use non-adherent cells, or adherent cells maintained in suspension, because cells are meant to have short residence durations within the device. In another flow regime, many devices are meant to apply very low flow rates for long durations. Typical examples of such microfluidic devices are those used for long-term static5 or perfusion cell culture6, 7.
Flows inevitably generate fluid shear stress (FSS) that may cause undesirable physiological cell stress. In the ‘short-but-intense’ flow category of devices, cells experience large FSS (~100s-1000s dynes/cm2) for a short duration (milliseconds-seconds). For the other category of ‘prolonged-and-gentle’ flow-based devices, cells experience lower FSS (0.001-10 dynes/cm2) for long durations (~hours-days). Several other microfluidic technologies fall in between these two extremes, where cells could experience moderate shear stresses (~10s-100s dynes/cm2) for moderate durations (~minutes-hours). The choice of fluid flow conditions (FSS intensity and duration) may not only depend on the device application, but also on the chosen cell phenotype1. Fluid shear stress may not always be detrimental to cell health because in some cases it is required for beneficial outcomes such as endothelial cell maintenance8. Nevertheless, in the context of cell-based microsystems and technologies, shear stress is generally viewed as a stress stimulus.
It is in fact challenging to quantify how intentionally or unintentionally imparted FSS may affect cell physiology. Cells demonstrate a complex combination of responses towards external stress stimuli. The exact set of cellular decisions depends on the bio-chemical and bio-physical cellular environment, the cell type, as well as on the type, intensity, and duration of the applied FSS9. Often, particle velocity profile or energy dissipation rate calculations/measurements are presented to first estimate the FSS profile around the cellular microenvironment10, 11. The consequent impact on cell physiology is typically reported via assessments of calcium uptake11, protein production12, gene expression6, 13, morphology11, proliferation14, migration15, cell adhesion16 or viability17, 18.
However, there are notable limitations of these approaches. Currently reported assays can either be too general such as in the measurement of calcium signalling, growth rate, adhesion or viability where the results may not directly point towards pathology specifically induced by shear. On the other hand, more specific assays of protein or gene expression can be technically difficult and are therefore rarely reported. The underlying challenge is that to achieve molecular specificity, one has to compromise the convenience of measurement and also develop sophisticated strategies for cell collection, handling and post-processing needed for standard assays. Furthermore, as there is no current ‘standard metric’ for quantifying physiological shear stress in microsystems, the choice of defining and using a relevant assay becomes subjective. Hence, there is currently no straightforward way to compare platforms to each other for evaluating cell physiology.
To address this challenge, we have developed a methodology that combines molecular specificity with a simple and convenient assay. Here we introduce a cell-based sensor intended for use by the microsystems community for evaluation of FSS effects on cells. The cell sensor connects applied stress exposure to shear stress pathway activation, subsequent gene expression, and finally fluorescence. The sensor allows for a visual and non-destructive assessment of gene expression through quantitative fluorescence using commonly available equipment and without requiring additional reagents, large numbers of cells, or overly sophisticated microscopy. We present the development and characterization of this cell sensor along with use of the sensor in an inertial microfluidic device.
Results
Chemical Induction of the Shear-Stress Pathway
We chose to develop a transcriptional reporter primarily because changes in gene expression present a reasonable balance between molecular specificity and the later downstream changes in cell physiology that dictate morphology, proliferation and viability. Examining upstream signalling (Ca++, phosphorylation) allows for faster readout but has less specificity (Ca++) or is more technically challenging (phosphorylation via genetically encoded sensors19). To minimize assay complexity we chose a visual reporter (assayed via flow cytometry or microscopy) rather than assaying expression with quantitative RT-PCR (which typically requires off-chip processing). This approach also allows for simultaneous monitoring of other observable parameters under microscopy (such as, cell morphology, proliferation, etc.) that can complement as generic markers of overall cell health.
Further, rather than develop the reporter in primary endothelial cells, which are the common model for studying FSS20, we used a common cell line that would be easy for microsystems engineers to culture and also easy to genetically manipulate. In this work, the shear stress sensor is embedded within NIH3T3 cells, which are mesenchymal cells that are known to show a response to FSS21.
Fluid shear causes a complicated mechanotransduction response which is highly dependent on its intensity, duration and profile (laminar/turbulent/pulsatile/oscillatory etc.). To the best of our knowledge, there is no single pathway or gene known to be induced exclusively due to fluid shear stress. We thus first investigated shear pathway activation in NIH3T3s to determine an appropriate node at which to target our sensor. Our goal was to create a sensor that would be sensitive to the short-duration shear stresses commonly found in microsystems, which could be applicable for high-throughput sorters as well. This motivated the need to specifically focus on genes induced by short-term FSS (min-hr duration). We further focused on genes induced by laminar shear stress, as laminar flows are prevalent in microfluidic environments. With this context, we screened for genes that had an immediate-early response to FSS, as those would capture the FSS induced stress in short term experiments. We thus chose as a gene panel: PDGFA, MCP-1, c-Fos, EGR-1, PDGFB and TF, ICAM-122. These genes have known shear stress response elements within their promoters, suggesting an ability to be upregulated by FSS23. Representative genes from other stress pathways such as DNA damage and heat shock pathways were chosen as p53 and HSF1 in order to assess cross-talk between FSS and other cell stress pathways.
As a convenient chemical surrogate to induce shear stress pathway activation and help determine an appropriate node to target for our shear sensor, we chose to induce the PKC-MAPK-ERK pathway, which is part of the shear stress response pathway, using the small molecule phorbol myristate acetate (PMA). It is known that short-term FSS generates reactive oxygen species that act upon redox-sensitive signalling kinase PKC24. To mimic this mechanism and its downstream transcriptional activation, PKC is commonly activated through PMA treatment25-27. Upon PMA treatment, we found that EGR-1 and c-Fos were significantly upregulated (p<0.05, Figure 1).
Figure 1.
