

Abstract
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer and the leading cause of cancer-related death in children and adolescents. Minimal residual disease (MRD) is a strong, independent prognostic factor. The objective of this study was to identify molecular signatures distinguishing patients with positive MRD from those with negative MRD in different subtypes of ALL, and to identify molecular networks and biological pathways deregulated in response to positive MRD at day 46. We compared gene expression levels between patients with positive MRD and negative MRD in each subtype to identify differentially expressed genes. Hierarchical clustering was applied to determine their functional relationships. We identified subtype-specific gene signatures distinguishing patients with positive MRD from those with negative MRD. We identified the genes involved in cell cycle, apoptosis, transport, and DNA repair. We also identified molecular networks and biological pathways dysregulated in response to positive MRD, including Granzyme B, B-cell receptor, and PI3K signaling pathways.
Keywords: minimal residual disease, B-cell acute lymphoblastic leukemia, gene expression
Introduction
B-precursor acute lymphoblastic leukemia (ALL) is a heterogeneous disease with several subtypes that differ markedly in cellular and molecular characteristics.1,2 Approximately 10%–20% of patients do not respond well to the current treatment protocols.3–5 Attempts to improve survival rate with hematopoietic stem-cell transplantation have improved outcome for some6 but not all subtypes, suggesting that intensification of existing treatment strategies is unlikely to improve the cure rate. Therefore, there is an urgent need to identify biomarkers to assess treatment response and guide treatment protocols.
End of induction minimal residual disease (MRD) is a powerful predictor of overall treatment response and is now routinely used to determine treatment intensity in contemporary treatment centers.6–9 MRD after induction therapy is a very strong, independent prognostic factor for ALL even after adjusting for other clinical or biologic features.6–9 A study from the Children’s Oncology Group revealed that having positive MRD at 0.01% is strongly associated with a lower 5-year event-free survival.8 The effect is more pronounced in the higher risk patients.8 Furthermore, even low-level MRD (0.001%–0.01%) at the end of induction has been associated with increased risk of relapse.9
Because ALL is a heterogeneous disease entity involving many subtypes, understanding the molecular mechanisms underpinning MRD for each subtype could allow better risk estimation in children treated for ALL and have profound impacts on future clinical management strategies. Therefore, the challenge is to comprehensively identify all genetic alterations driving leukemogenesis and MRD to guide risk stratification and targeted therapies.
Advances in microarray technology have identified molecular signatures for classification of subtypes of ALL and improved outcome prediction.1,2,10–20 Several studies have attempted to predict MRD in ALL,21–25 but the results are inconsistent and have not been reproducible. To date, no study has shown whether positive MRD and negative MRD patients can be stratified in different subtypes of childhood ALL to guide treatment. With the cure rate of childhood ALL approaching 90% and varying clinical outcomes in different subtypes, further improvement in the treatment outcome and quality of life will require discovery of subtype-specific prognostic markers for patient stratification to guide treatment. The objectives of this exploratory study were twofold: 1) to determine whether gene expression levels significantly differ between positive MRD and negative MRD patients in different subtypes of ALL and to identify a signature of functionally related genes distinguishing the two patient groups, and 2) to identify molecular networks and biological pathways that are dysregulated in response to positive MRD. Our working hypotheses were the following: 1) molecular perturbation in patients with positive MRD differs from those with negative MRD; and 2) genes dysregulated in response to positive MRD are functionally related and affect entire molecular networks and biological pathways, which in turn affect the severity of the disease. We have tested these hypotheses by analyzing gene expression data on patients diagnosed with positive and negative MRD in five subtypes of childhood ALL.
Materials and Methods
Gene expression data
We used publicly available gene expression data generated using RNA derived from leukemic blast samples obtained from pediatric patients with ALL. Gene expression data was downloaded from NCBI’s Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/gds/) under accession number GSE33315. Gene expression profiling was performed using Affymetrix U133A arrays. The details about the samples have been described by the data originators.26 Briefly, the dataset consisted of gene expression data from 484 B-ALL samples. Out of 484 ALL samples, 189 contained information about the MRD status at days 19 and 46. The remainder of the samples did not contain information on MRD and thus were not used. The 189 samples were classified as hyperdiploid, TCF-PBX1, ETV6-RUNX1, 11q23 (MLL) rearrangement, BCR-ABL1, hypodiploid, and B-ALL without detectable recurrent molecular abnormalities (others). The distribution of the samples according to MRD status by subtype is provided in Table 1. Unfortunately, the information on other clinical variables (gender, age, white blood cell counts at diagnosis, ethnicity, treatment protocol, response to treatment, and the level of MRD) and the details of the karyotypes in hyperdiploid and hypodiploid cases were not available. The samples from patients with BCR-ABL1 were excluded from the study because of a known prognosis and a different treatment approach. Patients with TCF3-PBX1 were excluded due to a very small sample size. This left 165 samples used in the analysis: 35 with positive day 46 MRD and 130 with negative MRD (Table 1). Gene expression data were normalized to log 2.
