

Abstract
Postinjury systemic fibrinolysis has been recognized as a biologic process for more than 200 years, but the specific mechanisms of regulation and their clinical implications remain to be elucidated. By the 1950s, the plasminogen-plasmin-antiplasmin system was established as critical in preserving microvascular patency during blood clotting to maintain hemostasis. The challenges in modulating systemic fibrinolysis became evident soon thereafter. In the 1960s systemic fibrinolysis was identified by thrombelastography (TEG) during the anhepatic phase of liver transplantation, prompting the recommendation for intraoperative antifibrinolytics. But the administration of antifibrinolytic was associated with fatal postoperative pulmonary emboli. During the same period, there was experimental evidence that antifibrinolytics prevented irreversible hemorrhagic shock. More recently, a randomized trial indicated that plasmin inhibition during coronary artery bypass grafting was associated with increased mortality. The interest in antifibrinolytic therapy for trauma induced coagulopathy (TIC) is a relatively recent event, largely driven by the increasing use of viscoelastic hemostatic assays. The CRASH-2 trial, published in 2010, stimulated worldwide enthusiasm for tranexamic acid (TXA). However, the limitations of this study were soon acknowledged, raising concern for the unbridled use of TXA. Most recently, the documentation of fibrinolysis shutdown soon after injury has highlighted the potential adverse effects due to the untimely administration of TXA. A recent retrospective analysis in severely injured patients supports this hypothesis. But final clarity of this volatile topic awaits the completion of the current ongoing randomized clinical trials throughout the world.
Postinjury systemic fibrinolysis has been recognized as a biologic process for more than 200 years, but the mechanisms of regulation and their clinical implications remain unclear. In 1794, John Hunter from Edinburgh observed that the last blood exiting from fatal gunshot wounds did not clot (1). Albert Dastre from Paris proposed the term fibrinolysis in 1893 (Archives de Physiologie) based on experimental work demonstrating digestion of fibrin. In 1927, interest in fibrinolysis was piqued by a Russian report that victims of sudden death were preferred as blood donors because their blood “reliquified” within a few hours, permitting transfusion without an anticoagulant. Scientific knowledge of physiologic fibrinolysis improved rapidly over the ensuing two decades and, by the 1950s, the plasminogen-plasmin-antiplasmin system was established as critical in preserving microvascular patency during clotting to maintain hemostasis (1, 2). Thus, in parallel to the highly regulated clot formation system, clot stabilization and physiologic degradation by the fibrinolysic system was also appreciated to be highly regulated.
The Challenges of Modifying Fibrinolysis
In 1963, Starzl et al (3) identified systemic fibrinolysis by thrombelastography (TEG) during the anhepatic phase of liver transplantation, and advocated routine antifibrinolytics (aminocaproic acid, ACA). Three years later (4), however, this Colorado transplant team reversed their recommendation when three of their four transplant survivors given ACA developed multiple pulmonary emboli. Interestingly, during the same period, Hardaway et al (5) demonstrated the benefits of fibrinolytic administration to prevent irreversible experimental hemorrhagic shock. Over the ensuing three decades, fibrinolytic therapy became the standard for arterial thromboemboli in the coronary, cerebral, mesenteric, and peripheral vasculative, with selective use in the venous system. By the late 1980s recombinant tissue plasminogen activator (tPA) became the fibrinolytic of choice. On the other side, with the widespread availability of TEG, excessive fibrinolysis was incriminated in post coronary artery bypass grafting (CABG) mediastinal bleeding, presumably due to contact activation. But the enthusiasm for antifibrinolytics was dampened after the Blood Conservation using Antifibrinolytics in a Randomized Trial (BART) indicated increased renal failure, myocardial infarction, and mortality following CABG when a plasmin inhibitor (aprotinin) was given (6).
