

 Ruthenium photolinkers provide a versatile method of using visible light to control structure and function of biopolymers.
Ruthenium photolinkers provide a versatile method of using visible light to control structure and function of biopolymers.
Abstract
Photochemical approaches afford high spatiotemporal control over molecular structure and function, for broad applications in materials and biological science. Here, we present the first example of a visible light responsive ruthenium-based photolinker, Ru(bipyridine)2(3-ethynylpyridine)2 (RuBEP), which was reacted stoichiometrically with a 25mer DNA or morpholino (MO) oligonucleotide functionalized with 3′ and 5′ terminal azides, via Cu(i)-mediated [3+2] Huisgen cycloaddition reactions. RuBEP-caged circular morpholinos (Ru-MOs) targeting two early developmental zebrafish genes, chordin and notail, were synthesized and tested in vivo. One-cell-stage zebrafish embryos microinjected with Ru-MO and incubated in the dark for 24 h developed normally, consistent with caging, whereas irradiation at 450 nm dissociated one 3-ethynylpyridine ligand (Φ = 0.33) and uncaged the MO to achieve gene knockdown. As demonstrated, Ru photolinkers provide a versatile method for controlling structure and function of biopolymers.
Introduction
Photochemical methods for regulating the structure, function, and/or localization of molecular species enable the manipulation of advanced materials (e.g., silicon computer chips) as well as complex biological systems. For example, channelrhodopsin—a single component, light-activated cation channel protein from algae—was co-opted in the development of optogenetic approaches for manipulating the activity of specific neurons and controlling animal behaviour.1 More generally, “caged” molecules,2 whose latent biological activity can be revealed with light, have been widely adopted, particularly for the study of amino acids,3 peptides,4 neurotransmitters,5 and metal ions.6 In each case, photoactivation with high spatiotemporal control can be achieved using a focused laser beam of suitable wavelength. Less investigated are caged oligonucleotides, despite the central roles played by DNA and RNA in biology and the tantalizing potential for being able to turn genes “on” or “off” with light. Synthetic challenges of site-specifically incorporating one or more photolabile moieties within a large oligonucleotide, and limitations arising from the available organic caging moieties, have slowed such development.
A particular focus for caged oligo development has been antisense morpholinos (MOs), which are commonly used to block mRNA translation and modify pre-mRNA splicing in a variety of model organisms, including mouse, zebrafish, frog, sea urchin, and chick.7 Initial caged antisense oligos from our lab,8–10 the Chen lab,11–13 and Tomasini et al. 14 employed a complementary sense strand and photocleavable linker. Deiters et al. subsequently presented caged MOs where multiple caged nucleotide monomers were incorporated during solid-phase synthesis.15 In this example, MO-mRNA hybridization was sterically blocked until the caging groups were released from the nucleobases.15 A newer design strategy, presented by the labs of Chen16 and Tang17,18 has involved linking the 5′ and 3′ ends with a photocleavable moiety. The covalent linkage enforces the closed circular conformation, which prevents efficient MO hybridization to target mRNA until photocleavage restores the linear, biologically active MO. All of these approaches employed an organic photocleavable linker, such as o-nitrobenzyl or hydroxycoumarin, which yielded optimally to near-UV irradiation.19
To expand in vivo applications using caged oligos, there is need for synthetically versatile photolinkers that can be activated at visible or near-IR wavelengths, as near-UV light has poor tissue penetration and can be toxic at high exposure levels.20–22 The Deiters and Chen labs recently advanced this concept by employing a red-shifted organic caging moiety, [7-(diethylamino)coumarin-4-yl]-methyl (DEACM).23 By co-injecting zebrafish embryos with 470 nm responsive DEACM-caged MO targeting flh and 365 nm responsive 2-nitrobenzyl-caged MO targeting spt, discrete spatiotemporal control was retained over each gene. Previous inorganic caging strategies included the use of near-IR-to-UV upconversion nanoparticles to achieve siRNA photoactivation in cells and tissues.21 Here, we exploit versatile ruthenium photochemistry and conjugation chemistries to generate caged oligos that are efficiently activated with visible light.