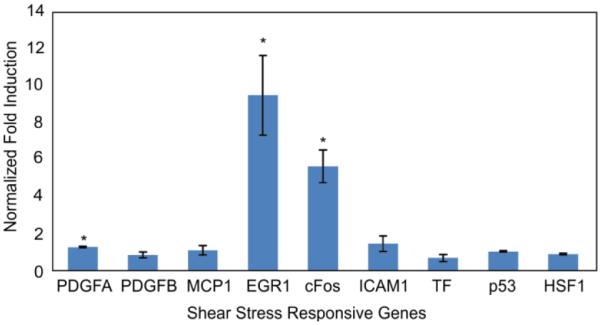
Chemical induction of shear pathways in NIH3T3 cells. GAPDH-normalized gene expression of PMA-induced cells after 2 hours of exposure of PMA at 100 ng/ml. N = 3 experiments, error bar: standard error of mean.
This makes sense because PMA can trigger upregulation of both c-Fos and EGR-1 genes through their common upstream PKC-ERK pathway28, 29. PMA is also known to trigger EGR-1 replacement of transcription factor SP1 from its binding site in the PDGFA promoter, in a manner similar to the mechanism proposed for shear-dependent up-regulation for that gene30. We found no significant induction of HSF1 or p53, representative of other cell stress pathways. Complementary serum-induction experiments, which target immediate-early pathway induction, also illustrated upregulation of EGR-1 and c-Fos (SI Figure 1).
From these experiments, we chose to target EGR-1 transcription for our shear stress sensor. EGR-1 expression can occur within minutes31, 32, making it suitable as an immediate induction sensor. Its sensitivity has been reported in a variety of cell types experiencing shear stress33, 34, thereby making its promoter a suitable candidate to report on shear stress, that also has applicability to other phenotypes. Also, in contrast to c-Fos, EGR-1 is known to be stimulated with mechanical injuries and stresses in endothelial cells33, 34.
Sensor Creation
In order to pick the appropriate section of the EGR-1 promoter to include in our sensor, we took advantage of prior work using promoter deletion assays35, which showed the highest fold activation (via luciferase reporter assay) with a minimal promoter length of 425 base pairs. Our shear stress reporter plasmid included 527 base pairs of the native murine EGR-1 promoter upstream to its transcription start site. We chose a red fluorescence protein (RFP) as the inducible fluorescent protein. Several versions of RFP exist36 and we chose TurboRFP as it had a good combination of being bright and having long expression lifetime. The resulting reporter plasmid had TurboRFP under control of a relevant portion of the inducible EGR-1 promoter and a constitutive antibiotic selection marker. This reporter plasmid was transfected into NIH3T3 cells, which could then be induced to express RFP fluorescence by an appropriate stressor, such as FSS (Figure 2A).
Figure 2.

Validating reporter plasmid functionality in transfected NIH3T3 cells using PMA induction of the FSS pathway. A. Schematic of approach, showing the reporter plasmid with TurboRFP under control of the EGR-1 promoter along with an antibiotic selection marker. The plasmid was introduced into NIH3T3 cells, which will then turn red upon stress. B. Images and flow cytometry histograms of transfected cells before and after PMA exposure. At left are phase + fluorescence overlay images of the cells and at right are the RFP histograms. The circle shows the mean fluorescence of the population, which increases upon PMA exposure.
As mentioned previously, PMA, a commonly used agonist to mimic short-term FSS, generates reactive oxygen species that act upon PKC26, which can then initiate EGR-1 transcription. Therefore, after antibiotic selection we verified plasmid functionality by inducing the stably transfected cells with PMA. In comparison to unexposed control cells, PMA-exposed cells exhibited higher population red fluorescence observed both by microscopy and flow cytometry (Figure 2B). Using the appropriate flow-cytometry gating (SI Methods and SI Figure 2), we proceeded with PMA induction of the stable parent population and FACS-sorted the induced population into 48 single cell clones. Our goal was to select a clone with the highest fold fluorescence induction of red cells and a high fraction of cells induced. By these metrics, we chose Clone 47, which had the highest increase in percent activated cells (1.8 fold) and highest normalized fold change in the population mean fluorescence (2.14 fold change, SI Figure 3) among all 48 clones assayed. Subsequent to the first clonal screen, Clone 47 was cultured under low antibiotic selection pressure to maximize the number of sensors having an inducible promoter, thereby ensuring a larger population response to PMA or FSS (SI Methods). We maintained these culture conditions for subsequent characterization and FSS experiments. These stable clonal sensors expressed modest background fluorescence, likely due to basal activity of the EGR-1 promoter driving expression of RFP. This basal promoter activity is not surprising, as many tissues are known to constitutively express EGR-1 at low levels, and is consistent with prior work using an EGR-1-based luciferase reporter in NIH3T3 cells37.
Sensor Characterization
In order to determine the sensitivity and dynamic range of the sensor to PMA, we exposed the cells to logarithmic concentrations of PMA. The maximum induction of the population fluorescence level (Figure 3A) as well as the percent of activated cells (SI Figure 4) occurred at 100 ng/ml. In evaluating these two potential parameters, we found both to be statistically sensitive to the same range of PMA concentrations (> 10 ng/ml).
Figure 3.

Chemical characterization of cell sensors. A. Induced population mean fluorescence 24h after PMA exposure. B. Map of fluorescence fold induction across PMA concentration and time after exposure. C. Inhibition of PMA induction using MEK inhibitor PD98059. N = 3, error bars: standard error of mean.
However, the dynamic range in the population mean fluorescence was higher than that from fraction of activated cells. Specifically, the population mean fluorescence could be induced up to 2 fold (Figure 3A), whereas the percent activated cells could only be induced around 1.03 fold (SI Figure 4A). The modest change in percent activated cells arises because the majority (94%) of cells in the clonal population expressed basal RFP even when uninduced, which limits the dynamic range of this metric. In contrast, the entire distribution underwent a rightward shift upon stimulus (SI Figure 4B), resulting in the 2-fold increase in the population mean fluorescence. Importantly, we observed that the sensor response maintained a normal distribution before and after induction, suggesting a homogeneous population response (SI Figure 4B) undertaken by a majority of cells. This response was consistent over several passages, indicative of robust performance. In fact, the resolution of the metric, computed by the ratio of the dynamic range to the standard error, was higher for the mean fluorescence. Therefore due to its higher dynamic range of induction and improved resolution, assessing the population mean fluorescence was a preferable metric to represent stress. Because our cell sensor population expressed background fluorescence, likely due to basal activity of the RFP-driven EGR-1 promoter, we chose to assess the entire population fluorescence shift from the control to induced population to best capture the induction (SI Methods and SI Figure 2).
