

 β-Lactam derivatives are produced through donor–acceptor cyclopropene intermediates in high yield with exclusive cis-diastereoselectivity, and high enantiocontrol.
β-Lactam derivatives are produced through donor–acceptor cyclopropene intermediates in high yield with exclusive cis-diastereoselectivity, and high enantiocontrol.
Abstract
β-Lactam derivatives are produced through intermediate donor–acceptor cyclopropene intermediates in high yield, exclusive cis-diastereoselectivity, and high enantiocontrol in a chiral dirhodium carboxylate catalyzed intramolecular C–H functionalization reaction of enoldiazoacetamides.
Introduction
The importance of β-lactam compounds (2-azetidinones) in biology and medicine is undisputed since the discovery of the antibiotic activity of the bicyclic penicillin in 1928,1,2 and monocyclic β-lactams that include the plasma cholesterol-lowering Ezetimibe3 also show biological activity.4 Considerable effort has been focused on chemical catalysis for the construction of β-lactams, including those through catalytic intramolecular amide N–H insertion reactions of diazo compounds,5 and asymmetric synthesis has been a primary focus.2,6 However, the same C–H functionalization methodology of diazo compounds that has provided exceptional selectivities in intermolecular reactions7 and intramolecular syntheses of γ-lactones and γ-lactams8 has been limited in efforts to synthesize β-lactams.
Direct intramolecular C–H functionalization of diazoacetamides catalyzed by transition metal catalysts is straightforward.9 The amide nitrogen activates the adjacent C–H bond for insertion. Although β-lactam formation by photocatalysis was described fifty years ago,10 and the first enantioselective process was reported in 1992,2c,11 there have only been a few examples that have overcome a majority of the electronic, steric, and conformational factors which control the selectivity of this process.12,13 Competing reactions include intramolecular cycloaddition to an aromatic ring of an aryl- or heterocycle attachment (Buchner reaction), addition to a carbon–carbon multiple bond of an allylic or propargylic system, or regioselective formation of a γ-lactam from C–H functionalization.14 Product selectivity is highly dependent on the diazo compound that is employed;12,14 for example, acceptor or donor–acceptor diazoamides form aromatic cycloaddition products when catalyzed by dirhodium catalysts (Scheme 1, Path A, R2 = H or EDG), but acceptor–acceptor diazoamides produce β-lactam products by a C–H functionalization reaction (Path B, R2 = EWG).
Scheme 1. Diazoacetamide substrate dependence on chemoselectivity.

To achieve high selectivity in C–H functionalization reactions that form β-lactam products, acceptor–acceptor diazoamides (R2 = EWG) have been constructed to avoid side reactions, but high regio- and enantiocontrol (>90% ee) has only been demonstrated in constrained cyclic systems in which the aliphatic γ-position has been made inaccessible for insertion.12a,b We have developed enoldiazoacetates as a new class of stable donor–acceptor diazo compounds with extensive applications for cycloaddition reactions.15 An attractive feature of this vinyldiazo compound in catalytic reactions with dirhodium(ii) carboxylates is the apparent donor–acceptor cyclopropene intermediate that serves as a resting state for the apparent reactive metal carbene intermediate.16 Could enoldiazoacetamides form intermediate donor–acceptor cyclopropenes and be precursors to donor–acceptor metal carbene intermediates on the pathway to C–H functionalization? Earlier work by Müller suggested that enantioselectivity in cyclopropanation reactions of styrene with an enoldiazoacetate is significantly greater than that with the corresponding diazoacetoacetate.17 We wish to report that asymmetric catalysis with donor–acceptor N-benzyldiazoamides proceeds through donor–acceptor cyclopropene intermediates to form cis-disubstituted β-lactams by intramolecular C–H functionalization in high yields and stereoselectivities (Path C).