Ruthenium complexes of the general type [Ru(bipyridine)2(X)2]2+, where X = amine,24 nitrile,25 pyridine,26 or thioether27 ligands, have been shown to undergo facile X ligand exchange with solvent upon irradiation with visible one-photon or near-IR two-photon excitation.28 Biologically active small molecules can be directly ligated to the Ru2+ center, and then released with visible light.29 In 2003 Etchenique and co-workers first applied this Ru-ligand exchange property by caging a potassium channel blocker, 4-aminopyridine,26 and have since caged several neurotransmitters.3,30,31 More recently, the Turro lab investigated ruthenium polypyridyl complexes for their potential as photodynamic drugs.32,33 Building on these and other Ru-caging examples,3,25,34–36 we set out to develop a Ru-photolinker amenable to caging oligos and other large biomolecules, with the goals of bypassing the harsh synthetic conditions typically required for ligand substitution at Ru2+, and avoiding direct reaction between biomolecules and Ru2+.
Here, we report the synthesis, characterization, and application of the first Ru-photolinker, [Ru(bipyridine)2(3-ethynylpyridine)2]Cl2 (RuBEP, Scheme 1). The bis-alkyne functionality enabled circularization of an oligonucleotide containing azides at both 5′ and 3′ termini via [3+2] azide-alkyne copper(i)-mediated cycloaddition reactions.37 In this way, the octahedral Ru2+ center remained coordinatively saturated, and side-reactions between Ru2+ and the nucleobases were avoided. Photolysis at 450 nm restored the linear, biologically active oligo (Scheme 1).
Scheme 1. RuBEP photolinker conjugated with 25mer bis-azido morpholino formed “caged” antisense MO; subsequent 450 nm irradiation restored biologically active MO.

Results and discussion
Synthesis and characterization of RuBEP
RuBEP was synthesized via a triflate intermediate from commercially available cis-Ru(bipyridine)2Cl2 (Acros Organics) and 3-ethynylpyridine (3EP) (Scheme S1†).25 Reaction progress was monitored by UV-Vis spectroscopy until an MLCT band at 450 nm was observed (Fig. S1†). The PF6 – salt (RuBEP[PF6]2), synthesized by metathesis with ammonium hexafluorophosphate in cold water, was purified in the dark by silica column chromatography using 1 : 9 acetonitrile : dichloromethane as the eluent. The water-soluble chloride salt (RuBEP) was then generated by metathesis with TBACl in cold acetone. Final yield was 60–70%.
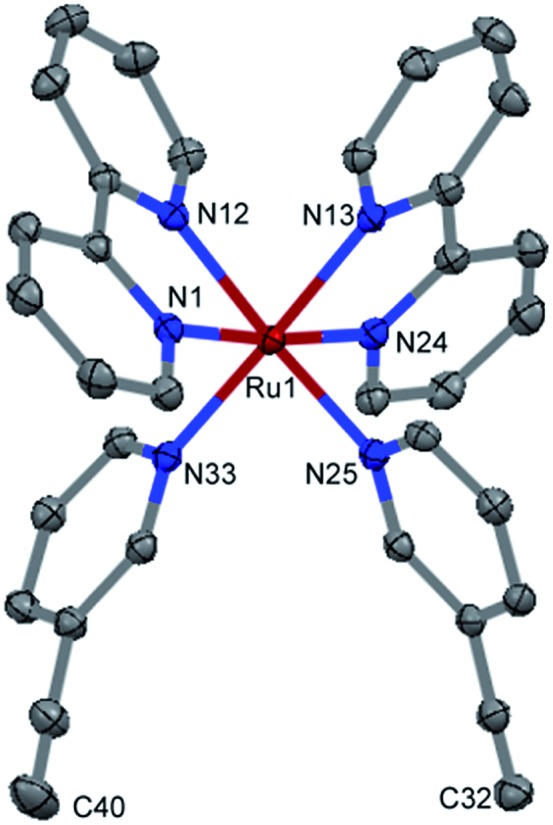
The identity and purity of RuBEP was confirmed by 1H and 13C NMR spectroscopy, ESI-MS, and elemental analysis. An X-ray crystal structure (Fig. 1, S2 and Tables S1–S6†) showed standard Ru2+–N bond lengths for the bipyridine and pyridine ligands. The N3EP–Ru–N3EP bond angle was 92.5° and twisting of the two 3EP ligands positioned the alkynes (C40–C32 = 6.188(3) Å) for subsequent cycloaddition reactions.