Enthusiasm for Tranexamic Acid (TXA) in Trauma Management
Acknowledging the potential role of the plasminogen-plasmin-system in trauma is a relatively recent event and largely due to the implementation of TEG (7) and rotational thromboelastometry (ROTEM) (8). The stage was set by Hoffman and Monroe in 2001 (9) who proposed the cell based model of hemostasis. Based on this construct Brohi, Cohen et al introduced the provocative concept that Trauma Induced Coagulopathy (TIC) was mediated via the activation of protein C (aPC) resulting in the degradation of clotting factors V and VIII (10). Embedded within this novel proposal was the consumption of plasminogen activator inhibitor-1 (PAI-1) by aPC, thus, indirectly enhancing fibrinolysis (11). Within a year, our group in Denver documented systemic hyperfibrinolysis by TEG in 18% of acutely injured patients requiring a massive transfusion (12). These data were further supported by contemporary reports from the United States (13) and Europe (14). The Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage (CRASH-2) trial, reported in 2010 (15), provided the ultimate impetus for the widespread adoption of an antifibrinolytic (tranexamic acid) for trauma management.
However, the significant limitations of this prospective randomized trial were soon emphasized by a number of groups (16,17). Although 20,211 patients were enrolled in this study designed to reduce mortality due to coagulopathy, only half the patients required a red blood cell (RBC) transfusion. Furthermore, there was no reduction in transfused blood products; each group received six units of RBCs. Finally, an additional analysis of the data indicated a 1.44 increased risk for mortality when TXA was given greater than three hours postinjury (18). While the Military Application of Tranexamic Acid in Trauma Emergency Resuscitation Study (MATTERS) suggested a benefit of TXA in combat casualty care, this was a retrospective analysis confounded by the administration of fibrinogen (19). Recently, Valle et al (20) in a retrospective analysis of civilian data, employing propensity score matching, found an increased mortality in severely injured patients administered TXA and Harvin et al (21) in a retrospective study of civilian patients confirmed to have fibrinolysis by TEG, observed no benefit from TXA.
Postinjury Hyperfibrinolysis, Physiologic Fibrinolysis, and Fibrinolysis Shutdown
Principal component analyses of patients with TIC performed by the Denver group (22) and the San Francisco General group (23) indicated that clotting factor deficiencies and systemic hyperfibrinolysis are mechanistically distinct. These analyses stimulated us to investigate the mechanistic regulation, and consequent manifestation, of fibrinolysis in our animal shock/trauma models. Although systemic fibrinolysis is difficult to replicate in animal models, we developed a tissue plasminogen activator (tPA) challenge assay to unmask latent hyperfibrinolysis versus fibrinolysis shutdown (24). We define shutdown as a relative resistance to tPA, due to a physiologic dysregulation of the plasminogen-plasmin system. Interestingly in both our rodent and swine models, shock (ischemia/reperfusion) produced consistent systemic hyperfibrinolysis; whereas, tissue injury (thoracotomy/laparotomy/femur fracture) provoked physiologic fibrinolysis shutdown (25).
Stimulated by these experimental findings, we then interrogated our prospectively collected TEG database from 2010 to 2013. Patients were eliminated if the first TEG was obtained greater than 12 hours postinjury or the patient was taking preinjury anticoagulants. A citrated kaolin TEG (CK-TEG) was conducted by standard TEG 5000 methods (Haemonetics), and used because of its greater accuracy for identifying lysis compared to the rapid TEG. Systemic fibrinolysis was quantified as the % clot lysis at 30 minutes after maximum strength was achieved (LY30). Patients were then stratified into three groups by LY30 criteria: hyperfibrinolysis (≥ 3%); physiologic fibrinolysis (0.81–2.9%); and fibrinolysis shutdown (≤ 0.08%). The hyperfibrinolysis cutoff was based on previous clinical studies indicating increased blood product consumption and mortality in acutely injured patients (13, 26). Because there were no previous reports addressing fibrinolysis shutdown, we employed a receiver operating characteristic (ROC) curve for mortality in the remaining patients with a LY30 < 3%. The point of greatest specificity and sensitivity was 0.8% based on a Youden index. Our study population consisted of 180 severely injured patients who were 43 years old (IQR:28–55), 70% male, and 79% sustained blunt trauma. The median injury severity score (ISS) was 29 (IQR:22–36), median initial base deficit (BD) was nine (IQR: 6–3), and the mortality was 20% with two-thirds occurring within 24 hours. Fibrinolysis shutdown (as defined above) was the most common phenotype, accounting for 64% (n=115) with physiologic fibrinolysis (n=32) and hyperfibrinolysis (n=33) representing 18% each. Interestingly, the three phenotypes could not be distinguished by age, sex, ISS, or BD. When considering patients who required RBCs, the systemic hyperfibrinolysis patients required more RBCs and fresh frozen plasma (FFP), and this correlated with an increased need for massive transfusion (Figure 1).