Results and discussion
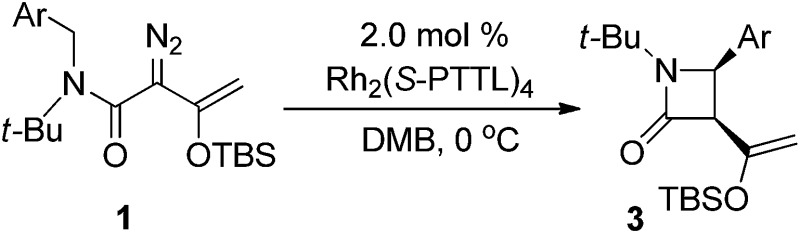
In initial studies we selected the N-tert-butyl-N-(p-methoxy-benzyl)enoldiazoacetamide 1a as a model substrate since previous studies have shown that the tert-butyl group fixed the reactant in the conformation in which the benzyl and diazo functional groups are syn to each other.13 In this conformation C–H insertion into the tert-butyl group is prevented, but aromatic cycloaddition into the anisyl group could be competitive with C–H insertion into its benzyl group, and indeed this competition was observed (Table 1). The Buchner product 2a was dominant in reactions catalyzed by sterically unencumbered Rh2(OAc)4 or the electrophilic Rh2(pfb)4, but with the sterically restrictive Rh2(tpa)4 or Rh2(esp)2 the sole product was the cis-disubstituted β-lactam 3a, formed by C–H insertion into the benzylic position. With asymmetric catalysts similar steric influences were operative so that increasing the steric bulk of the chiral Hashimoto dirhodium(ii) carboxylate catalyst ligand increased the 3a/2a ratio and, also, enhanced enantioselectivity (entries 5–9) for both products. Both Rh2(S-PTTL)4 and Rh2(S-PTAD)4 gave β-lactam 3a as the only product in high yield with 64% ee (entries 8 and 9). We chose Rh2(S-PTTL)4 for further optimization and, after screening solvents and reaction temperatures, the reaction carried out at 0 °C in 2,2-dimethylbutane (DMB) gave the optimum result with 85% isolated yield and 92% ee of 3a (entry 17).18 The more Lewis acidic Rh2(S-TCPTTL)4 gave 3a in higher yield but slightly lower % ee. Chiral dirhodium carboxamidates were not effective as catalysts for this transformation, but the more reactive Rh2(S-DOSP)4 produced a mixture of 2a and 3a with low enantioselectivities (entry 5).
Table 1. Optimization of reaction conditions for the enantioselective C–H functionalization of enlodiazoacetamide 1a a .

| |||||
| Entry | Rh(ii) | Solvent | 2a : 3a e | Yield f , (%) 2a + 3a | ee g (%) 2a/3a |
| 1 b | Rh2(OAc)4 | DCCl3 | 80 : 20 | 87 | —/— |
| 2 b | Rh2(pfb)4 | DCCl3 | 80 : 20 | 89 | —/— |
| 3 b | Rh2(tpa)4 | DCCl3 | <5 : 95 | 92 | —/— |
| 4 b | Rh2(esp)2 | DCCl3 | <5 : 95 | 87 | —/— |
| 5 b | Rh2(S-DOSP)4 | DCCl3 | 37 : 63 | 90 | 27/10 |
| 6 b | Rh2(S-PTA)4 | DCCl3 | 25 : 75 | 92 | 35/30 |
| 7 b | Rh2(S-PTPA)4 | DCCl3 | 33 : 67 | 90 | 53/41 |
| 8 b | Rh2(S-PTTL)4 | DCCl3 | <5 : 95 | 93 | —/64 |
| 9 b | Rh2(S-PTAD)4 | DCCl3 | <5 : 95 | 93 | —/64 |
| 10 | Rh2(S-PTTL)4 | DCM | <5 : 95 | 89 | —/65 |
| 11 | Rh2(S-PTTL)4 | DCE | <5 : 95 | 88 | —/68 |
| 12 | Rh2(S-PTTL)4 | Toluene | <5 : 95 | 76 | —/75 |
| 13 | Rh2(S-PTTL)4 | CF3Ph | <5 : 95 | 72 | —/67 |
| 14 | Rh2(S-PTTL)4 | Cyclohexane | <5 : 95 | 90 | —/82 |
| 15 | Rh2(S-PTTL)4 | TBME | <5 : 95 | 91 | —/71 |
| 16 | Rh2(S-PTTL)4 | DMB | <5 : 95 | 83 | —/88 |
| 17 c | Rh2(S-PTTL)4 | DMB | <5 : 95 | 85 | —/92 |
| 18 d | Rh2(S-PTTL)4 | DMB | <5 : 95 | 82 | —/91 |
| 19 c | Rh2(S-TCPTTL)4 | DMB | <5 : 95 | 88 | —/89 |
aReactions were carried out at room temperature on a 0.2 mmol scale in 1.0 mL solvent with 2.0 mol% dirhodium catalyst in 5 hours.