Fig. 1. X-ray structure of RuBEP photolinker showing alkynes (bottom) available for subsequent cycloaddition reactions.
Photodissociation of 3EP from RuBEP was monitored by UV-Vis (Fig. 2), LCMS (Fig. S3†), and NMR (Fig. S4†) spectroscopies. Upon continuous irradiation with 450 nm laser (53 mW cm–2, focused to 0.5 cm2), the λ max red-shifted from 450 nm to 473 nm (Fig. 2). Complete photolysis of the bulk RuBEP solution (80 μM, 1.5 mL, stirred) occurred in 5 min. The orange photo-product [Ru(bpy)2(3EP)(OH2)]2+ was consistent with previously characterized [Ru(bpy)2(pyr)(OH2)]2+ complexes.38 Isosbestic points were observed at 450 nm and in the near-UV, as expected for the exchange of one pyridine ligand without formation of rate-limiting intermediates.34 1H NMR also showed the exchange of only one 3EP ligand with a solvent water molecule, based on a shifted alkyne peak and change in integration (Fig. S4†). HR-MS also confirmed the photoproduct assignment (Fig. S3†). The quantum yield of ligand exchange in water in ambient conditions (φ = 0.33 ± 0.06) was determined by fitting the initial kinetics of the photoreaction (Fig. S5†). This was comparable to the quantum yield of ligand exchange reported for Ru(bpy)2(pyr)2Cl2 (φ = 0.4).26 The uncaging efficiency for RuBEP (ε 450 times φ) was determined to be 2.0 × 103 M–1 cm–1 at 450 nm, which is much higher than measured for typical organic chromophores activated at near-UV wavelengths. Commonly used nitrobenzyl derivatives, for example, have uncaging efficiencies less than 100 M–1 cm–1 at 365 nm.4,39
Fig. 2. (A) Irradiation of RuBEP with 450 nm laser induced ligand exchange. (B) UV-Vis absorption spectroscopy showed isosbestic points at 364, 385, and 450 nm, consistent with the exchange of one 3EP ligand for solvent (water).

Circularization of 25mer oligonucleotides
Circularization protocols were investigated initially using a bis-azido 25mer DNA oligonucleotide. The [3+2] cycloaddition reaction was performed at a stoichiometry of 1.2 DNA : 1 RuBEP, in the presence of 10× CuBr and 20× chelator tris(benzyltriazolylmethyl)amine (TBTA) and monitored by polyacrylamide gel electrophoresis (PAGE). A band migrating faster than bis-azido DNA appeared within the first 15 min of reaction (Fig. S6†) consistent with RuBEP-circularization inducing a more compact structure and contributing positive charge. The reaction was complete within 3 h and quenched by NAP-5 desalting column. As a control, mono-azido DNA was subjected to the same reaction conditions, which resulted in a slower migrating band (Fig. S7†). The desired circular Ru–DNA product was isolated by reverse-phase HPLC in 20–25% yield (Fig. S8, S9 and Table S7†), and confirmed by MALDI-TOF MS (Fig. S10 and Table S8†). After 3 min irradiation with 450 nm light emitting diode (14 mW cm–2, beam area = 12.6 cm2) this band migrated at the same rate as the linear DNA, suggesting full conversion to the active, linear species. No changes to the photoproduct (including reversion to the circular Ru–DNA) were observed after 24 h in solution under ambient conditions (Fig. S9†).