Figure 1. Blood Product Transfusions between Systemic Fibrinolysis Phenotypes.

The blood component transfusion among the systemic fibrinolysis phenotypes summarized; Y axis represents the number of units within the first six hours postinjury. RBC and plasma transfusions were higher in the systemic hyperfibrinolysis phenotype. RBC= Red blood cell; Cyro = Cryoprecipitate; * p<0.05 after pair wise adjustment. Reproduced from Moore et al24 with permission of Lippincott Williams & Wilkins.
Of note, mortality among the fibrinolysis phenotypes had a U-shaped distribution (Figure 2), with the nadir in the physiologic group ( 3%) compared to systemic hyperfibrinolysis ( 44%) and fibrinolysis shutdown (17%). The cause of death was also substantially different between the phenotypes (Figure 3). Acute blood loss accounted for 66% of the mortality in the hyperfibrinolysis group compared to 15% in the fibrinolysis shutdown patients. Conversely, death due to multiple organ failure occurred in 7% of the hyperfibrinolysis group compared to 40% in the shutdown patients.
Figure 2. U Shaped Distribution of Mortality Related to Systemic Fibrinolysis Phenotype.
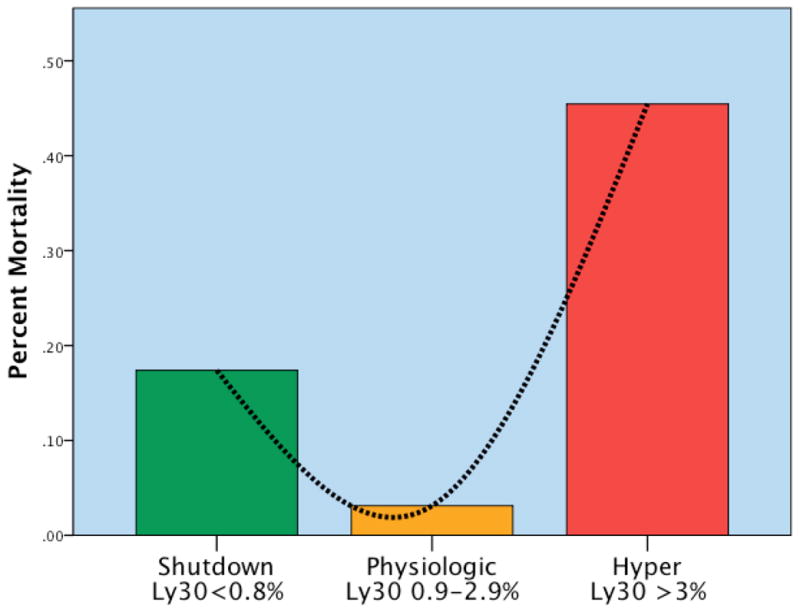
The mortality among the systemic fibrinolysis phenotypes was U-shaped; Y axis represents mortality by phenotype L30 = % lysis at 30 minutes by TEG Shutdown = Fibrinolysis shutdown; Physiologic = Physiologic fibrinolysis; Hyper= Hyperfibrinolysis; Ly30 = the percent fibrinolysis 30 minutes after reaching maximum amplitude measured by thombelastography. Y axis represents the percent mortality per phenotype. There is a U shaped distribution of mortality with a nadir in mortality identified in the physiologic group (Ly30 between 0.9 and 2.8%). Ly30% above and below this range had statistical increases in mortality. Reproduced from Moore et al24 with permission of Lippincott Williams & Wilkins.
Figure 3. Distribution of Mortality According to Systemic Fibrinolysis Phenotype.