Table 1.
Distribution of samples according to ALL subtypes. Due to small sample sizes, BCR-ABL1 and TCF-PBX1 samples were not used in the analysis.
| B-ALL SUBTYPES | POSITIVE MRD | NEGATIVE MRD | TOTAL |
|---|---|---|---|
| Hyperdiploid | 7 | 37 | 44 |
| t(12;21)(ETV6-RUNX1) | 7 | 44 | 51 |
| MLL (11q23) rearrangement | 6 | 5 | 11 |
| Hypodipoloid | 4 | 11 | 15 |
| Others | 11 | 33 | 44 |
| t(9;22)(BCR-ABL1) | 11 | 2 | 13 |
| t(1;19) (TCF-PBX1) | 1 | 10 | 11 |
| Total | 47 | 142 | 189 |
Data analysis
Because B-ALL is a heterogeneous disease entity and both gene expression and treatment outcomes have been shown to be subtype-specific,19,20 we elected to use a subclass mapping analysis strategy treating each subtype as a different disease entity. Under this approach, total gene expression data was partitioned according to individual subtypes of B-ALL. Within each B-ALL subtype, the data was further partitioned into two patient groups, the patient group with positive day 46 MRD and the group with negative MRD. Analysis was then performed within each subtype. We did not use ANOVA to adjust for different subtypes since the sample sizes were too small in some subtypes to avoid sampling errors.
To obtain a more robust analysis of gene expression data, we used a combination of different analysis strategies. First, we used supervised analysis comparing gene expression levels between positive MRD and negative MRD patients for each subtype of B-ALL to determine whether gene expression levels significantly differed between the two groups and to identify a signature of genes distinguishing the two patient groups. A t-test was used to identify significantly differentially expressed genes. We used the false discovery rate (FDR) to correct for multiple hypothesis testing.27 A P-value of less than 0.005 was used as the threshold for selecting significantly differentially expressed genes. Due to the small sample sizes, we did not divide the data into test and validation sets to identify genes that are predictive of MRD. Instead, we used an out- of-sample (leave-one-out) validation procedure, which has been shown to be effective when samples sizes are small.28
The differences in gene expression levels between patient groups are important in stratifying patients, but they alone are insufficient to explain the dynamics of the molecular mechanisms underpinning MRD. Our next step was to determine whether genes deregulated in response to positive day 46 MRD are functionally related and are involved in the same biological processes and cellular components. To achieve this goal, we used the gene ontology (GO) information to identify functionally related genes among the genes differentially expressed between the two patient groups.29 We considered all three GO categories: molecular function, biological process, and cellular component. Molecular function defines what a gene product does at the biochemical level without specifying where or when the event actually occurs. Biological process describes the contribution of the gene product to the biological objective. Cellular component refers to where in the cell a gene product functions.
Next, for each subtype, we performed unsupervised analysis using gene expression data on differentially expressed genes with P-values less than 0.005. The goal of this analysis was to determine whether the identified genes are co-expressed and have similar patterns of expression profiles. It has been shown that co-expression is correlated with functional relationships, though this does not necessarily imply a causal relationship among transcript levels.30 Importantly, co-expression of genes of known function with novel genes may provide leads to the functions of novel genes.30 We computed the Pearson correlation coefficient for each pair of genes as a distance measure to assess the similarity and dissimilarity in patterns of gene expression profiles between pairs of genes. The genes were then ordered into clusters using hierarchical clustering via the complete linkage method. Again, because of the heterogeneity of the disease, the analyses were performed for each subtype of B-ALL. Prior to clustering, all the data were normalized, standardized, and centered.30 Supervised and unsupervised analyses were performed using the GenePattern software package.31
Finally, we performed network and pathway analysis and visualization using the Ingenuity Pathway Analysis (IPA) System (Ingenuity Systems, www.ingenuity.com). The goal was to determine whether the genes dysregulated in response to positive MRD interact in gene regulatory networks and biological pathways. A set of differently expressed genes from each subtype with P-value less than 0.002 was used for this analysis. We chose this lower threshold to reduce the number of genes and to increase the reliability of identified gene networks and biological pathways by focusing on the set of genes with good evidence of discriminating between positive MRD and negative MRD patients. The Human Genome Organization (HUGO) gene identifiers/symbols were mapped onto the networks and biological pathways using the network and pathway design modules as implemented in IPA, and the probability score was computed and used as an indicator of the likelihood for correctly assigning the genes to the networks. Using a 99% confidence interval, Z-scores of ≥3 are considered significant, although in this study we used much more stringent levels. Validation of predicted pathways and identification of other downstream target genes were achieved through the literature and database mining module implemented in the Ingenuity System. This module allows identification of other functionally related genes.