We next created a map of the sensor response across PMA concentration and time (Figure 3B). The sensor exhibited a graded response, with increasing response at higher PMA concentrations, and time responses that generally peaked at 24h, in accordance with previous reports of PMA induction of the FSS pathway38. The observed dynamics are consistent with those of the TurboRFP protein, which is known to be detectable in mammalian cells after 8-10 hours and fully matures around 22 hours (Evrogen, FP231). The minimum detectable concentration of PMA was 10 ng/ml across all timepoints, with a maximum response to a PMA concentration of 100 ng/ml. Higher concentrations of PMA had lower fold induction than the 100 ng/ml concentration, indicating that the maximum effective concentration of PMA is 100 ng/ml, which we chose for subsequent sensor characterization.
To assess the specificity of our sensor we first verified that the sensor is due to the EGR-1 promoter activation. Specifically, we treated untransfected NIH3T3 cells and those transfected with a minimal (non-inducible) promoter plasmid with varying PMA concentrations for 24h along with the EGR-1 sensor. We found no increase in RFP levels due to FSS pathway activation via PMA in either the untransfected cells or cells transfected with the minimal promoter (SI Figure 9). Thus, we concluded that the RFP induction is due to the EGR-1 promoter part of the plasmid. To examine sensor specificity to other stress pathways, we intentionally activated the heat shock and DNA damage stress pathways and measured sensor response. Specifically, we found that while an FSS agonist induced RFP expression, small molecule agonists of these other pathways did not significantly induce the sensor (SI Figure 10). Thus, in the context of stresses commonly encountered in microfluidics, the sensor has appropriate specificity.
To assess the specificity of our sensor to PKC-MAPK pathway activation, we used the small-molecule inhibitor PD9805939, 40, which has been shown to abolish shear-dependent induction of EGR-1 by blocking MEK1 activation of ERK1/2 needed for EGR-1 transcription35. We found that addition of this inhibitor significantly reduced the induced fluorescence response in a dose dependent manner, and significantly abolished the induction at 75 µM concentration (p<0.05, Figure 3C). We also used the small-molecule inhibitor staurosporine that blocks PKC activation needed for a FSS response41, and found it to indeed block RFP induction due to PMA (p<0.05, SI Figure 5). Then we pretreated our cells with both these inhibitors at their potent concentrations and again observed no significant RFP induction due to PMA (SI Figure 6). Taken together, these inhibition studies demonstrate that PMA is inducing RFP through the PKC-MAPK pathway rather than via a non-specific means, thus confirming the specificity of the induction mechanism of our cell sensors.
Next, we evaluated the sensitivity of the sensors to FSS. We fabricated a simple parallel-plate PDMS-on-glass microfluidic culture device (SI Figure 7) using standard soft-lithography techniques. After introducing cells into the device and allowing them to attach, we perfused them at different levels of FSS and recovered the cells for assay via microscopy and flow cytometry (SI Figure 8). As controls, we used cells in petri dishes and cells from devices without perfusion. Interestingly, we found no observable change in commonly assayed gross parameters of cell health, such as cell adhesion, morphology, alignment between control and exposed cells (Figure 4A). However, when analysed under flow cytometry, we observed induction in population mean fluorescence between the device and dish control as well as between the device control and perfused cells (Figure 4B). Importantly, this and subsequent data compare between cells in devices that were exposed to FSS versus cells in devices not exposed to FSS. Any potential RFP induction due to on-chip culture, etc., would be equally applicable to the two device populations and any remaining induction would be that due to FSS alone. This ensures that the experiments assessed the effect of FSS only upon cells. These results highlight the utility of using cell sensors to assess FSS-induced stress pathway induction that may not result in morphological changes.
Figure 4.

Microscopy and flow cytometry analysis of cells following FSS. A. Phase microscopy images of cells in a dish (top), in a device without perfusion (middle) or cells in a device with perfusion (bottom), B. Flow cytometry histograms of the same three conditions, along with the population means (circles).
Building on the knowledge that physiological shear stresses typically range from 1-20 dyne/cm2, we characterized the cell sensors across FSS intensity from 1 to 32 dynes/cm2 (Figure 5A). These conditions are known to elicit a gene expression response in vascular flows and are prevalent in many microfluidic environments1, 42. Importantly, for our sensors, we needed to verify that under all operating conditions the system would be convection-dominated, and thus observed changes would be unlikely to be due to altered soluble signalling or nutrient transport. By carefully choosing device dimensions and flow conditions, we have previously shown the ability to operate in one regime or the other6, 7. Using well-established nondimensional numbers, we calculated the Peclet number (Pe, comparing convection/diffusion) and Damkohler Type-II number (DaII, comparing reaction/diffusion) in our conditions, where Pe/DaII then compares convection/reaction. Modelling the chambers as we have previously done7, we found that in the lowest FSS condition (1 dyne/cm2), Pe ~ 2300 and the ratio of Pe/DaII ~ 900, indicating convection-dominant flow. Since these nondimensional numbers scale linearly with the channel fluid velocity, all higher FSS conditions would also be convection-dominated. These calculations confirm that our sensor response would likely be due to convection-dominant shear activation with the chosen FSS range. We normalized induced fluorescence of perfused cells to those in devices without perfusion in order to isolate RFP induced by FSS rather than the device environment. The population mean fluorescence demonstrated a significant positive correlation (slope>0, p<0.05) to FSS applied from 1 to 32 dynes/cm2, with a statistically significant difference starting at 2 dynes/cm2 (p<0.05), which was the limit of detection in the tested conditions. We found maximum (1.3 fold) induction at our maximum tested condition of 32 dynes/cm2. These results demonstrate that the sensors are capable of quantitative assessment of FSS-induced cellular responses.
Figure 5.

FSS characterization of cell sensors. A. Induced population mean fluorescence from various FSS intensities applied for 30 minutes. B. Induced population mean fluorescence for various FSS durations at 16 dynes/cm2. N = 3, error bars: standard error of mean.