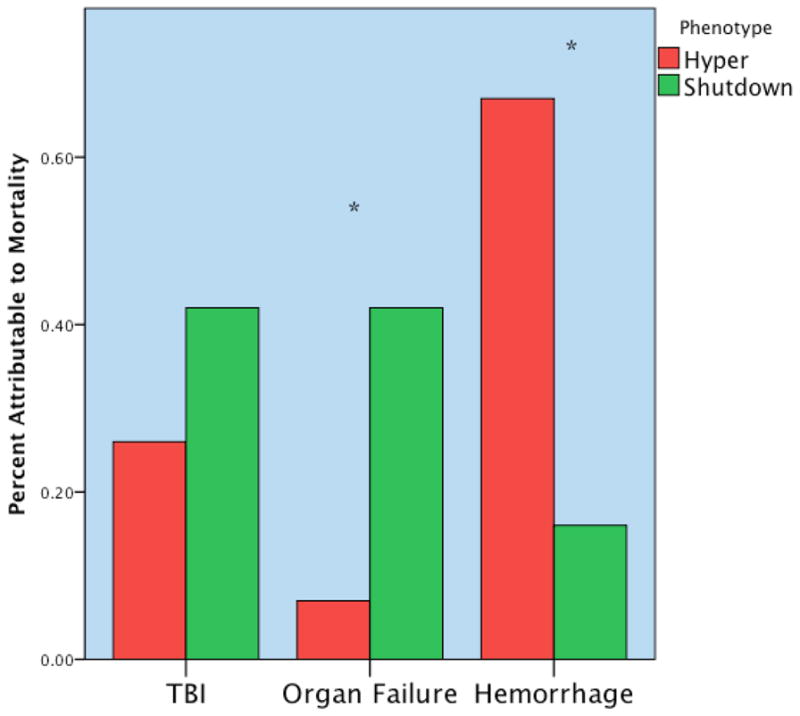
The distribution of mortality among the systemic fibrinolysis phenotypes was substantially different; Y axis represents % of total mortality per systemic fibrinolysis phenotype. The systemic hyperfibrinolysis phenotype died primarily due to hemorrhage; whereas the systemic fibrinolysis shutdown phenotype succumbed to multiple organ failure. TBI= Traumatic brain injury; Hyper = hyperfibrinolysis; Shutdown= fibrinolysis shutdown; * p<0.05. The Y-axis represents the percent of total mortality per phenotype. The hyperfibrinolytic phenotype had a high frequency of mortality associated with hemorrhage. The shutdown phenotype has a high frequency of organ failure related death. TBI did not reach statistical difference between phenotypes but was more common in the shutdown cohort. Reproduced from Moore et al24 with permission of Lippincott Williams & Wilkins.
In sum, our clinical study identified three distinct phenotypes of fibrinolysis after severe injury. Unfortunately, it was not possible to predict which phenotype would be manifest based on injury severity (ISS) and magnitude of shock (BD). The majority of these patients (64%) had fibrinolysis shutdown, and typically had delayed mortality from multiple organ failure. These patients should not improve with further blockade of the fibrinolytic system, and may even be harmed, by the untimely administration of TXA. Conversely, the 18% with documented systemic hyperfibrinolysis will presumably benefit from timely TXA delivery, as mortality in this group was early and due to acute blood loss. We believe these findings argue against the empiric use of TXA in acutely injured patients, and support the routine monitoring for lysis with TEG or ROTEM in high risk patients. There are limitations to our initial clinical study identifying fibrinolysis phenotype due to the delay in acquiring the TEG analysis up to 12 hours (24). Consequently, we have an ongoing prospective study to define the timing of fibrinolysis shutdown employing our graduated tPA challenge assay. The experience to date indicates that the majority of severely injured patients manifest tPA resistance/fibrinolysis shutdown within three hours of injury. Considering the half-life of two hour for TXA, we believe our data support our recommendation for the selective administration of TXA(16).