Results
Differences in gene expression between patient groups
We performed subclass mapping, as explained in the Methods section, by comparing gene expression levels between positive day 46 MRD and negative MRD patients within each subtype of B-ALL. We identified five subtype-specific signatures totaling 691 highly significantly (P < 0.005) differentially expressed genes, which distinguished positive MRD from negative MRD patients at FDR <1%. This confirmed our hypothesis that molecular perturbations significantly differ between the two patient groups. The number of significantly differentially expressed genes identified in hyperdiploid, ETV6-RUNX1, MLL rearrangement, hypodiploid, and other subtypes was 93, 82, 87, 140, and 289, respectively. Among the significantly differentially expressed genes identified were BCL2, BECN1, CBFB, IKZF1, PAX5, SH2B3, and TOX, which have been implicated in ALL.2,10,17 A complete list of all the 691 genes along with their estimates of P-values classified by subtype is presented in Supplementary Table 1. Interestingly, none of the 691 highly significantly differentially expressed genes from the analysis of each subtype was overlapping between all subtypes. This observation, along with the small sample size for some subtypes, spurred our confidence not to perform comparisons between subtypes.
One of the critical needs in a clinical setting is the identification of prognostic markers for risk stratification of positive and negative MRD patients to guide treatment. To identify gene signatures predictive of MRD in each subtype, we evaluated the P-values of sets of genes with discriminative power (P < 10−4) identified after performing an out- of-sample (leave-one-out) validation procedure, as explained in the Methods section. For each subtype, we selected the top 10 most highly significantly differentially expressed genes as assessed by the P-value (P < 10−4) after correcting for multiple hypothesis testing (FDR <1%). The results showing sets of the most highly significantly differentially expressed genes that are predictive of positive MRD for each subtype are shown in Table 2. Analysis involving the hyperdiploid subtype revealed the genes CMMD10, DHX29, CAMK2G, NRF1, LOC100132832, PMS2P1, WISP1, CANX, ALPL, and EMP1 (P < 8.0 × 10−4). For the MLL rearrangement subtype, we identified a signature consisting of the genes BHLHE40, BIRC5, C2ORF27, C7ORF25, CC2D1A, CD8A, CDK16, CES2, and CHAT (P < 2.0 × 10−4). The same analysis for the ETV6-RUNX1 subtype produced the genes FAM204A, ICOS, RYBP, CLIP3, ZHX2, BMP8A, MPL, MYH11, TCL6, and SLC7A6 (P < 8.0 × 10−4). Analysis involving the hypodiploid subtype identified a gene signature consisting of the genes ANKRD40, ATF7IP, ATG4B, C15ORF63, CEPT1, DNAJC13, DOCK2, FAM48A, FTO, and GUCY1A3 (P < 2.0 × 10−4). Analysis involving the “other” subtype produced a signature consisting of the genes CTDSPL, FGF17, HIST1H2AB, IL8, ITGB3, KDM3A, MYL6, NPDC1, ST8SIA3, and TSPYL2 (P < 2.0 × 10−4).
Table 2.
List of highly significantly differentially expressed genes with predictive power as assessed by the estimated P-value in each subtype of childhood ALL.
| GENE NAME | Chr. POSITION | P-VALUE |
|---|---|---|
| Hyperdiploid subtype | ||
| COMMD10 | 5q23.1 | 2.0 × 10−4 |
| DHX29 | 5q11.2 | 2.0 × 10−4 |
| CAMK2G | 10q22 | 2.0 × 10−4 |
| NRF1 | 7q32 | 2.0 × 10−4 |
| LOC100132832 | 7q11.23 | 4.0 × 10−4 |
| PMS2P1 | 7q22.1 | 4.0 × 10−4 |
| WISP1 | 8q24.22 | 4.0 × 10−4 |
| CANX | 5q35 | 6.0 × 10−4 |
| ALPL | 1p36.12 | 6.0 × 10−4 |
| EMP1 | 12p12.3 | 8.0 × 10−4 |
| MLL rearrangement subtype | ||
| BHLHE40 | 3p26 | 2.0 × 10−4 |
| BIRC5 | 17q25.3 | 2.0 × 10−4 |
| C20orf27 | 20p13 | 2.0 × 10−4 |
| C7orf25 | 7p14.1 | 2.0 × 10−4 |
| CC2D1A | 19p13.12 | 2.0 × 10−4 |
| CD8A | 2p12 | 2.0 × 10−4 |
| CDK10 | 16q24.3 | 2.0 × 10−4 |