Caging was confirmed through a molecular beacon assay (Fig. 3) in which a stem-loop, reverse complementary probe with fluorophore-quencher pair was incubated with oligo samples for 20 min at 25 °C. The degree of DNA–beacon hybridization, determined by relative fluorescence intensity, was nearly zero for a mismatched sequence, and scaled to 100% for the linear, fully complementary DNA. Only 5% beacon fluorescence was observed with Ru–DNA vs. fully restored fluorescence after 3 min irradiation with 450 nm light (14 mW cm–2), consistent with complete uncaging.
Fig. 3. Molecular beacon assay showing near complete caging of circular Ru–DNA and Ru–MO, with restoration of fluorescence intensity after photoactivation.

Circularization of 25mer morpholino
The circularization conditions were subsequently applied to antisense MOs, in order to photoregulate gene expression in living zebrafish embryos (Scheme 1). Two early developmental zebrafish genes were targeted, chordin (chd) and notail (ntl), due to their well characterized and easily recognizable knockdown phenotypes with antisense MOs.40 Bis-azido MOs were purchased from Gene Tools (Philomath, OR) and added to RuBEP in a 1.05 : 1 ratio with 10× CuBr and 20× TBTA. The reaction proceeded at rt for 18 h. These conditions promoted reaction of one RuBEP per MO, thus favouring intramolecular reaction and circularization. Higher molecular weight Ru-MO polymers precipitated under the reaction conditions and were removed by centrifugation. Excess reagents (RuBEP, Cu, TBTA, and solvents) were removed with NAP-5 column, leaving pure Ru-MO in water (isolated yield = 20–30%). Mass confirmation was obtained by MALDI-TOF MS (Table S8†).
Ru-MO formation was assessed by gel-shift assay employing a 25mer complementary DNA strand (Fig. 4). Due to the neutral charge of morpholinos, Ru-MO-chd and Ru-MO-ntl could not be analysed using standard PAGE or HPLC as Ru–DNA was. Thus, a Ru-MO : DNA hybrid was formed by heating to 80 °C and cooling to 4 °C, run on a 15% native polyacrylamide gel on ice (100 V, 120 min) and stained with ethidium bromide (Fig. 4). The complementary DNA (lane 1) ran slower when hybridized to linear MO (lane 2). Upon circularization (lane 3), the Ru-MO-chd : DNA hybrid migrated even slower, which was due to its secondary structure and reduced affinity for complementary DNA. Photoactivation at 450 nm (14 mW cm–2, 3 min) resulted in complete uncaging, yielding a mono-Ru-functionalized linear MO that was hybridized to DNA (lane 4) and ran comparably to the linear MO : DNA hybrid (lane 2); the pendant Ru2+ moiety (in lane 4) had no apparent effect. All lanes contained a slight excess of complementary DNA (lowest band) to promote hybridization. QuantIT band quantification showed less than 5% unreacted bis-azido MO after 18 h RuBEP reaction. A molecular beacon assay was similarly used to confirm caging of the Ru-MO construct, with only 28% fluorescence intensity observed relative to the linear control (Fig. 3). Different beacon designs produced varying levels of background fluorescence for the Ru-MO-chd constructs, but in all cases significant modulation of fluorescent signal was observed, consistent with Ru-oligo caging and uncaging.
Fig. 4. 15% native PAGE gel-shift assay with 25 pmol of complementary 25mer DNA (lane 1) and DNA hybridized to 20 pmol MO-chd (lane 2), Ru-MO-chd (lane 3) and its subsequent photo-product (lane 4).