Regulatory Mechanisms of Postinjury Fibrinolysis
At a simplistic level, the regulation of postinjury fibrinolysis can be viewed as a set of activators and inhibitors of the plasminogen-plasmin system (Figure 4), but the molecular events are more complex and yet to be fully elucidated (27). A perusal of the molecular structure of fibrinogen underscores the complexities of this regulation (28, 29). Our experimental work and clinical studies (11, 24, 25, 26, 30, and 31) indicate that circulatory arrest provokes systemic hyperfibrinolysis. Our experimental work further confirms that shock (ischemia) stimulates systemic fibrinolysis (24, 25). Collectively, these studies suggest a primary mechanism for systemic hyperfibrinolysis is the release of tPA that overwhelms the counter-regulatory mechanisms (30). At this point, we believe a major component is that tPA release exceeds the capacity of its cognate inhibitor, plasminogen activator inhibitor-1 (PAI-1, Serpin E1). TPA and PAI-1 form a mutually inhibitory covalent complex with 1:1 stoichiometry, and are cleared by the liver. PAI-1 activity may be further impaired due to the actions of other proteolytic enzymes, including activated protein C (aPC) and neutrophil elastase, which are known to be upregulated by acute injury and inflammation. While the endothelium is a major source of tPA, the precise mediators in trauma are unclear and production is organ specific ( manuscript submitted ). Furthermore, there may be other direct contributors such as neuronal tissue (27). Regulation of the plasminogen-plasmin system involves a myriad of molecular events at multiple steps, including overt and covert protein domains that modulate the binding of tPA and plasminogen to fibrinogen (27, 32). For example, our experimental work, employing proteomics, indicates that partners bearing accessible lysine residues enhance plasmin fibrinolysis independent of tPA and PAI-1 levels (31, 32). Furthermore, our ongoing investigation with metabolomics indicates a number of potential modifiers of the plasmin system (manuscript submitted).
Figure 4. Regulation of Postinjury Systemic Fibrinolysis.
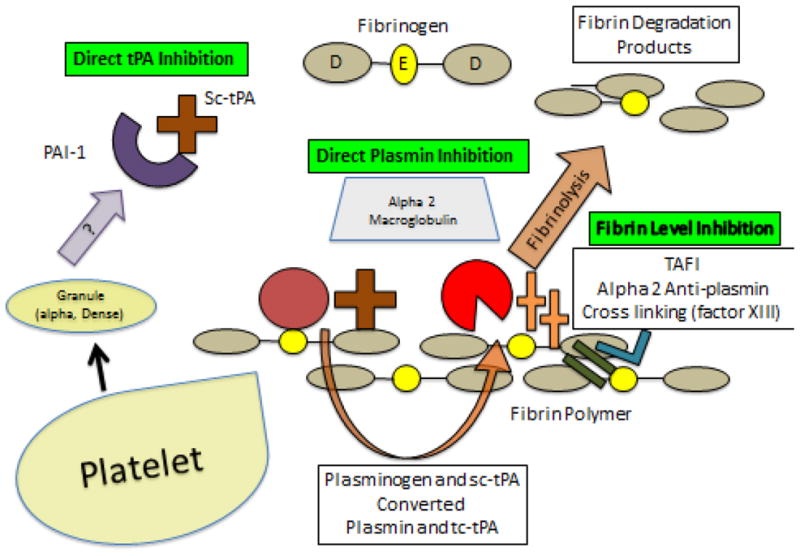
The control of systemic fibrinolysis driven by tPA is controlled at multiple levels, including the direct inhibition of tPA, the direct inhibition of plasmin, and the inhibition at the fibrin cross-linking stage.
The pathogenesis of postinjury systemic fibrinolysis shutdown remains even further mysterious. There are a number of potential regulatory events following severe trauma. Plasminogen ( PLG) activation on fibrin is initiated when tPA binds to fibrin followed by the binding of plasminogen. Once this trimolecular complex is formed, plasmin cleaves fibrin and exposes carboxy terminal lysine residues. Kringle 2 of tPA and kringles 1 and 4 of PLG contain lysine binding sites. Their counter-binding sites in fibrin (Aa 148–160 and c 312–324) are cryptogenic in fibrinogen but become exposed during fibrin crosslinking due to intermolecular D:E interactions that result in conformational changes in the D region. The Aa 148–160 site binds tPA and plasminogen with similar affinity, while the c 312–324 site binds tPA exclusively. Fibrin polymerization occurs with considerable diversity, and the resulting viscoelastic properties are generally referred to as clot stability. Fibrin structure affects the rate of fibrinolysis; thinner fibrin strands with frequent branch points are more resistant to plasmin disassembly. Disaggregation of the fibrin fibers proceeds by lateral transection rather than surface erosion. When plasmin is generated, it converts single chain tPA to a double chain form which has much greater activity. In degrading cross-linked fibrin, plasmin initially cleaves the C termini of the α and β chains within the D region resulting in a variety of fibrin degradation products (FDP). Degradation of fibrin crosslinked by FXIII releases the FDP known as D-dimer, which consists of fragments containing two D regions and one E region.