| CDK16 | Xp11 | 2.0 × 10−4 |
| CES2 | 16q22.1 | 2.0 × 10−4 |
| CHAT | 10q11.2 | 2.0 × 10−4 |
| Other subtypes | ||
| CTDSPL | 3p21.3 | 2.0 × 10−4 |
| FGF17 | 8p21.3 | 2.0 × 10−4 |
| HIST1H2AB | 6p22.1 | 2.0 × 10−4 |
| IL8 | 4q13–q21 | 2.0 × 10−4 |
| ITGB3 | 17q21.32 | 2.0 × 10−4 |
| KDM3A | 2p11.2 | 2.0 × 10−4 |
| MYL6 | 12q13.13 | 2.0 × 10−4 |
| NPDC1 | 9q34.3 | 2.0 × 10−4 |
| ST8SIA3 | 18q21.31 | 2.0 × 10−4 |
| TSPYL2 | Xp11 | 2.0 × 10−4 |
| ETV6-RUNX1 subtype | ||
| FAM204A | 10q26.12 | 2.0 × 10−4 |
| ICOS | 2q33 | 2.0 × 10−4 |
| RYBP | 3p14.2 | 4.0 × 10−4 |
| CLIP3 | 19q13.12 | 6.0 × 10−4 |
| ZHX2 | 8q24.13 | 6.0 × 10−4 |
| BMP8A | 1p35–p32 | 6.0 × 10−4 |
| MPL | 1p34 | 6.0 × 10−4 |
| MYH11 | 16p13.11 | 6.0 × 10−4 |
| TCL6 | 14q32.1 | 6.0 × 10−4 |
| SLC7A6 | 16q22.1 | 8.0 × 10−4 |
| Hypoploid subtype | ||
| ANKRD40 | 17q21.33 | 2.0 × 10−4 |
| ATF7IP | 12p13.1 | 2.0 × 10−4 |
| ATG4B | 2q37.3 | 2.0 × 10−4 |
| C15orf63 | 15q14 | 2.0 × 10−4 |
| CEPT1 | 1p13 | 2.0 × 10−4 |
| DNAJC13 | 3q22.1 | 2.0 × 10−4 |
| DOCK2 | 5q35.1 | 2.0 × 10−4 |
| FAM48A | 13q13 | 2.0 × 10−4 |
| FTO | 16q12.2 | 2.0 × 10−4 |
| GUCY1A3 | 4q31.3–q33 | 2.0 × 10−4 |
As expected, we observed significant variability in expression levels between B-ALL subtypes. The variability and differences in the number of genes exhibiting significant differences can be partially explained by the genetic heterogeneity and the small sample sizes. The significant differences in gene expression levels between positive MRD and negative MRD patients suggest that gene expression signatures could potentially be used to stratify the two patient groups in each subtype of B-ALL. The lack of overlapping in significantly differ entially expressed genes across the subtypes indicates that, for patients with positive day 46 MRD, no single molecular signature could predict response to treatment across subtypes of B-ALL and that molecular stratification of patients in response to treatment should be subtype-specific. Overall, the results suggest that gene expression profiling could become a clinically relevant tool for treatment stratification for B-ALL patients.
Functional analysis and patterns of gene expression profiles for the two patient groups
Another import aspect of this investigation was the quantification of the functional relationship of the identified gene signatures in each subtype of ALL. Genes with similar patterns of expression and those with similar functions are likely to be regulated via the same mechanisms.32 In fact, genes with strongly correlated mRNA expression profiles and those with similar functional annotation are likely to be bound by common transcription factors.32 Using this knowledge, along with the understanding that gene expression profiles exhibited subtype-specific expression, we performed functional analysis using clustering and GO information for each subtype.
GO analysis revealed functionally related up- and down-regulated genes with multiple overlapping functions. Functionally related genes dysregulated in response to positive MRD were predominantly associated with DNA recombination and repair, chromosome organization, apoptosis, transcription factors, and cell cycle (Fig. 1A and B, indicating up- and downregulated, respectively). The pattern of the results suggests impaired apoptosis, impaired cell proliferation, and impaired DNA damage repair in ALL samples with suboptimal response to induction chemotherapy.
Figure 1.
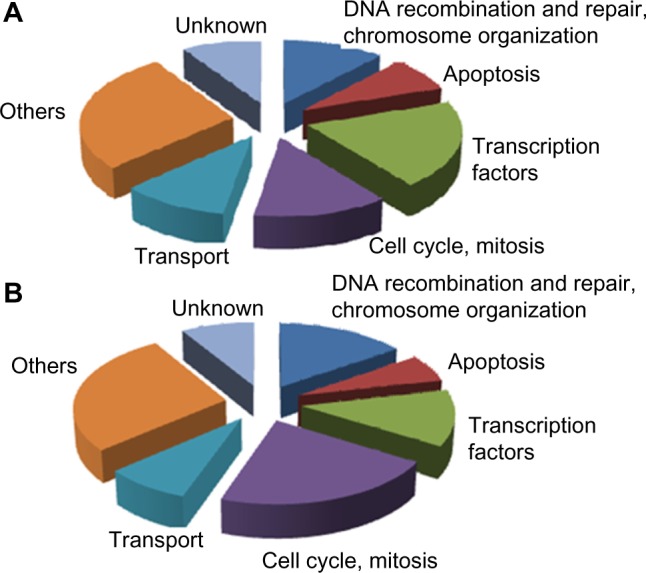
Pie charts showing distribution of the biological processes in which the identified significantly differentially expressed genes are involved as assessed by GO analysis. (A) A set of genes that are upregulated in patients with positive MRD at day 46. (B) Genes downregulated in patients with positive MRD at day 46.