Ru-MO in vivo studies
MO-chd or Ru-MO-chd (514 pmol μL–1) was microinjected into 1-cell-stage zebrafish embryos, which were incubated at 28 °C in the dark and at 24 hours post-fertilization (hpf) scored for phenotypic response and imaged by confocal microscopy.41 Embryos scored as normal had V-shaped somites, and normal head and tail development. The chordin knockdown phenotype ranged from severe to mild where severe was identified by decreased head size, U-shaped somites, and a large blood island on the tail. The moderate and mild phenotypes were identified by U-shaped somites and a medium or small blood island on the tail. Zebrafish experiments were performed following a protocol approved by the University of Pennsylvania IACUC.
Representative images of the three levels of chd knockdown phenotypic response compared to wildtype are shown in Fig. S11.† Half of the Ru-MO-chd embryos were irradiated with 450 nm light (14 mW cm–2, 5 min) at 1 hpf and returned to dark incubation. Fig. 5(A–D) shows representative images of (A) uninjected control, (B) Ru-MO-chd-injected embryos incubated in the dark, (C) Ru-MO-chd-injected embryos irradiated with 450 nm light, and (D) positive control embryos injected with MO-chd. A graph of phenotypic responses, Fig. 5E, confirms that Ru-MO-chd was significantly caged in vivo, with only 14% of embryos showing some level of MO-chd activity. After irradiation, 92% of embryos developed with the expected chd knockdown phenotype, showing that the retained Ru moiety did not affect MO activity in vivo. The 8% of embryos with normal development can be attributed to injection variability, as this was consistent with the MO-chd control injections (∼5% normal phenotype). To confirm sequence specificity, identical experiments were performed with Ru-MO targeting ntl,42 and similar caging/uncaging results were obtained (Fig. S12–S14†). Ru-MO-ntl showed increased background activity likely due to the slight impurities in the injection sample. The ntl bis-azido MO was received in lower purity than chd bis-azido MO, which decreased the yield and purity of the desired circular product. RuBEP was injected as a control with and without irradiation, and no toxicity or phenotypic response was observed (Fig. S15†). Additionally, a scramble morpholino was injected into 1-cell stage embryos and resulted in normal development (Fig. S16 and Table S9†).
Fig. 5. Representative confocal micrographs of 24–28 hpf zebrafish embryos, showing chd knockdown phenotypes depending on experimental protocol. (A) Wildtype embryo, uninjected. (B) Ru-MO-chd, incubated in the dark, showing normal development. (C) Ru-MO-chd, irradiated for 5 min at 1 hpf with 450 nm light, showing chd knockdown phenotype. (D) Bis-azido MO-chd showing chd knockdown phenotype. All embryos were injected at 1-cell stage and imaged with a 10× air objective (Olympus UPlanSApo, NA = 0.40). (E) Percentage of embryos showing wildtype or degree of phenotypic response.

Conclusions
We synthesized and characterized the first ruthenium photolinker, RuBEP, and demonstrated reaction with bis-azide-functionalized oligonucleotides to form circular, caged oligos in good yields and purity. The RuBEP photolinker is extremely versatile and will support a variety of caged oligo designs, including lariat, stem-loop, and hairpin structures. The compact geometry and structural rigidity of RuBEP are distinguishing features of this inorganic photolinker, and likely contributed to very effective caging in all circular Ru-oligos that were tested.
Ru-caged antisense MOs underwent efficient Ru2+-ligand exchange upon 450 nm irradiation, to reveal the biologically active, linear structures. The pendant Ru2+ moiety did not adversely affect target hybridization (Fig. 3 and 4) nor biological activity (Fig. 5E). In addition to the broad in vivo applications for Ru-caged MOs, we expect that RuBEP can be used to cage or crosslink many other azide-modified biomolecules, e.g., peptides, lipids, and oligosaccharides. Finally, the versatile inorganic photochemistry of ruthenium polypyridyl complexes32,34 will allow the development of numerous Ru photolinkers for multiplexed photocontrol of diverse applications in biology and materials science.