Finally, we evaluated the sensitivity of the reporter to different durations of FSS (Figure 5B) at a fixed intensity of 16 dynes/cm2. The shortest duration we tested was 30 seconds, which we found to induce a small but statistically significant induction (p<0.05). All longer durations of flow also resulted in significant inductions, with a maximum induction at 45 minutes of flow, which decreased at 60 min duration. The EGR-1 gene follows immediate-early induction dynamics that peaks around 30 minutes and then decreases in expression levels afterwards. Since we used the native EGR-1 promoter, this may explain the decreased expression at 60 min. Taken together, the characterization of our cell sensor sensitivity to FSS intensity and duration verifies the ability of our sensor to respond to the actual levels of FSS found in many microfluidic devices.
Since many microsystems naturally use microscopy-based measurement, we investigated if sensor response could be quantified using microscopy. We added a whole-cell stain to the cells to allow masking and quantification of per-cell fluorescence using methods similar to Desai et al.43 (Figure 6A).
Figure 6.

Quantifying sensor response using fluorescence microscopy. A. Images of cells showing phase, whole-cell masking channel fluorescence, and sensor fluorescence. B. Fold induction of sensor response for cells with no flow (static), 30 or 60-min FSS at 20 dynes/cm2, and 24h PMA (100 ng/ml) induction control, as measured via quantitative microscopy. Data taken from at least 3 devices from the same culture, with at least 250 cells/device; error bars: standard error of mean.
Cells exposed to FSS and PMA had fluorescence significantly higher than static controls (Figure 6B), and similar in magnitude to those obtained by flow cytometry. These results highlight that the sensors can be adapted to microfluidic applications that employ microscopy-based readout.
Case Study: Shear Stress in a Microfluidic Margination Device
Our primary motivation for creating the sensor is to use it to evaluate FSS in various microfluidic systems. One interesting class of devices uses high flow rates, such as inertial microfluidic devices and microfluidic margination devices used for sorting cells3. Cells in inertial microfluidic systems experience shear and normal stresses acting on the surfaces that generate drag and lift forces44, 45. In Poiseuille flow, a shear gradient-induced lift force pushes cells away from the center of the channel towards the wall until the force is balanced by an opposing wall-induced lift force. The equilibrium force balance positions the cells in focused streams around the channel perimeter18, 44. In such equilibrium positions, cells are free to move and rotate with the fluid which may minimize cell deformations, and supposedly any adverse effects on cell health. To support this statement, several researchers have performed viability studies to show that the cells remain highly viable after traversing such environments17, 18, 46, 47.
Although forces at equilibrium may be minimal, transient shear forces of substantial magnitude may act on cells during the movement to equilibrium, which can impact their physiology. Researchers have specifically investigated such ‘short-but-intense’ FSS effects on non-adherent CHO cell viability within a single-pass high-shear microfluidic device48, but did not investigate any sub-lethal stress-induced changes. We decided to investigate whether this operating regime could induce the shear stress pathway in an margination device3.
The dynamic flow profiles in the inertial devices are not straightforward to calculate analytically. Without having to model the intricate flow profile of this device, we estimated the average wall shear stress in the device and used it as a parameter to assess our sensor response. In the fabrication of such devices, we created modified outlets just after the high-shear region such that we only assess the influence of that part of the device (Figure 7A) and compared to cells that transited through a similar setup without device (Figure 7B). After overnight culture we assessed our sensor fluorescence response as a function of multiple flow rates using flow cytometry. We normalized the fluorescence expression of cells that had gone through the device to the control cells (no device, just tubing).
Figure 7.
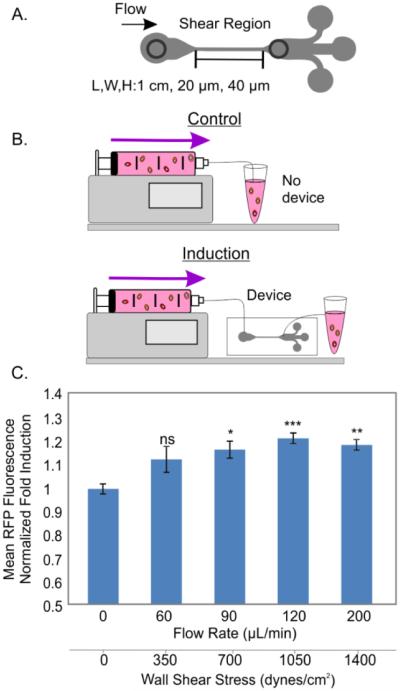
Characterization of inertial microfluidic margination device. A. High-shear region within the margination device. B. Experimental setup: cells flowing through the setup without the device (controls) and with the device (induced) C. Normalized fold inductions of mean fluorescence at various flow rates. N =3, error bars: standard error of mean.
We found that exposing sensor cells to flow rates at or above 90 µL/min led to significant inductions of ~1.2 fold (p<0.05, Figure 7C), whereas a flow rate of 60 µL/min did not induce significant fluorescence. The corresponding wall shear stress at 90 µL/min was 700 dynes/cm2, with total transit time through the high-shear region of about 5.3 ms. In principle, after focusing into the low shear streamlines, the cells should not experience further FSS, and the FSS-induced signal should arise from the local/transient shear experienced in the part of the device where the flow is not fully developed. Our results here indeed demonstrate that such transient FSS conditions activate FSS pathway in our sensors. In this manner, our sensor reports ‘local’ shear profile of a device as an integrated response, which is sensitive to the flow conditions. Furthermore, to the best of our knowledge, these results provide the first evidence of FSS pathway activation within an inertial microfluidic device. Additionally, these results suggests that the cell sensors, though developed in an adherent cell line, can be used to characterize acute FSS while being used in a non-adherent ‘flow-through’ fashion.
Discussion
In this paper we present a genetically encoded live-cell sensor that provides information on shear-stress pathway activation via expression of fluorescence. This ability eliminates the need for disruptive cellular processing and does not require any additional reagents for fluorescence assessment. This sensor provides a convenient alternative to techniques requiring to study global gene expression via sequencing or DNA microarrays to assess cell physiology13, and also offers an extra, more specific, dimension of information about cell heath alongside common live-cell measurements of morphology, growth rate and viability that are typically performed in evaluating microtechnologies for their impact on cell health.