bReactions were carried out at room temperature on a 0.2 mmol scale in 0.5 mL DCCl3 with 2.0 mol% dirhodium catalyst in an NMR tube.
cThe reaction was carried out at 0 °C in 3 hours.
dThe reaction was carried out at –20 °C overnight.
eThe ratio was determined by integration of characteristic 1H NMR absorptions from the spectrum of the reaction mixture.
fIsolated yield after chromatography.
gEnantioselectivity was determined by chiral HPLC analysis, see ESI for details. TBME = tert-butyl methyl ether; DMB = 2,2-dimethylbutane.
The observed high enantioselectivity obtained from catalytic intramolecular C–H insertion of 1a was not observed with the corresponding diazoacetamide. β-Lactam 5a was obtained in only 60% ee for when diazoacetamide 4a was reacted under the same conditions (eqn (1)) and, in contrast to the exclusive cis-selectivity observed in the formation of β-lactam 3a, 5a was formed with exclusive trans-selectivity.19 The trans β-lactam product was also obtained in high yield in reactions catalyzed by achiral [RuCl2(p-cymene)]2 reported by Chi.14f The reason for this difference in diastereoselectivity is probably isomerization of the β-ketoamide and, indeed, cis-3c is converted to trans-5c upon hydrolysis (eqn (2)) without loss of enantioselectivity.
 |
1 |
 |
2 |
The scope of the C–H functionalization reaction of representative diazoacetamides 1 was investigated with Rh2(S-PTTL)4 catalysis under the optimum conditions established for 1a (Table 2). In all examples, cis-β-lactam 3 was generated exclusively and in high yield (80–92%) and with high enantioselectivities (83–99% ee). The % ee of β-lactam 3 was lower when strong electron-withdrawing substituents were on the aromatic ring, and these reactions also required longer times for completion than did enoldiazoacetamides with electron-donating substituents (entries 6 and 7). Lower enantioselectivities were obtained with substrates having m- or o-substituents, and the lowered % ee was independent of a second substituent at the para position (entries 10 and 11) or of the size of the ortho substituent (entries 12 and 13). Using the N,N-diisopropylamide instead of the benzyl-t-butylamide also resulted in a β-lactam with lower enantioselectivity but good yield. Rh2(S-NTTL)4 gave higher enantiocontrol by 7% ee in the reaction of unsubstituted enoldiazo acetamide 1b (entry 2), but the same or lower enantioselectivities were observed in reactions with 1a, 1f, 1g, and 1k. Product mixtures were obtained when Ar = the heteroaryl 3-furanyl group and included products from [3 + 4]-cycloaddition.7 It is noteworthy that β-lactam 3i with the p-dimethylamino substituent was obtained with 99% ee in 80% yield (entry 9). And TIPS protected substrate 1c′ gave similar results as 1c (entry 3 vs. entry 15). When an N-aryl substituent was used instead of the t-butyl group diazo compound 1o, the reaction gave both Buchner reaction and C–H functionalization products in a 1 : 2 ratio with 78% total yield, and β-lactam 3o was formed with 71% ee (eqn (3)).
 |
3 |
Table 2. Enantioselective C–H functionalization of 1 a .