Acknowledgments
This work was supported by National Institutes of Health (R01 GM083030) and a McKnight Foundation Technological Innovations Award to IJD and JE. The authors would like to thank Sean Yeldell and Brittany Riggle for helpful discussions. Bruker Ultraflex III MALDI-TOF/TOF and Waters LCT Premier XE LC/MS ESI-TOF were supported by NSF CHE-0820996 and NIH 1S10RR023444. Olympus FV1000 confocal microscope was supported by NIH 1S10RR021113.
Footnotes
References
- Boyden E. S., Zhang F., Bamberg E., Nagel G., Deisseroth K. Nat. Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Young D. D., Deiters A. Org. Biomol. Chem. 2007;5:999–1005. doi: 10.1039/b616410m. [DOI] [PubMed] [Google Scholar]
- Salierno M., Fameli C., Etchenique R. Eur. J. Inorg. Chem. 2008;2008:1125–1128. [Google Scholar]
- Gatterdam V., Ramadass R., Stoess T., Fichte M. A. H., Wachtveitl J., Heckel A., Tampe R. Angew. Chem., Int. Ed. 2014;53:5680–5684. doi: 10.1002/anie.201309930. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M., Hayama T., Kasai H., Ellis-Davies G. C. R. Nat. Chem. Biol. 2010;6:255–257. doi: 10.1038/nchembio.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis-Davies G. C. R. Chem. Rev. 2008;108:1603–1613. doi: 10.1021/cr078210i. [DOI] [PubMed] [Google Scholar]
- Eisen J. S., Smith J. C. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- Tang X., Dmochowski I. J. Angew. Chem., Int. Ed. 2006;45:3523–3526. doi: 10.1002/anie.200600954. [DOI] [PubMed] [Google Scholar]
- Tang X., Dmochowski I. J. Mol. BioSyst. 2007;3:100–110. doi: 10.1039/b614349k. [DOI] [PubMed] [Google Scholar]
- Tang X., Maegawa S., Weinberg E. S., Dmochowski I. J. J. Am. Chem. Soc. 2007;129:11000–11001. doi: 10.1021/ja073723s. [DOI] [PubMed] [Google Scholar]
- Ouyang X., Shestopalov I. A., Sinha S., Zheng G., Pitt C. L., Li W. H., Olson A. J., Chen J. K. J. Am. Chem. Soc. 2009;131:13255–13269. doi: 10.1021/ja809933h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestopalov I. A., Chen J. K. Methods Cell Biol. 2011;104:151–172. doi: 10.1016/B978-0-12-374814-0.00009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestopalov I. A., Sinha S., Chen J. K. Nat. Chem. Biol. 2007;3:650–651. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- Tomasini A. J., Schuler A. D., Zebala J. A., Mayer A. N. Genesis. 2009;47:736–743. doi: 10.1002/dvg.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiters A., Garner R. A., Lusic H., Govan J. M., Dush M., Nascone-Yoder N. M., Yoder J. A. J. Am. Chem. Soc. 2010;132:15644–15650. doi: 10.1021/ja1053863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazoe S., Shestopalov I. A., Provost E., Leach S. D., Chen J. K. Angew. Chem., Int. Ed. 2012;51:6908–6911. doi: 10.1002/anie.201201690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wu L., Wang P., Lv C., Yang Z., Tang X. Nucleic Acids Res. 2012;40:11155–11162. doi: 10.1093/nar/gks840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Wang Y., Wu J., Lv C., Wang J., Tang X. Nucleic Acids Res. 2013;41:677–686. doi: 10.1093/nar/gks996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H., Furuta T., Tsien R. Y., Okamoto H. Nat. Genet. 2001;28:317–325. doi: 10.1038/ng583. [DOI] [PubMed] [Google Scholar]
- Pawley J. B., Handbook of Biological Confocal Microscopy, Springer, 2006. [Google Scholar]