One of the main contributions here is the successful development of the first cell-based shear-stress sensor based on quantitative fluorescence. Our transcriptional sensor is based on immediate-early gene expression that retains specificity to FSS-pathway activation and also maintains sensitivity to short-term FSS in microsystems. Choosing a transcriptional sensor presents a balance between specificity and dynamics. For example, a sensor targeting upstream signaling (calcium activation, phosphorylation, etc.) would allow for almost immediate readout, but have even lower specificity and, may require more elaborate instrumentation (ratiometric imaging) that is not common in the user community. Alternatively, events further downstream such as cell alignment to flow may require continual exposure to FSS, which limits the use space of the sensor. Immediate-early genes activation represents a balance between specificity, experimental complexity, and temporal response49
Any transcriptional sensor based on a single node will have limited specificity. For example, many shear-responsive genes are downstream of MAP kinase activation50, for example EGR-1 is downstream of MEK-ERK1/2, c-fos is downstream of MEKK-JNK and KLF2 is downstream of MEK5/ERK551. Therefore agonists that activate common upstream pathways (such as mitogens, inflammatory cytokines and reactive oxygen species) could also induce the shear-responsive genes through common upstream pathways. We performed experiments examining common cross-reactivity (SI Figure 10) and did not find substantial reaction to DNA damage and heat shock. However, it is possible that other stressors that upregulate EGR-1 transcription, or its upstream events in a manner similar to FSS, could induce our sensor. Thus, proper usage of the sensor in turn requires a specific context, such as comparison of conditions where only FSS is the varying stimulus (e.g., Figure 4B, Figure 5A,B).
In the context of EGR-1 based shear inductions, prior efforts have been made on constructing transiently transfected HeLa cells which increase luciferase activity upon shear. Specifically, Schwachtgen and others reported normalized luciferase inductions of 4.5-6 luciferase fold over a comparable range and duration of shear stress. However they noted a large variance in expression levels35, and only transiently infected the plasmid in the cells. Use of a stable cell line can allow for nearly infinite propagation and therefore, the ability to maintain shear stress sensitivity. Additionally, embedding the sensor into NIH3T3 cells strikes a balance as the cells are neither cancerous (and thus would have dramatically altered physiology) nor a primary cell line (which cannot typically be propagated in culture and is difficult to genetically engineer). Also, successful fluorescence induction and its correlation to applied chemical or FSS stimuli, along with inhibition experiments, validated the specificity and sensing ability of our cells.
In terms of their performance, our cell sensors were able to reveal subtle FSS-induced physiological stress through their fluorescence expression. For example, the FSS characterization in the laminar-flow device (Figure 4) is an example of applying shear stress that does not result in gross morphological changes in the cells, but still activates the shear-stress pathway, resulting in quantitative fluorescence changes. The ability to uncover subtle but significant FSS-induced stress pathway activation with a convenient assay illustrates the utility of our cell sensors.
Our sensor response to FSS is in coherence to what has been reported in literature38. With half-hour flows, our sensors can detect FSS as low as 2 dynes/cm2, with maximal response at 32 dynes/cm2, which was the highest tested condition. However, since our sensors integrate the FSS response to a given intensity over the applied duration, their sensitivity, operating range, and limit of detection is contingent upon the overall flow exposure in a particular device environment. Moving forward, one possible approach to enhance the sensor sensitivity would be through optimizing inducible promoter region. It is important to note that shear stress pathways are complicated and the complex interplay of between shear stress response elements in shear sensitive gene promoters has not been fully elucidated52. However we anticipate that careful construction of the promoter using a concoction of shear inducible elements could develop the sensitivity for finer resolution sensing of shear induced pathway activation.
To extend the applicability of our sensor, we were interested in characterizing its response to scenarios where cells are used in a flow-through fashion, instead of being used in adherent culture. As an example of a use case for the sensor, we examined whether a common inertial microfluidic device would cause FSS-induced fluorescence induction (Figure 7). Although cells in these devices may transiently experience high shear before equilibrating into low-shear streamlines45, such effects have not been previously explored and may not be significant enough to affect viability, which is a typical assay performed to investigate cell health in inertial microfluidic devices18, 46, 47. Such sub-lethal FSS-induced effects are of high relevance to overall cell health. For instance, sub-lethal FSS is known to induce numerous changes in intracellular molecular and pathway activation in adherent endothelial cells53, and is also reported to affect protein production, signalling pathways, glycosylation and cell membrane integrity of a variety of non-adherent cells54. In spite of its importance, there is minimal knowledge on the sub-lethal high FSS experienced in inertial microfluidics and therefore, its relevance on cell health. Our finding that ‘short-but-intense’ FSS can indeed induce FSS pathway activation is the first evidence of such FSS-induced stress pathway activation in inertial microfluidic devices.
We have distributed the reporter plasmid in Addgene for users wishing to create the sensor in different background cells, and will provide the cells upon request to interested users. We anticipate potential users to culture our cell-sensors in their microenvironments and devices to evaluate the physiological impact of their operational procedures. For example, reference cell population fluorescence levels could be established based on cell culture and handling protocols and would be used as a control for the post-experimental fluorescence levels. If the cells could be recovered from the device in adequate numbers (1000s), they could be cultured and assessed using flow cytometry (as done for Figure 7). Alternatively, smaller numbers of cells (100s) could be imaged in a device to attain a quantitative response to the FSS conditions (as done for Figure 6). It should be noted however, that the exact number of cells required in establishing significance of results would depend not only on the statistical methodology used, but also on the experimental context, systematic error of the assay, and biological variance (see SI note for details). Hence, it would be recommended to establish an understanding of the variance arising from all such factors while using our sensors. While a particular fluorescence level may not necessarily be absolutely good or bad, the relative levels between the control and post-experimental state would give users an idea about potentially stressful operating conditions. Mapping our cell sensor response to a variety of device operating conditions will then allow microfluidic engineers to design and compare platforms for desirable cell physiological states.