| |||||
| Entry | Ar (1) | t (hours) | 3 | Yield c (%) | ee d (%) |
| 1 | 4-MeO6H4 (1a) | 3 | 3a | 85 | 92 |
| 2 | C6H5(1b) | 12 | 3b | 82 (81) b | 80 (87) b |
| 3 | 4-ClC6H4 (1c) | 5 | 3c | 88 | 93 |
| 4 | 4-MeC6H4 (1d) | 5 | 3d | 92 | 93 |
| 5 | 4-FC6H4 (1e) | 5 | 3e | 88 | 91 |
| 6 | 4-BrC6H4 (1f) | 12 | 3f | 89 | 89 |
| 7 | 4-NO2C6H4 (1g) | 12 | 3g | 88 | 83 |
| 8 | 4-PhC6H4 (1h) | 5 | 3h | 89 | 91 |
| 9 | 4-Me2NC6H4 (1i) | 5 | 3i | 80 | 99 |
| 10 | 3,4-(MeO)2C6H3 (1j) | 5 | 3j | 81 | 77 |
| 11 | 3-MeOC6H4 (1k) | 5 | 3k | 92 | 78 |
| 12 | 2-MeOC6H4 (1l) | 12 | 3l | 85 | 25 |
| 13 | 1-Naphthyl (1m) | 5 | 3m | 85 | 24 |
| 14 e | N,N-Diisopropyl (1n) | 5 | 3n | 81 | 67 |
| 15 f | 4-ClC6H4 (1c′) | 5 | 3c′ | 84 | 92 |
aReactions were carried out on a 0.2 mmol scale in 1.0 mL DMB with 2.0 mol% Rh2(S-PTTL)4.
bResults in parentheses was catalyzed by Rh2(S-NTTL)4.
cIsolated yield.
dEnantioselectivity was determined by chiral HPLC analysis, see ESI for details.
e N,N-Diisopropyl instead of N-tert-butyl-N-benzyl diazoamide was used.
fTIPS protection instead of TBS was used.
As is suggested by the reaction conditions, these reactions are relatively rapid. However, close spectroscopic inspection of reactions with 1c revealed that donor–acceptor cyclopropene 6c was formed at a much faster rate than was the product from C–H insertion. To determine if the donor–acceptor cyclopropene is a precursor to the donor–acceptor chiral metal carbene intermediate whose intramolecular C–H insertion produces highly enantiomerically enriched β-lactam, reaction of 1c was performed in DMB with 2.0 mol% Rh2(OAc)4 and at 5 min was filtered through Celite to remove the Rh2(OAc)4. Spectral analysis of the residue showed <1% 1c, 8 ± 1% 3c, and 92 ± 2% donor–acceptor cyclopropene 6c. This mixture was then submitted to the same reaction conditions as reported in Table 2 with Rh2(S-PTTL)4 catalysis to produce 3c in 73% isolated yield and 83 ± 3% ee. Subtracting racemic product formed from Rh2(OAc)4 gives 3c formed from donor–acceptor cyclopropene 6c with the same selectivity as that formed from 1c (Scheme 2).
Scheme 2. Donor–acceptor cyclopropenes are reaction intermediates in metal carbene formation.

That donor–acceptor cyclopropene 6 can serve as a precursor to an intermediate metal carbene of dirhodium(ii) which undergoes C–H insertion prompted us to investigate if other transition metals, particularly those of copper(i) and silver(i), could undergo the same transformation. Although both of these catalytically active metal ions are known to form metal carbenes directly from diazo compounds,7,8,20 they undergo Lewis acid catalyzed reactions with enoldiazoacetates,15 and they are distinctly different from dirhodium(ii) carboxylates in cycloaddition reactions with nitrones.18c Since C–H insertion is notably unique to metal carbenes, reactions of metal catalysts with enoldiazoacetamides or their derivative cyclopropenes would be a demonstration of metal carbene involvement with these catalysts. To undertake this investigation, reactions were performed on both enoldiazoacetamide 1c and cyclopropene 6c that was prepared from 1c by treatment with Rh2(OAc)4 in DMB as previously described, and these results are described in Table 3. The copper(i) and silver(i) catalysts are distinctly different from each other and from Rh2(OAc)4 in their reactions with enoldiazoacetamide 1c: aromatic cycloaddition is favored over C–H-insertion in reactions of 1c in the catalyst order: Ag(SbF6) > Cu(MeCN)4PF6 > Rh2(OAc)4 (entries 1 and 2); and this difference is also reflected in the results from reactions with cyclopropene 6c. Surprisingly, the Cu(MeCN)4PF6 and AgSbF6 catalyzed reactions with enoldiazoacetamide 1c provide more of the aromatic cycloaddition product, which is reported to be due to a more electrophilic metal carbene intermediate,13 than do their reactions with donor–acceptor cyclopropene 6c. The observed differences in the 2c : 3c ratios from reactions with 1c and 6c suggest that there may be some dependence on the carbene source among the catalysts employed for aromatic cycloaddition and C–H insertion, and that donor–acceptor cyclopropene 6c and enoldiazoacetate 1c may not form the same conformationally identical metal carbene intermediate. Use of a box ligand effectively inhibits dinitrogen extrusion from enoldiazoacetamide 1c, but metal carbene formation from cyclopropene 6c occurs without this limitation (entries 3 and 5). Other Lewis acids or under the thermal condition did not give any Buchner reaction or C–H insertion product, and only slowly decomposition of 6c was observed (entries 8–11).