Once plasmin is generated, there are a number of inhibitors that can attenuate its activity. The most active is α2 – antiplasmin (α2-AP), a 70 kDA single chain glycoprotein that is a serpin. α2-AP is made in the liver and has a circulating half-life exceeding two days. α2-AP forms a lysine binding site dependent α2-PI plasmin (PAP) complex which is cleared by the liver. α2-AP can a be crosslinked to fibrin α chains, and this further enhances resistance to fibrinolysis. FXIII is also capable of incorporating α2-AP into fibrinogen. α2 – macroglobulin is a 72 kDA dimeric protein synthesized by endothelial cells and macrophages. α2 macroglobulin, like a α2-AP, is also in the platelet granule. Thrombin-activatable fibrinolysis inhibitor (TAFI), also known as procarboxypeptidase B, is a 60 kDA polypeptide that is generated in the liver and present in platelets. Thrombin activation of TAFI is enhanced 1250 fold in the presence of thrombomodulin. TAFI inhibits fibrinolysis by cleaving lysine residues on fibrin that bind tPA and PLG. There are additional proteins released after platelet activation that can influence fibrinolysis. Polyphosphate, a negatively charged polymer of inorganic phosphate, is secreted from the dense granules of platelets and promotes tighter fibrin aggregates with reduced sites for tPA and PLG binding. Enhanced interactions with the extracellular matrix by binding fibrin to fibronectin also impairs fibrinolysis.
Our clinical studies indicate the majority of severely injured patients manifest fibrinolysis shutdown within three hours of injury, but predicting that response based on injury pattern has been challenging. Our experimental work indicates that tissue injury provokes fibrinolysis shutdown, but the precise mechanisms remains unclear(25). While the release of PAI-1 exceeding tPA activity is an intuitive mechanism(30, 33), the process appears to involve other molecular interactions. Plasmin activity on fibrin might be obstructed by differential expression of cellularly produced regulators or incur interactions with novel inhibitors released by tissue injury. Structurally, it appears that modulators of the kringle domains in plasmin and tPA can both be activators and inactivators (27). With tissue injury as an emerging unifying stimulus, danger signaling could be another partial explanation. Recently, we have shown experimentally that lysed platelets strongly inhibit systemic fibrinolysis. Although the mechanism is unknown, the data suggest platelet activation and the release of granule contents may have a role (31). Finally, we have now documented that fibrinolysis shutdown is prevalent in the postinjury recovery period, a dominant phenotype in the surgical intensive care unit (SICU), and may explain sequelae ranging from acute lung injury to venous thromboembolism (34).
Acknowledgments
Research supported by National Institutes of Health grants: T32-GM008315 (NIGMS), P50-GM049222 (NIGMS), and UMIHL120877 (NHLBI), and Department of Defense contract W81XWH-12-2-0028.
Footnotes
Presented at the 3rd annual Remote Damage Control Resuscitation symposium in Bergen, Norway June 9–11, 2014.
Contributor Information
Ernest E. Moore, Email: ernest.moore@dhha.org.
Hunter B. Moore, Email: hunter.moore@ucdenver.edu.
Eduardo Gonzalez, Email: eduardo.gonzalez@ucdenver.edu.
Michael P. Chapman, Email: michael.chapman@ucdenver.edu.
Angela Sauaia, Email: angela.sauaia@ucdenver.edu.
Christopher C. Silliman, Email: christopher.silliman@ucdenver.edu.
Anirban Banerjee, Email: anirban.banerjee@ucdenver.edu.