Conclusions
Cell handling, culturing and processing requires fluidic flows within microsystems that inevitably generate shear stress and potentially harm cells. We have created an EGR-1 based cell-sensor that fluoresces in response to shear stress as a way to assess the physiological impact of FSS within microfluidics using quantifiable fluorescence in a simple, reagent-free assay. We assayed the performance of the sensor using chemical induction of the shear pathway (verifying functionality), and also by perfusion at various FSS conditions (verifying shear sensitivity). Additionally, we demonstrated its applicability for both long-term (~hrs) and short-term (~ms) shear-induced activation in a variety of microsystems. In particular, the sensor cells were inducible in the high flows found in a microfluidic margination device, illustrating the utility of the technology.
Materials and Methods
Cell culture
NIH3T3 cells were expanded from a continuing cell stock in our laboratory, with the parental cell line originating from ATCC cell bank (ATCC® CRL-1658™). The culture conditions were maintained according to the proposed ATCC protocol. The cell culture media ATCC-formulated Dulbecco's Modified Eagle's Medium, with high glucose content (Life Technologies, No. 30-2002) was supplemented with bovine calf serum (10% v/v), L-glutamine (2% v/v) and penicillin-streptomycin (1% v/v).
Cell line construction
The promoter region was chosen as 527 base-pairs upstream of the transcription start site of the native murine EGR-1 promoter (GeneBank ID: NM_007913.5). Using this sequence, an EGR1-pTurboRFP plasmid was synthesized by Genewiz Inc., and deposited in Addgene (52306). Cells were transfected with the recommended protocol of NIH3T3 cells, using the Superfect transfection reagent (Qiagen, 301305). Constitutively expressing RFP plasmid (Evrogen, FP231) was used as a positive control, and untransfected cells as a selection control. Transfected cells were incubated overnight and then exposed to antibiotic selection (G-418) at 1 mg/mL for 7 days. Clonal screening was performed with the first culture of each single cell clone which had not been expanded under antibiotic selection, in order to assist in clonal growth. This chosen clonal population was subsequently cultured under low antibiotic selection (G-418 at 200 µg/ml) to maintain sensor functionality.
Chemical inductions
For PKC-MAPK induction of NIH3T3s, cells were seeded in 12-well plates to reach 80% confluence over 24 hours. They were exposed for 2h, following by washing with regular culture media. Cells were lysed immediately for qRT-PCR. For comparing clones of the parent population, clones were serum starved using serum free media (0.15% serum) and treated with regular media with PMA at 100 ng/ml for 24h before flow cytometry analysis. Chemical induction and characterization of the chosen clone was performed by incubation with respective concentrations of PMA (diluted with media) for 24h, with control cells incubated with media alone. For cross-sensitivity analysis cells were exposed to 4h of methyl-methanosulfate (for DNA damage) in serum free media for various concentrations, with controls treated with serum free media for 4h only. Cells were treated with varying concentrations of sodium arsenite in normal cell culture medium for 30 minutes to activate heat shock pathway.
MEK and PKC inhibition
Cells were seeded overnight in culture dishes in regular culture media. The media was then replaced with the respective concentrations of PD98059 (Sigma Aldrich, P215), or staurosporine (Sigma Aldrich, 62996-74-1) for 30 min. The supernatant was then then replaced without washing cells, to media containing PMA at 100 ng/ml. The cells were in this condition for 1h before the cells were washed with media and then incubated with the same overnight prior to flow cytometry analysis. Cells treated with inhibitors and PMA were compared to populations induced with PMA only.
Quantitative RT-PCR
Cellular mRNA was extracted from the collected lysate using Qiagen RNeasy Micro kit (catalog #:74004) using its recommended protocol. 1 µg mRNA was measured for conversion to cDNA using random hexamer primers (DyNAmo cDNA synthesis kit, Thermo Scientific, F-470L). We performed real-time qPCR (Bio Rad CFX96) using the Bio-Rad iQ SYBR Green Supermix (170-8882) according to the recommended protocol. Gene expression profiles were quantified and normalized relative to GAPDH expression using its standard curve. See SI Methods for the PCR primers.
Microfluidic device fabrication and operation
The microfluidic culture devices were cast in PDMS from a mold (Fineline Inc.). Each device consisted of 8 chambers (20 mm length × 2.5 mm width × 0.15 mm height, SI Figure 7). Each device was plasma-bonded to a borosilicate glass slide. Subsequently, a 5% APTES solution (Sigma Aldrich, 440140) in pure ethanol was introduced for 5 min and rinsed 5X with pure ethanol and 10X with DI water. A 6% aqueous solution of glutaraldehyde (Sigma Aldrich G5882) was introduced for 5 min and rinsed 10X with water. A 0.1% aqueous gelatin solution was introduced for 30 min and subsequently, cell culture media was introduced into each chamber to prime the device. The entire disconnected device was stored in a cell culture incubator for 16-24h to equilibrate the device with media constituents and the cell culture environment. Cells in suspension (5 × 106 cells/ml) in media were introduced into each chamber. The device with cells was placed in an incubator for 24h.
For perfusions, chambers experiencing flow were connected to media containing syringes mounted on a syringe pump. Reference controls consisted of cells in devices experiencing no flow and static controls consisted of cells in culture dishes. For characterizing sensitivity to FSS intensity, 30 min perfusions were performed (Figure 5A). For characterizing sensitivity to FSS durations, 16 dynes/cm2 of FSS was applied within devices (Figure 5B). Following perfusion, Tryple Select 10X (Life Technologies, A12177-01) was used to remove cells. The recovered cells were then transferred to 12-well culture plate with culture media for overnight culture prior to flow cytometry analysis.
For the inertial microfluidic device, cells in suspension (5 × 104 cells/ml) were used either for direct collection from syringe pump as control, or through the device under various flow rates. Total volume collected was approximately 1 ml in each case. Recovered cells were cultured overnight prior to flow cytometry analysis on the following day.
FSS Calculations
We assumed that cells in the culture devices experience the wall shear stress in a parallel plate flow chamber according to , where η is the DMEM viscosity (0.84 Pa-s at 37°C), Q is the volumetric flowrate, h is the channel height, and w is the channel width.
Quantitative Imaging Analysis
Cells were seeded and perfused in devices accordingly to Figure 6, with some treated with PMA (100 ng/ml, 24h) on-chip. After 24h, all cells were stained with Cell Tracker Blue (Life Technologies, C2110) according to manufacturer’s protocols for 15 min. Devices channels were washed twice with PBS and cells were imaged using blue and red fluorescence channels. The blue channel was thresholded to create a binary mask and the red intensity was quantified within each cell area after background subtraction.