Table 3. Comparison of catalysts in C–H insertion and aromatic cycloaddition reactions of 1c and 6c a .

| ||||||
| Entry | Catalyst | Reactant | t (hours) | 2c : 3c | Yield d (%) 2c + 3c | ee e (%) 3c |
| 1 b | AgSbF6 | 1c | 12 | >95 : 5 | 87 | — |
| 2 b | Cu(MeCN)4PF6 | 1c | 12 | 85 : 15 | 85 | — |
| 3 c | Cu(MeCN)4PF6/(S)-t-BuBox | 1c | 48 | <5 : 95 | 10 | 28 |
| 4 b | Cu(MeCN)4PF6 | 6c | 12 | 75 : 25 | 87 | — |
| 5 c | Cu(MeCN)4PF6/(S)-t-BuBox | 6c | 12 | <5 : 95 | 89 | 30 |
| 6 b | AgSbF6 | 6c | 12 | 85 : 15 | 82 | — |
| 7 c | AgSbF6/(S)-t-BuBox | 6c | 12 | 75 : 25 | 91 | 24 |
| 8 | Sc(OTf)3 | 6c | 12 | — | NR f | — |
| 9 | La(OTf)3 | 6c | 12 | — | NR f | — |
| 10 | BF3·Et2O | 6c | 12 | — | NR f | — |
| 11 g | (—) | 6c | 12 | — | NR f | — |
aReactions were carried out on a 0.2 mmol scale in 1.0 mL DCM.
bReactions were carried out with 10 mol% Lewis acid catalyst.
cReactions were carried out with 10 mol% Lewis acid catalyst and 12 mol% ligand.
dIsolated yield.
eThe enantioselectivity was determined by chiral HPLC analysis, see ESI for details.
fNeither 2c nor 3c was observed, and only slowly decomposition of 6c was observed.
gThe reaction was carried out in 70 °C.
The assignment of relative stereochemistry for β-lactam 3 was made from 1H NMR coupling constants,21 and the observed cis-configuration is consistent with a steric influence of the catalyst attachments and rhodium surface on the aryl substituent of the intermediate metal carbene. The chiral Rh2(S-PTTL)4 catalyst was reported to exist in a crown conformation by Fox,22 which means all of the ligands in an “all-up” orientation, and the bulky t-butyl and TBS substituents of the carbene are enclosed within the crown. This crowded transition state defines conformational preference for the aryl group to approach the carbene center for C–H insertion and the Buchner reaction. The (3S,4R)-configuration of the generated chiral centers in β-lactam 3 was confirmed by single-crystal X-ray diffraction analysis of 3i, and the configurations of other compounds were assigned by analogy.23
Conclusions
In conclusion, we have discovered a highly selective asymmetric intramolecular C–H functionalization reaction of enoldiazoacetamides catalyzed by Rh2(S-PTTL)4 that occur via an intermediate donor–acceptor cyclopropene. The Buchner reaction was totally excluded in reactions catalysed by steric bulky dirhodium carboxylate catalyst, and β-lactam derivatives are obtained from intramolecular C–H insertion as one diastereoisomer in high yield with up to 99% enantiomeric excess. Furthermore, reactions of enoldiazoacetamides and their corresponding donor–acceptor cyclopropenes performed with copper(i) and silver(i) catalysts validate their formation of metal carbene intermediates, but they show differences in reactivity and selectivity. The high enantiocontrol that is achieved relies on both electronic and steric influences of the unique silylenol group in these metal carbene transformation.