References
- 1.Macfarlane RG, Biggs R. Fibrinolysis. Its mechanism and significance. Blood. 1948;3:167–87. [PubMed] [Google Scholar]
- 2.Sherry S, Fletcher AP, Alkjaersig N. Fibrinolysis and fibrinolytic activity in man. Physiological Reviews. 1959;39:343–382. doi: 10.1152/physrev.1959.39.2.343. [DOI] [PubMed] [Google Scholar]
- 3.Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet. 1963;117:659–76. [PMC free article] [PubMed] [Google Scholar]
- 4.Groth CG, Pechet L, Starzl TE. Coagulation during and after orthotopic transplantation of the human liver. Arch Surg. 1969;98:31–4. doi: 10.1001/archsurg.1969.01340070049006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardaway RM, Drake DC. Prevention of “irreversible” hemorrhagic shock with fibrinolysin. Ann Surg. 1963;157:39–47. doi: 10.1097/00000658-196301000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fergusson DA, Hebert PC, Mazer CD, Fremes S, MacAdams C, Murkin JM, Teoh K, Duke PC, Arellano R, Blajchman MA, et al. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. N Engl J Med. 2008;358:2319–31. doi: 10.1056/NEJMoa0802395. [DOI] [PubMed] [Google Scholar]
- 7.Plotkin AJ, Wade CE, Jenkins DH, Smith KA, Noe JC, Park MS, Perkins JG, Holcomb JB. A reduction in clot formation rate and strength assessed by thrombelastography is indicative of transfusion requirements in patients with penetrating injuries. J Trauma. 2008;64(2 Supp):S64–8. doi: 10.1097/TA.0b013e318160772d. [DOI] [PubMed] [Google Scholar]
- 8.Gorlinger KJ, Dirkmann D, Solomon C, Hanke AA. Fast interpretation of thromboelastometry in non-cardiac surgery: Reliability in patients with hypo-, normo-, and hypercoagulability. Br J Anaesth. 2013;110:222–30. doi: 10.1093/bja/aes374. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman M, Monroe DM. A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–65. [PubMed] [Google Scholar]
- 10.Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittel JF. Acute traumatic coagulopathy: Initiated by hypoperfusion modulated through the protein C pathway? Ann Surg. 2007;245:812–818. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, Pittet JF. Acute coagulopathy of trauma: Hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64:1211–1217. doi: 10.1097/TA.0b013e318169cd3c. [DOI] [PubMed] [Google Scholar]
- 12.Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, Barnett C, Biffl WL, Burlew CC, Johnson JL, Sauaia A. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252:434–42. doi: 10.1097/SLA.0b013e3181f09191. [DOI] [PubMed] [Google Scholar]
- 13.Cotton BA, Harvin JA, Kostousouv V, Minei KM, Radwan ZA, Schochl H, Wade CE, Holcomb JB, Matijevic N. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J Trauma Acute Care Surg. 2012;73:365–70. doi: 10.1097/TA.0b013e31825c1234. [DOI] [PubMed] [Google Scholar]
- 14.Schochl H, Frietsch T, Pavelka M, Jambor C. Hyperfibrinolysis after major trauma: Differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67:125–131. doi: 10.1097/TA.0b013e31818b2483. [DOI] [PubMed] [Google Scholar]
- 15.CRASH-2 collaborators. Shakur H, Roberts I, Bautista R, Caballero J, Coast T, Dewan Y, El-Sayed H, Gogichaishvili T, Gupta S, et al. Effects of Tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): A randomized, placebo-controlled trial. Lancet. 2010;376:23–32. doi: 10.1016/S0140-6736(10)60835-5. [DOI] [PubMed] [Google Scholar]
- 16.Napolitano LM, Cohen MJ, Cotton BA, Schreiber MA, Moore EE. Tranexamic acid in trauma: How should we use it? J Trauma Acute Care Surg. 2013;74:1575–86. doi: 10.1097/TA.0b013e318292cc54. [DOI] [PubMed] [Google Scholar]
- 17.Pusateri AE, Weiskopf RB, Bebarta V, Butler F, Cestero RF, Chaudry IH, Deal V, Dorlac WC, Gerhardt RT, Given MB, et al. Tranexamic acid and trauma: Current status and knowledge gaps with recommended research priorities. Shock. 2013;39:121–6. doi: 10.1097/SHK.0b013e318280409a. [DOI] [PubMed] [Google Scholar]
- 18.CRASH-2 collaborators. Roberts I, Shakur H, Afolabi A, Brohi K, Coats T, Dewan Y, Gando S, Guyatt G, Hunt BJ, et al. The importance of early treatment with Tranexamic acid in bleeding trauma patients: An exploratory analysis of the CRASH-2 randomised controlled trial. Lancet. 2011;377:1096–101. doi: 10.1016/S0140-6736(11)60278-X. [DOI] [PubMed] [Google Scholar]