The results showing patterns of expression profiles for both positive and negative MRD patients for the 93 gene signatures in the hyperdiploid and the 82 gene signatures in the ETV6-RUNX1 subtypes are presented in Figure 2A and B, respectively. The patterns of expression profiles for the 87 gene signatures representing the MLL rearrangement, the 140 gene signatures representing the hypodiploid, and the 289 gene signatures representing the others subtypes are presented in Figure 3A–C, respectively. Additional information on gene names/symbols represented in the heat maps in Figures 1 and 2 is provided in Supplementary Table 1. Remarkably, in all the five subtypes, including those without recurrent mole cular abnormalities (herein defined as “others”), we identified functionally related up- and downregulated genes with similar patterns of expression profiles. However, there was significant heterogeneity in patterns of gene expression profiles within subtypes. The analysis in the two patient groups across all the subtypes revealed significant heterogeneity and inconsistencies in the patterns of expression profiles (results not presented). Many factors could explain these heterogeneity and inconsistencies. B-ALL is a heterogeneous disease entity and gene expression can be subtype-specific; therefore, such an outcome should be expected. Other factors such as age and ethnicity, both of which were not accounted for in the analysis, could contribute to the observed patterns in gene expression profiles, although sampling errors cannot be ruled out given the small sample sizes. Our recent study on childhood ALL involving race and ethnicity revealed that gene expression varies among and differs across patient populations.33
Figure 2.

Pattern of gene expression profiles for the significantly differentially expressed genes distinguishing ALL patients with positive MRD at day 46 from patients with negative MRD. (A) Hyperdiploid: Represents a signature of 93 significantly differentially expressed genes, of which 41 genes are upregulated in positive MRD and 46 genes downregulated. (B) ETV6-RUNX1: Represents a signature of 82 significantly differentially expressed genes, of which 22 genes are upregulated in positive MRD and 60 genes downregulated. The red color indicates upregulation and blue color downregulation.
Figure 3.

Pattern of gene expression profiles for the significantly differentially expressed genes distinguishing ALL patients with positive MRD at day 46 MRD from patients with negative MRD. (A) MLL rearrangement: Represents a signature of 87 significantly differentially expressed genes, all downregulated in positive MRD. (B) Hypodiploid: Represents a signature of 140 significantly differentially expressed genes, of which 36 genes are upregulated in positive MRD and 104 genes downregulated. (C) Others: Represents a signature of 287 significantly differentially expressed genes, of which 130 genes are upregulated in positive MRD and 159 genes downregulated. The red color indicates upregulation and blue color downregulation.
Network and pathway analysis
The second objective of this investigation was to identify molecular networks and biological pathways that are dysregulated in response to positive MRD. Our working hypothesis was that genes dysregulated in response to positive MRD affect entire molecular networks and biological pathways, which in turn affect the severity of the disease. To address this hypothesis, we mapped 270 highly significantly (P-value <0.002) differentially expressed genes onto the networks and canonical pathways using the network and pathway prediction modules built into IPA. This analysis produced many molecular networks. We selected the top five networks with the highest scores (ranging from 35 to 72) and merged them into one large network using the build, design, and merge modules as implemented in IPA. In the networks, the genes are represented by the nodes and functional relationships by vertices. The genes are color-coded, with each color representing a subtype. The results of network analysis are presented in Figure 4. Network analysis revealed genes with multiple overlapping functions, including genes involved in cell morphology, cellular function and maintenance, developmental disorder, hematological disease, cell cycle and embryonic development, cancer, dermatological diseases, cellular movement, cell-to-cell signaling and interaction, hematological system development/function, and skeletal/muscular system development/function. In addition, we also identified the upstream regulator genes RB1, CDKN2A, E2F4, TP53, and HIF1A. These genes are tumor suppressor genes and are known to be associated with poor prognosis of B-ALL.34,35 Specifically, the genes TP53, RB1, and CDKN2A have been implicated in hypodiploid ALL, which has a poor outcome.36 The complete list of the 270 genes used in the network analysis, their predicted scores for correctly assigning them to the appropriate molecular networks, and information on the molecular functions in which the genes are involved are presented in Supplementary Table 2 provided as supplementary data to this report.
Figure 4.