Supplementary Material
Acknowledgments
Support to M.P.D. from the National Institutes of Health (GM 46503) and from the National Science Foundation (CHE-1212446) is gratefully acknowledged. Michael Mandler provided spectral information. X.F.X. thanks the startup funding from Soochow University and Key Laboratory of Organic Synthesis of Jiangsu Province.
Footnotes
†Electronic supplementary information (ESI) available: Experimental details of substrates preparation, catalytic experiments, identification of the products and crystal data for 3i. CCDC 1018978. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03991b
References
- Fleming A. Rev. Infect. Dis. 1980;2:129. [PubMed] [Google Scholar]
- (a) Raviña E., The Evolution of Drug Discovery, Wiley-VCH, Weinheim, Germany, 2011, pp. 254–272. [Google Scholar]; (b) Magriotis P. A. Eur. J. Org. Chem. 2014:2647. [Google Scholar]; (c) Pitts C. R., Lectka T. Chem. Rev. 2014;114:7930. doi: 10.1021/cr4005549. [DOI] [PubMed] [Google Scholar]; (d) France S., Weatherwax A., Taggi A. E., Lectka T. Acc. Chem. Res. 2004;37:592. doi: 10.1021/ar030055g. [DOI] [PubMed] [Google Scholar]
- (a) Clader J. W. J. Med. Chem. 2004;47:1. doi: 10.1021/jm030283g. [DOI] [PubMed] [Google Scholar]; (b) Blazing M. A., Giugliano R. P., Cannon C. P., Musliner T. A., Tershakovec A. M., White J. A., Reist C., McCagg A., Braunwald E., Califf R. M. Am. Heart J. 2014;168:205. doi: 10.1016/j.ahj.2014.05.004. [DOI] [PubMed] [Google Scholar]; (c) Turley S. D., Dietschy J. M. Curr. Opin. Lipidol. 2003;14:233. doi: 10.1097/00041433-200306000-00002. [DOI] [PubMed] [Google Scholar]
- For reviews: ; (a) Galletti P., Giacomini D. Curr. Med. Chem. 2011;18:4265. doi: 10.2174/092986711797200480. [DOI] [PubMed] [Google Scholar]; (b) Pierrat O. A., Strisovsky K., Christova Y., Jonathan L. J., Ansell K., Bouloc N., Smiljanic E., Freeman M. ACS Chem. Biol. 2011;6:325. doi: 10.1021/cb100314y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mann P. A. ACS Chem. Biol. 2013;8:2442. doi: 10.1021/cb400487f. [DOI] [PubMed] [Google Scholar]; (d) Vandekerckhove S., D'hooghe M. Bioorg. Med. Chem. 2013;21:3643. doi: 10.1016/j.bmc.2013.04.033. [DOI] [PubMed] [Google Scholar]
- (a) Davis F. A., Yang B., Deng J. J. Org. Chem. 2003;68:5147. doi: 10.1021/jo030081s. [DOI] [PubMed] [Google Scholar]; (b) Ratcliffe R., Salzmann T., Christensen B., Tetrahedron Lett., 1980, 21 , 31 , , intermolecular amide N–H insertion reactions: . [Google Scholar]; (c) Lee E. C., Fu G. C. J. Am. Chem. Soc. 2007;129:12066. doi: 10.1021/ja074483j. [DOI] [PubMed] [Google Scholar]; (d) Xu B., Zhu S., Xie X., Shen J., Zhou Q. Angew. Chem., Int. Ed. 2011;50:11483. doi: 10.1002/anie.201105485. [DOI] [PubMed] [Google Scholar]; (e) Chuprakov S., Worrell B. T., Selander N., Sit R. K., Fokin V. V. J. Am. Chem. Soc. 2014;136:195. doi: 10.1021/ja408185c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recent advances in asymmetric catalytic syntheses of β-lactam: ; (a) Pedroni J., Boghi M., Saget T., Cramer N. Angew. Chem., Int. Ed. 2014;53:9064. doi: 10.1002/anie.201405508. [DOI] [PubMed] [Google Scholar]; (b) Smith S. R., Douglas J., Prevet H., Shapland P., Slawin A. M. Z., Smith A. D. J. Org. Chem. 2014;79:1626. doi: 10.1021/jo402590m. [DOI] [PubMed] [Google Scholar]; (c) Chen Z., Lin L., Wang M., Liu X., Feng X. Chem.