- Jayakumar M. K. G., Idris N. M., Zhang Y. Proc. Natl. Acad. Sci. U. S. A. 2012:8483–8488. doi: 10.1073/pnas.1114551109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoien J. D., Wang R. J. Proc. Natl. Acad. Sci. U. S. A. 1974;71:3961–3965. doi: 10.1073/pnas.71.10.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazoe S., Liu Q., McQuade L. E., Deiters A., Chen J. K. Angew. Chem., Int. Ed. 2014;53:10114–10118. doi: 10.1002/anie.201405355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayat L., Salierno M., Etchenique R. Inorg. Chem. 2006;45:1728–1731. doi: 10.1021/ic0512983. [DOI] [PubMed] [Google Scholar]
- Garner R. N., Gallucci J. C., Dunbar K. R., Turro C. Inorg. Chem. 2011;50:9213–9215. doi: 10.1021/ic201615u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayat L., Calero C., Albores P., Baraldo L., Etchenique R. J. Am. Chem. Soc. 2003;125:882–883. doi: 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]
- Garner R. N., Joyce L. E., Turro C. Inorg. Chem. 2011;50:4384–4391. doi: 10.1021/ic102482c. [DOI] [PubMed] [Google Scholar]
- Araya R., Andino-Pavlovsky V., Yuste R., Etchenique R. ACS Chem. Neurosci. 2013;4:1163–1167. doi: 10.1021/cn4000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayat L., Filevich O., Baraldo L. M., Etchenique R. Philos. Trans. R. Soc., A. 2013;371:20120330. doi: 10.1098/rsta.2012.0330. [DOI] [PubMed] [Google Scholar]
- Fino E., Araya R., Peterka D. S., Salierno M., Etchenique R., Yuste R. Front. Neural Circuits. 2009;3:1–9. doi: 10.3389/neuro.04.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filevich O., Etchenique R. Photochem. Photobiol. Sci. 2013;12:1565–1570. doi: 10.1039/c3pp25248e. [DOI] [PubMed] [Google Scholar]
- Albani B. A., Durr C. B., Turro C. J. Phys. Chem. A. 2013;117:13885–13892. doi: 10.1021/jp4085684. [DOI] [PubMed] [Google Scholar]
- Albani B. A., Pena B., Dunbar K. R., Turro C. Photochem. Photobiol. Sci. 2014;13:272–280. doi: 10.1039/c3pp50327e. [DOI] [PubMed] [Google Scholar]
- Sears R. B., Joyce L. E., Ojaimi M., Gallucci J. C., Thummel R. P., Turro C. J. Inorg. Biochem. 2013;121:77–87. doi: 10.1016/j.jinorgbio.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Goldbach R. E., Rodriguez-Garcia I., van Lenthe J. H., Siegler M. A., Bonnet S. Chem.–Eur. J. 2011;17:9924–9929. doi: 10.1002/chem.201101541. [DOI] [PubMed] [Google Scholar]
- Palmer A. M., Pena B., Sears R. B., Chen O., El Ojaimi M., Thummel R. P., Dunbar K. R., Turro C. Philos. Trans. R. Soc., A. 2013;371:1–10. doi: 10.1098/rsta.2012.0135. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B. Angew. Chem., Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Bryant J. R., Mayer J. M. J. Am. Chem. Soc. 2003;125:10351–10361. doi: 10.1021/ja035276w. [DOI] [PubMed] [Google Scholar]
- Furuta T., Wang S. S.-H., Dantzker J. L., Dore T. M., Bybee W. J., Callaway E. M., Denk W., Tsien R. Y. Proc. Natl. Acad. Sci. U. S. A. 1999;96:1193–1200. doi: 10.1073/pnas.96.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A., Ekker S. C. Nat. Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Westerfield M., The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), University of Oregon Press, 2000. [Google Scholar]
- Tallafuss A., Gibson D., Morcos P., Li Y., Seredick S., Eisen J., Washbourne P. Development. 2012;139:1691–1699. doi: 10.1242/dev.072702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.