Molecular networks for the top 270 highly significantly (P < 0.002) differentially expressed genes of different subtypes of ALL. Genes are represented by the nodes, and functional relationships by vertices/edges. Genes from each subtypes are color-coded as follows: red – hyperdiploid (15 genes), pink – ETV6-RUNX1 (4 genes), green – MLL rearrangement (22 genes), blue – hypodiploid (14 genes) and purple – others (58 genes), as depicted in the key below the figure.
To gain more insights into the broader biological context in which the identified genes operate, we performed pathway analysis and visualization using IPA. A log P < 1.2 was used as an indication of significance for the identified pathway. We identified many novel biological pathways. The most highly significant pathways that were found to be deregulated in response to positive MRD are presented in Figure 5. Among the highly significant pathways identified were the renal cell carcinoma signaling, granzyme B signaling, role of osteoblast, osteoclasts, and chrondrocytes in arthritis, the TCA cycle II, thrombopoietin signaling, rank signaling in osteoclasts, virus entry endocytic pathways, PI3K signaling in B lymphocytes, and the eNOS signaling (Fig. 5). These results confirmed our hypothesis that genes deregulated in response to positive MRD are functionally related and interact with one another in biological pathways. Interestingly, many of the identified pathways including granzyme B signaling,37,38 B-cell receptor signaling,39,40 thrombopoietin signaling,41 and PI3K signaling in B lymphocytes42,43 have been associated with hematologic malignancies.
Figure 5.
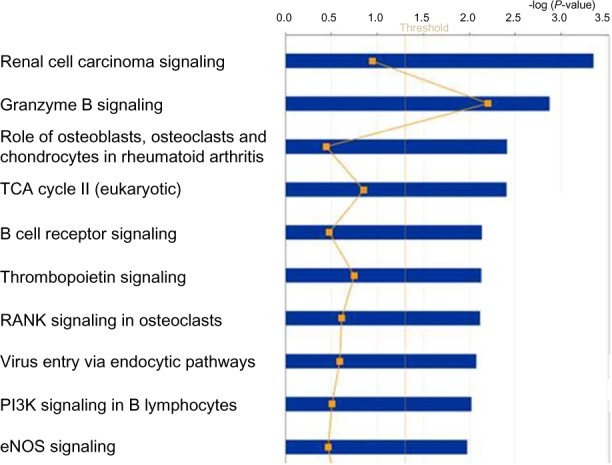
The top 10 highly significant biological pathways that were found to be deregulated in patients with positive day 46 MRD. The blue bars indicate the pathway. The straight yellow line indicates the threshold for assessing significant association as measured by the log P value.
Discussion
The prognostic value of end of induction MRD in childhood ALL has been firmly established by several groups worldwide.7–9 However, to date, no literature reports have shown whether molecular perturbation between patients with positive and negative MRD at post induction (day 46) differs in different subtypes of ALL and whether stratification of patients by MRD using transcription profiling could lead to measurable changes in therapeutic decision making. In this study, we addressed this knowledge gap by comparing gene expression levels between pediatric patients with positive day 46 MRD and negative MRD in different subtypes of B-ALL. Our analysis revealed molecular signatures unique to each subtype of childhood B-ALL. The clinical significance of these findings lies in the fact that, because MRD is a strong, independent prognostic factor,7–9 the identified molecular signatures predictive of positive MRD in different subtypes of B-ALL could serve as prognostic markers for stratifying patients and guiding treatment protocols. This is the first study to identify molecular signatures predictive of MRD that are unique to each subtype of childhood B-ALL.
Intriguingly, the results found in this study are consistent with the recent findings based on a randomized clinical trial (UKALL2003).44 Of note is the fact that, because persistence of MRD in the bone marrow is a key early prognostic indicator and is strongly associated with event-free survival,45 discovery of clinically actionable biomarkers that stratify patients could guide post-remission therapy.44 Indeed, early response to treatment that can be monitored by MRD is the most predictive marker for the risk of ALL relapse. Although we did not investigate how many of the patients with positive day 46 MRD eventually relapse in this study because of the lack of such information in the data used, a recent study demonstrated that MRD-based treatment is adequate for relapse-prone childhood ALL with an intra chromosomal amplification of chromosome 21.46 Because early response to treatment assessed by MRD testing provides a precise and objective measurement of drug sensitivity of leukemia cells and the efficacy of treatment as well as host pharmacogenomics and immune surveillance,7,47 the identified biomarkers could serve as targets for the development of novel therapeutics. The PI3K signaling in the B-lymphocyte pathway discovered in this study is associated with the natural killer (NK) cells involved in cancer control.47 The NK cell genotype and phenotype at diagnosis of ALL has been recently shown to correlate with post-induction residue disease,47 which was the main focus of this study. This suggests that the PI3K signaling in the B-lymphocyte pathway could be a potential therapeutic target.