–Eur. J. 2013;19:7561. doi: 10.1002/chem.201204373. [DOI] [PubMed] [Google Scholar]; (d) Huang L., Zhao W., Staples R. J., Wulff W. D. Chem. Sci. 2013;4:622. [Google Scholar]; (e) Chen S., Salo E. C., Wheeler K. A., Kerrigan N. J. Org. Lett. 2012;14:1784. doi: 10.1021/ol3003783. [DOI] [PubMed] [Google Scholar]; (f) Vaske Y. S. M., Mahoney M. E., Konopelski J. P., Rogow D. L., McDonald W. J. J. Am. Chem. Soc. 2010;132:11379. doi: 10.1021/ja1050023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Davies H. M. L., Morton D. Chem. Soc. Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; (b) Davies H. M. L., Manning J. R. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cui X., Xu X., Jin L., Wojtas L., Zhang X. P. Chem. Sci. 2015;6:1219. doi: 10.1039/c4sc02610a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tran D. N., Battilocchio C., Lou S., Hawkins J. M., Ley S. V. Chem. Sci. 2015;6:1120. doi: 10.1039/c4sc03072a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) McGrath N. A., Andersen K. A., Davis A. K. F., Lomax J. E., Raines R. T. Chem. Sci. 2015;6:752. doi: 10.1039/c4sc01768d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hansen J. H., Davies H. M. L. Chem. Sci. 2011;2:457. [Google Scholar]
- For review: ; (a) Doyle M. P., Liu Y., Ratnikov M. Org. React. 2013;80:1. [Google Scholar]; (b) Doyle M. P., Duffy R., Ratnikov M., Zhou L. Chem. Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (c) Doyle M. P., Ratnikov M., Liu Y. Org. Biomol. Chem. 2011;9:4007. doi: 10.1039/c0ob00698j. [DOI] [PubMed] [Google Scholar]
- For reviews: ; (a) Doyle M. P., Forbes D. C. Chem. Rev. 1998;98:911. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]; (b) Gois P. M. P., Afonso C. A. M. Eur. J. Org. Chem. 2004:3773. [Google Scholar]
- (a) Corey E. J., Felix A. M. J. Am. Chem. Soc. 1965;87:2518. doi: 10.1021/ja01089a055. [DOI] [PubMed] [Google Scholar]; (b) Burke S. D., Grieco P. A. Org. React. 1979;26:361. [Google Scholar]
- Doyle M. P., Protopopova M. N., Winchester W. R., Daniel K. L. Tetrahedron Lett. 1992;33:7819. [Google Scholar]
- (a) Doyle M. P., Kalinin A. V. Synlett. 1995:1075. [Google Scholar]; (b) Anada M., Watanabe N., Hashimoto S. Chem. Commun. 1998:1517. [Google Scholar]; (c) Candeias N. R., Carias C., Gomes L. F. R., André V., Duarte M. T., Gois P. M. P., Afonso C. A. M. Adv. Synth. Catal. 2012;354:2921. [Google Scholar]
- Padwa A., Austin D. J., Price A. T., Semones M. A., Doyle M. P., Protopopova M. N., Winchester W. R., Tran A. J. Am. Chem. Soc. 1993;115:8669. [Google Scholar]
- Advances in regioselectivity: ; (a) Lo V. K., Guo Z., Choi M. K., Yu W., Huang J., Che C. J. Am. Chem. Soc. 2012;134:7588. doi: 10.1021/ja3006989. [DOI] [PubMed] [Google Scholar]; (b) Snyder J. P., Padwa A., Stengel T., Arduengo A. J., Jockisch A., Kim H. J. Am. Chem. Soc. 2001;123:11318. doi: 10.1021/ja016928o. [DOI] [PubMed] [Google Scholar]; (c) Gois P. M. P., Afonso C. A. M. Eur. J. Org. Chem. 2003:3798. [Google Scholar]; (d) Yoon C. H., Nagle A., Chen C., Gandhi D., Jung K. W. Org. Lett. 2003;5:2259. doi: 10.1021/ol0345834. [DOI] [PubMed] [Google Scholar]; (e) Doyle M. P., Shanklin M. S., Oon S., Pho H. Q., van der Heidet F. R., Veal W. R. J. Org. Chem. 1988;53:3384. [Google Scholar]; (f) Choi M. K., Yu W., Che C. Org. Lett. 2005;7:1081. doi: 10.1021/ol050003m. [DOI] [PubMed] [Google Scholar]; (g) Moody C. J., Miah S., Slawin A. M. Z., Mansfield D. J., Richards I. C. J. Chem. Soc., Perkin Trans. 