- 19.Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military application of tranexamic acid in trauma emergency resuscitation (MATTERs) study. Arch Surg. 2012;147:113–9. doi: 10.1001/archsurg.2011.287. [DOI] [PubMed] [Google Scholar]
- 20.Valle EJ, Allen CJ, Van Haren RM, Jouria JM, Li H, Livingstone AS, Namias N, Schulman CI, Proctor KG. Do all trauma patients benefit from Tranexamic acid? J Trauma Acute Care Surg. 2014;76:1373–8. doi: 10.1097/TA.0000000000000242. [DOI] [PubMed] [Google Scholar]
- 21.Harvin JA, Mims MM, Hudson J, Podbielski J, Wade CE, Holcomb JB, Cotton BA. The impact of Tranexamic acid on mortality in injured patients with hyperfibrinolysis. J Trauma Acute Care Surg. doi: 10.1097/TA.0000000000000612. In Press. [DOI] [PubMed] [Google Scholar]
- 22.Chin TL, Moore EE, Moore HB, Gonzalez E, Chapman MP, Stringham JR, Ramos CR, Banerjee A, Sauaia A. A principal component analysis of Postinjury viscoelastic assays: Clotting factor depletion versus fibrinolysis. Surgery. 2014;156:570–7. doi: 10.1016/j.surg.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutcher ME, Ferguson AR, Cohen MJ. A principal component analysis of coagulation after trauma. J Trauma Acute Care Surg. 2013;74:1223–1228. doi: 10.1097/TA.0b013e31828b7fa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin TL, Silliman CC, Banerjee A, Sauaia A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: The spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J Trauma Acute Care Surg. 2014;77:811–7. doi: 10.1097/TA.0000000000000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore HB, Moore EE, Lawson P, Gonzalez E, Fragoso M, Morton AP, Gamboni F, Chapman MP, Sauaia A, Banerjee A, Silliman CC. Fibrinolysis shutdown phenotype masks changes in rodent coagulation in tissue injury versus hemorrhagic shock. Surgery. doi: 10.1016/j.surg.2015.04.008. In Press. [DOI] [PMC free article] [PubMed]
- 26.Chapman MP, Moore EE, Ramos CR, Ghasabyan A, Harr JN, Chin TL, Stringham JR, Sauaia A, Silliman CC, Banerjee A. Fibrinolysis greater than 3% is the critical value for initiation of antifibrinolytic therapy. J Trauma Acute Care Surg. 2013;75:961–7. doi: 10.1097/TA.0b013e3182aa9c9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cesar-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Brit J Haem. 2005;129:307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 28.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haem. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 29.Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haem. 2007;5:116–24. doi: 10.1111/j.1538-7836.2007.02504.x. [DOI] [PubMed] [Google Scholar]
- 30.Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Ghasabyan A, Chin T, Sauaia A, Banerjee A, Silliman CC. Overwhelming tPA, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured patients. J Trauma Acute Care Surg. doi: 10.1097/TA.0000000000000885. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore HB, Moore EE, Gonzalez E, Hansen KC, Dzieciatkowska M, Chapman MP, Sauaia A, West B, Banerjee A, Silliman CC. Hemolysis exacerbates hyperfibrinolysis while platelolysis shuts down fibrinolysis: Evolving concepts of the spectrum of fibrinolysis in response to severe injury. Shock. 2015;43:39–46. doi: 10.1097/SHK.0000000000000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore HB, Moore EE, Gonzalez E, Chapman MP, Chin T, Banerjee A, Sauaia A, Silliman CC. Plasma is the physiologic buffer of tPA mediated fibrinolysis: Rational for plasma first resuscitation after life threatening hemorrhage. J Am Coll Surg. doi: 10.1016/j.jamcollsurg.2015.01.026. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Booth NA, Simpson AJ, Croll A, Bennett B, MacGregor IR. Plasminogen activator inhibitor-1 (PAI-1) in plasma and platelets. Brit J Haem. 1988;70:327–33. doi: 10.1111/j.1365-2141.1988.tb02490.x. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez e, Moore EE, Moore HB, Pieracci FC, Chin T, Chapman MP, Sauaia A, Silliman CC, Banerjee A. Is Fibrinolysis the missing link leading to postinjury hypercoagulability. J Am Coll Surg. 2014;219:s47. [Google Scholar]