In this study, we have used whole genome transcription profiling. In a clinical environment, the use of such profiling may be a daunting task. However, as demonstrated in this study, one could identify the signature of prognostic markers. In a clinical setting, MRD could then be monitored by assessing the expression of the identified signature of biomarkers using real-time quantitative polymerase chain reaction (RQ-PCR) to identify patients at high risk and to guide treatment.48 A recent study demonstrated convincingly that post-induction MRD investigated in this study could be monitored successfully using RQ-PCR in a clinical setting.48
In a clinical setting, most ALL induction treatments last 4–6 weeks,49–51 and MRD is measured at day 19 and day 46. In this study, we used day 46 as the checkpoint for assessing MRD because this was the only information available in the dataset used. But most importantly, MRD at day 46 is a good representation of end of induction MRD, which guides the intensification of treatment in current practice.44,48 The clinical significance lies in the fact that the molecular signatures deregulated in response to positive MRD at day 46 could be predictive of relapse,48 thus accelerating the planning of salvage therapy and/or hematopoietic stem-cell transplantation. In the scientific literature, many studies have demonstrated the prognostic significance of MRD levels in pediatric B-ALL.21–25 The novel feature of our study is that it identifies subtype- specific molecular signatures distinguishing positive MRD from negative MRD patients, not previously reported. With the heterogeneity inherent is childhood ALL, further improvement in the treatment outcome may depend on the discovery of subtype-specific prognostic markers and targets for the development of novel therapeutics.
Functional analysis produced genes involved in cell cycle and apoptosis. This is a significant finding, given that in all subtypes of B-ALL the treatment agents used for induction therapy affect cell proliferation and apoptosis. This difference in patterns of expression profiles between positive MRD and negative MRD patients could be indicative of resistance to treatment. The identification of genes involved in DNA damage and repair was expected since chemotherapy agents induce some form of DNA damage, the mechanism that likely activates the repair mechanisms. These results are consistent with earlier reports.21–25 The clinical significance of these findings is that discovery of DNA repair and apoptosis pathways and understanding the mechanisms of action could lead to development of novel and less toxic therapeutics.
Although the study shows appreciable differences in gene expression levels and patterns of gene expression profiles between MRD positive and MRD negative patients, limitations of the study must be acknowledged, the chief among them being the small sample size, which has the propensity to cause sampling errors. For this reason, and because it is very clear from previous studies that gene expression is subtype-specific,24,25 we restricted our analysis to within-subtype comparisons, treating each subtype as separate disease entity. It is worth noting that, due to the small sample size, we did not include all the subtypes. Notable also is the fact that our study did not consider other factors such as age, white blood cell count, obesity, and ethnicity, all of which could influence end of induction MRD and clinical outcomes, a weakness that we readily acknowledge. Information on these variables was not available in the dataset used in this study. In light of the acknowledged prognostic significance of age and white blood cell count,52 and more recently obesity,45 these variables should be considered in future studies. In addition, treatment results of ALL may depend not only on biological factors but also on ethnicity.53 These factors also should be considered in future studies.
In conclusion, this study revealed significant differences in gene expression levels between positive MRD and negative MRD patients at day 46 and identified subtype-specific molecular signatures. The study demonstrated that transcription profiling could be used to stratify patients on the basis of MRD. Because of the sample size per subtype used in this study, we view this study as exploratory and recommend that future studies should include larger sample sizes where it is possible to reliably identify subtype-specific predictive prognostic markers and assess their potential biological impact on clinical outcomes in all subtypes of pediatric B-ALL.
Supplementary Data
Supplementary Table 1. List of the 691 significantly (P < 0.005) differentially expressed genes and their estimates of P-values between positive day 46 MRD and negative MRD patients in each subtypes of B-ALL and their direction of expression values.
Supplementary Table 2. Complete list of the 270 genes used in network analysis, their predicted scores for correctly assigning them to the appropriate molecular networks and information on the molecular functions in which the genes are involved.