1. 1998:4067. [Google Scholar]; (h) Qu Z., Shi W., Jin X., Wang J. Chin. J. Org. Chem. 2003;23:988. [Google Scholar]; (i) Clarke L. A., Ring A., Ford A., Sinha A. S., Lawrencea S. E., Maguire A. R. Org. Biomol. Chem. 2014;12:7612. doi: 10.1039/c4ob01430h. [DOI] [PubMed] [Google Scholar]
- For reviews: ; (a) Xu X., Doyle M. P. Acc. Chem. Res. 2014;47:1396. doi: 10.1021/ar5000055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu X., Doyle M. P., Aust. J. Chem., 2014, 67 , 365 , , recent advances: . [Google Scholar]; (c) Xu X., Zavalij P. Y., Doyle M. P. Angew. Chem., Int. Ed. 2013;52:12664. doi: 10.1002/anie.201305539. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu X., Zavalij P. Y., Doyle M. P. Angew. Chem., Int. Ed. 2012;51:9829. doi: 10.1002/anie.201203962. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Qian Y., Xu X., Wang X., Zavalij P. Y., Hu W., Doyle M. P. Angew. Chem., Int. Ed. 2012;51:5900. doi: 10.1002/anie.201202525. [DOI] [PubMed] [Google Scholar]
- (a) Xu X., Zavalij P. Y., Doyle M. P. J. Am. Chem. Soc. 2013;135:12439. doi: 10.1021/ja406482q. [DOI] [PubMed] [Google Scholar]; (b) Xu X., Zavalij P. Y., Doyle M. P. Chem. Commun. 2013;49:10287. doi: 10.1039/c3cc46415f. [DOI] [PubMed] [Google Scholar]
- (a) Müller P., Allenbach Y. F., Grass S. Tetrahedron: Asymmetry. 2005;16:2007. [Google Scholar]; (b) Müller P., Bernardinelli G., Allenbach Y. F., Ferri M., Grass S. Synlett. 2005:1397. [Google Scholar]; (c) Müller P., Bernardinelli G., Allenbach Y. F., Ferry M., Flack H. D. Org. Lett. 2004;6:1725. doi: 10.1021/ol049554n. [DOI] [PubMed] [Google Scholar]
- Nonpolar solvents provide a significant increase in the enantioselectivity in some metal carbene reactions, for example, 2,2-DMB and TBME have previously been demonstrated to be an optimal solvent for enantioselective C–H functionalization and other metal carbene transformations: ; (a) Wang H., Li G., Engle K. M., Yu J., Davies H. M. L. J. Am. Chem. Soc. 2013;135:6774. doi: 10.1021/ja401731d. [DOI] [PubMed] [Google Scholar]; (b) Smith A. G., Davies H. M. L. J. Am. Chem. Soc. 2012;134:18241. doi: 10.1021/ja3092399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang X., Xu X., Zavalij P. Y., Doyle M. P. J. Am. Chem. Soc. 2011;133:16402. doi: 10.1021/ja207664r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The cis isomer was not detected, even at short reaction times.
- Díaz-Requejo M., Pérez P. J. Chem. Rev. 2008;108:3379. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]
- The relative chemistry could be distinguished by 1H NMR coupling constants, see ref. 14e: generally cis = 5–8 Hz; trans = 0–2 Hz.
- (a) DeAngelis A., Dmitrenko O., Yap G. P. A., Fox J. M. J. Am. Chem. Soc. 2009;131:7230. doi: 10.1021/ja9026852. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hashimoto S., Watanabe N., Sato T., Shiro M., Ikegami S. Tetrahedron Lett. 1993;34:5109. [Google Scholar]; (c) Boruta D. T., Dmitrenko O., Yap G. P. A., Fox J. M. Chem. Sci. 2012;3:1589. doi: 10.1039/C2SC01134D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.