Flow cytometry
Cells were analyzed using BD LSR II HTS flow cytometer using PE-TexasRed-YG-A filter (ex:561 nm, em:610 nm, BW: 20 nm) with a FITC reference (ex:488 nm, em:530 nm, BW:30 nm). The mean RFP fluorescence of the positive population was divided by that of the uninduced population and was termed as the normalized fold RFP induction (SI Methods and SI Figure 2).
Statistical Analysis
Student’s unpaired t-test was used to compare three independent induction conditions to independent controls. For FSS data, we performed ANOVA tests among the biological and device replicates of each condition and found no statistical difference among them and found statistical differences of the same against static controls. To validate positive correlation (increase in signal with increasing FSS) we performed linear regression analysis and found the slope to be positive and statistically significant (p<0.05). For all analysis: *: p-value < 0.05; **: p-value < 0.01 ***: p-value < 0.001.
Supplementary Material
Acknowledgements
This work was supported by the NIH (GM090194). We would like to thank staff of The Swanson Biotechnology Center Flow Cytometry Facility at MIT for their technical advice regarding flow cytometry. We would also like to acknowledge Dr. Yi-Chin Toh, Dr. Catherine T. Lo and Dr. Anna Fendyur for their insightful discussions. Lastly, we would like to thank Dr. Han-Wei Hou and Prof. Jongyoon Han for providing the mold for the inertial microfluidic device case study and for their discussions.
Footnotes
Electronic Supplementary Information (ESI) available [Supporting results, materials and methods, SI Figure 1-10]. See DOI: 10.1039/b000000x/
References
- 1.Kim L, Toh YC, Voldman J, Yu H. Lab on a chip. 2007;7:681–694. doi: 10.1039/b704602b. [DOI] [PubMed] [Google Scholar]
- 2.Xia N, Hunt TP, Mayers BT, Alsberg E, Whitesides GM, Westervelt RM, Ingber DE. Biomedical microdevices. 2006;8:299–308. doi: 10.1007/s10544-006-0033-0. [DOI] [PubMed] [Google Scholar]
- 3.Wei Hou H, Gan HY, Bhagat AA, Li LD, Lim CT, Han J. Biomicrofluidics. 2012;6:24115–2411513. doi: 10.1063/1.4710992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu L, Chen P, Dong Y, Feng X, Liu BF. Biomedical microdevices. 2013;15:553–560. doi: 10.1007/s10544-013-9754-z. [DOI] [PubMed] [Google Scholar]
- 5.Honegger T, Scott MA, Yanik MF, Voldman J. Lab on a chip. 2013;13:589–598. doi: 10.1039/c2lc41000a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toh YC, Voldman J. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:1208–1217. doi: 10.1096/fj.10-168971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Przybyla LM, Voldman J. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:835–840. doi: 10.1073/pnas.1103100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topper JN, Gimbrone MA., Jr. Mol Med Today. 1999;5:40–46. doi: 10.1016/s1357-4310(98)01372-0. [DOI] [PubMed] [Google Scholar]
- 9.Davies PF. Physiological reviews. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green JV, Kniazeva T, Abedi M, Sokhey DS, Taslim ME, Murthy SK. Lab on a chip. 2009;9:677–685. doi: 10.1039/b813516a. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Heo J, Hua SZ. Lab on a chip. 2010;10:235–239. doi: 10.1039/b914874d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chau L, Doran M, Cooper-White J. Lab on a chip. 2009;9:1897–1902. doi: 10.1039/b823180j. [DOI] [PubMed] [Google Scholar]
- 13.Hur SC, Henderson-MacLennan NK, McCabe ER, Di Carlo D. Lab on a chip. 2011;11:912–920. doi: 10.1039/c0lc00595a. [DOI] [PubMed] [Google Scholar]
- 14.Kim L, Vahey MD, Lee HY, Voldman J. Lab on a chip. 2006;6:394–406. doi: 10.1039/b511718f. [DOI] [PubMed] [Google Scholar]
- 15.Gutierrez E, Petrich BG, Shattil SJ, Ginsberg MH, Groisman A, Kasirer-Friede A. Lab on a chip. 2008;8:1486–1495. doi: 10.1039/b804795b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JY, Yoo SJ, Hwang CM, Lee SH. Lab on a chip. 2009;9:2194–2202. doi: 10.1039/b822006a. [DOI] [PubMed] [Google Scholar]
- 17.Edd JF, Di Carlo D, Humphry KJ, Koster S, Irimia D, Weitz DA, Toner M. Lab on a chip. 2008;8:1262–1264. doi: 10.1039/b805456h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuntaegowdanahalli SS, Bhagat AA, Kumar G, Papautsky I. Lab on a chip. 2009;9:2973–2980. doi: 10.1039/b908271a. [DOI] [PubMed] [Google Scholar]
- 19.Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minami T, Aird WC. Trends in cardiovascular medicine. 2005;15:174–184. doi: 10.1016/j.tcm.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Ji JY, Jing H, Diamond SL. Circulation research. 2003;92:279–285. doi: 10.1161/01.res.0000057753.57106.0b. [DOI] [PubMed] [Google Scholar]
- 22.Braddock M, Schwachtgen JL, Houston P, Dickson MC, Lee MJ, Campbell CJ. News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society. 1998;13:241–246. doi: 10.1152/physiologyonline.1998.13.5.241. [DOI] [PubMed] [Google Scholar]
- 23.Resnick N, Yahav H, Khachigian LM, Collins T, Anderson KR, Dewey FC, Gimbrone MA., Jr. Advances in experimental medicine and biology. 1997;430:155–164. doi: 10.1007/978-1-4615-5959-7_13. [DOI] [PubMed] [Google Scholar]
- 24.Matlung HL, Bakker EN, VanBavel E. Antioxidants & redox signaling. 2009;11:1699–1709. doi: 10.1089/ars.2008.2408. [DOI] [PubMed] [Google Scholar]
- 25.Chiu JJ, Wung BS, Shyy JY, Hsieh HJ, Wang DL. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:3570–3577. doi: 10.1161/01.atv.17.12.3570. [DOI] [PubMed] [Google Scholar]
- 26.Tai LK, Okuda M, Abe J, Yan C, Berk BC. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:1790–1796. doi: 10.1161/01.atv.0000034475.40227.40. [DOI] [PubMed] [Google Scholar]
- 27.Matsubara T, Ziff M. Journal of cellular physiology. 1986;127:207–210. doi: 10.1002/jcp.1041270203. [DOI] [PubMed] [Google Scholar]
- 28.Dieckgraefe BK, Weems DM. The American journal of physiology. 1999;276:G322–330. doi: 10.1152/ajpgi.1999.276.2.G322. [DOI] [PubMed] [Google Scholar]
- 29.Janknecht R, Cahill MA, Nordheim A. Carcinogenesis. 1995;16:443–450. doi: 10.1093/carcin/16.3.443. [DOI] [PubMed] [Google Scholar]
- 30.Khachigian LM, Williams AJ, Collins T. The Journal of biological chemistry. 1995;270:27679–27686. doi: 10.1074/jbc.270.46.27679. [DOI] [PubMed] [Google Scholar]
- 31.Treisman R. Trends in biochemical sciences. 1992;17:423–426. doi: 10.1016/0968-0004(92)90013-y. [DOI] [PubMed] [Google Scholar]
- 32.Sukhatme VP, Cao XM, Chang LC, Tsai-Morris CH, Stamenkovich D, Ferreira PC, Cohen DR, Edwards SA, Shows TB, Curran T, et al. Cell. 1988;53:37–43. doi: 10.1016/0092-8674(88)90485-0. [DOI] [PubMed] [Google Scholar]
- 33.Yan SF, Pinsky DJ, Mackman N, Stern DM. The Journal of clinical investigation. 2000;105:553–554. doi: 10.1172/JCI9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCaffrey TA, Fu C, Du B, Eksinar S, Kent KC, Bush H, Jr., Kreiger K, Rosengart T, Cybulsky MI, Silverman ES, Collins T. The Journal of clinical investigation. 2000;105:653–662. doi: 10.1172/JCI8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwachtgen JL, Houston P, Campbell C, Sukhatme V, Braddock M. The Journal of clinical investigation. 1998;101:2540–2549. doi: 10.1172/JCI1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shcherbo D, Merzlyak EM, Chepurnykh TV, Fradkov AF, Ermakova GV, Solovieva EA, Lukyanov KA, Bogdanova EA, Zaraisky AG, Lukyanov S, Chudakov DM. Nature methods. 2007;4:741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 37.Bhattacharyya S, Chen SJ, Wu M, Warner-Blankenship M, Ning H, Lakos G, Mori Y, Chang E, Nihijima C, Takehara K, Feghali-Bostwick C, Varga J. The American journal of pathology. 2008;173:1085–1099. doi: 10.2353/ajpath.2008.080382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu JJ, Wung BS, Hsieh HJ, Lo LW, Wang DL. Circulation research. 1999;85:238–246. doi: 10.1161/01.res.85.3.238. [DOI] [PubMed] [Google Scholar]
- 39.Hodge C, Liao J, Stofega M, Guan K, Carter-Su C, Schwartz J. The Journal of biological chemistry. 1998;273:31327–31336. doi: 10.1074/jbc.273.47.31327. [DOI] [PubMed] [Google Scholar]
- 40.Datta R, Rubin E, Sukhatme V, Qureshi S, Hallahan D, Weichselbaum RR, Kufe DW. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10149–10153. doi: 10.1073/pnas.89.21.10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morita T, Kurihara H, Maemura K, Yoshizumi M, Nagai R, Yazaki Y. Circulation research. 1994;75:630–636. doi: 10.1161/01.res.75.4.630. [DOI] [PubMed] [Google Scholar]
- 42.Zheng W, Xie Y, Zhang W, Wang D, Ma W, Wang Z, Jiang X. Integrative biology : quantitative biosciences from nano to macro. 2012;4:1102–1111. doi: 10.1039/c2ib20094e. [DOI] [PubMed] [Google Scholar]
- 43.Desai SP, Voldman J. Integrative biology : quantitative biosciences from nano to macro. 2011;3:48–56. doi: 10.1039/c0ib00067a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Carlo D. Lab on a chip. 2009;9:3038–3046. doi: 10.1039/b912547g. [DOI] [PubMed] [Google Scholar]
- 45.Zhou J, Papautsky I. Lab on a chip. 2013;13:1121–1132. doi: 10.1039/c2lc41248a. [DOI] [PubMed] [Google Scholar]
- 46.Di Carlo D, Irimia D, Tompkins RG, Toner M. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18892–18897. doi: 10.1073/pnas.0704958104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Carlo D, Edd JF, Irimia D, Tompkins RG, Toner M. Analytical chemistry. 2008;80:2204–2211. doi: 10.1021/ac702283m. [DOI] [PubMed] [Google Scholar]
- 48.Mollet M, Godoy-Silva R, Berdugo C, Chalmers JJ. Biotechnology and bioengineering. 2007;98:772–788. doi: 10.1002/bit.21476. [DOI] [PubMed] [Google Scholar]
- 49.Barakat A, Lieu D. Cell biochemistry and biophysics. 2003;38:323–343. doi: 10.1385/cbb:38:3:323. [DOI] [PubMed] [Google Scholar]
- 50.Traub O, Berk BC. Arteriosclerosis, thrombosis, and vascular biology. 1998;18:677–685. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- 51.Young A, Wu W, Sun W, Benjamin Larman H, Wang N, Li YS, Shyy JY, Chien S, Garcia-Cardena G. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1902–1908. doi: 10.1161/ATVBAHA.109.193540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silberman M, Barac YD, Yahav H, Wolfovitz E, Einav S, Resnick N, Binah O. Angiogenesis. 2009;12:231–242. doi: 10.1007/s10456-009-9143-7. [DOI] [PubMed] [Google Scholar]
- 53.Li YS, Haga JH, Chien S. Journal of biomechanics. 2005;38:1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 54.Hu W, Berdugo C, Chalmers JJ. Cytotechnology. 2011;63:445–460. doi: 10.1007/s10616-011-9368-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.