Footnotes
ACADEMIC EDITOR: William C. S. Cho, Editor in Chief
FUNDING: The authors wish to thank the Chew Children’s Research Fund, the Hyundai Hope on Wheels, and the University of Mississippi Medical Center’s Cancer Institute for providing funding support to conduct this research. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
JS, a pediatric hematology oncology fellow, led the project; BH, GM, JP, MD, CG, SH, and CH (residence supervisor) defined the project to address the unmet clinical needs. JS, TK, and CH conducted the data analysis and visualization. All authors contributed to the interpretation and writing of the manuscript, with JS taking the lead. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29:551–65. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122:3407–15. doi: 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabin KR. Attacking remaining challenges in acute leukemia. N Engl J Med. 2012;366:1445–6. doi: 10.1056/NEJMe1200989. [DOI] [PubMed] [Google Scholar]
- 4.Gaynon PS, Angiolillo AL, Carroll WL, et al. Children’s Oncology Group Long-term results of the children’s cancer group studies for childhood acute lymphoblastic leukemia 1983–2002: a Children’s Oncology Group Report. Leukemia. 2010;24:285–97. doi: 10.1038/leu.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salzer WL, Devidas M, Carroll WL, et al. Long-term results of the pediatric oncology group studies for childhood acute lymphoblastic leukemia 1984–2001: a report from the children’s oncology group. Leukemia. 2010;24:355–70. doi: 10.1038/leu.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hochberg J, Khaled S, Forman SJ, Cairo MS. Criteria for and outcomes of allogeneic haematopoietic stem cell transplant in children, adolescents and young adults with acute lymphoblastic leukaemia in first complete remission. Br J Haematol. 2013;161:27–42. doi: 10.1111/bjh.12239. [DOI] [PubMed] [Google Scholar]
- 7.Campana D. Progress of minimal residual disease studies in childhood acute leukemia. Curr Hematol Malig Rep. 2010;5:169–76. doi: 10.1007/s11899-010-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borowitz MJ, Devidas M, Hunger SP, et al. Children’s Oncology Group Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood. 2008;111:5477–85. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stow P, Key L, Chen X, et al. Clinical significance of low levels of minimal residual disease at the end of remission induction therapy in childhood acute lymphoblastic leukemia. Blood. 2010;115:4657–63. doi: 10.1182/blood-2009-11-253435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–84. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhojwani D, Kang H, Menezes RX, et al. Children’s Oncology Group Study, Dutch Childhood Oncology Group. German Cooperative Study Group for Childhood Acute Lymphoblastic Leukemia Gene expression signatures predictive of early response and outcome in high-risk childhood acute lymphoblastic leukemia: a Children’s Oncology Group Study. J Clin Oncol. 2008;26:4376–84. doi: 10.1200/JCO.2007.14.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinase, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115:5312–21. doi: 10.1182/blood-2009-09-245944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silveira VS, Scrideli CA, Moreno DA, et al. Geneexpressionpattern contributing to prognostic factors in childhoodacute lymphoblastic leukemia. Leuk Lymphoma. 2013;54(2):310–4. doi: 10.3109/10428194.2012.710330. [DOI] [PubMed] [Google Scholar]
- 15.Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group Study. Blood. 2012;119:3512–22. doi: 10.1182/blood-2011-11-394221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waanders E, van der Velden VH, van der Schoot CE, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. 2011;25:254–8. doi: 10.1038/leu.2010.275. [DOI] [PubMed] [Google Scholar]
- 17.Cario G, Zimmermann M, Romey R, et al. Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BMF 2000 protocol. Blood. 2010;115:5393–7. doi: 10.1182/blood-2009-11-256131. [DOI] [PubMed] [Google Scholar]
- 18.Haferlach T, Kohlmann A, Wieczorek L, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 20.Ross ME, Zhou X, Song G, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102:2951–9. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- 21.Cario G, Stanulla M, Fine BM, et al. Distinct gene expression profiles determine molecular treatment response in childhood acute lymphoblastic leukemia. Blood. 2005;105(2):821–6. doi: 10.1182/blood-2004-04-1552. [DOI] [PubMed] [Google Scholar]
- 22.Flotho C, Coustan-Smith E, Pei D, et al. Genes contributing to minimal residual disease in childhood acute lymphoblastic leukemia: prognostic significance of CASP8AP2. Blood. 2006;108:1050–7. doi: 10.1182/blood-2006-01-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flotho C, Coustan-Smith E, Pei D, et al. A set of genes that regulate cell proliferation predicts treatment outcome in childhood acute lymphoblastic leukemia. Blood. 2007;110:1271–7. doi: 10.1182/blood-2007-01-068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson A, Ritz C, Lindgren D, et al. Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia. 2007;21:1198–203. doi: 10.1038/sj.leu.2404688. [DOI] [PubMed] [Google Scholar]
- 25.Kang H, Chen IM, Wilson CS, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood. 2010;115:1394–405. doi: 10.1182/blood-2009-05-218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 28.Bautista D, Arana E, Martí-Bonmatí L, Paredes R. Validation of logistic regression models in small samples: application to calvarial lesions diagnosis. J Clin Epidemiol. 1999;52:237–41. doi: 10.1016/s0895-4356(98)00165-6. [DOI] [PubMed] [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–1. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 32.Allocco DJ, Kohane IS, Butte AJ. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics. 2004;5:18. doi: 10.1186/1471-2105-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hicks C, Miele L, Koganti T, et al. Analysis of patterns of gene expression variation within and between ethnic populations in pediatric B-ALL. Cancer Inform. 2013;12:155–73. doi: 10.4137/CIN.S11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwab CJ, Chilton L, Morrison H, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblasticleukemia: association with cytogenetics and clinical features. Haematologica. 2013;98:1081–8. doi: 10.3324/haematol.2013.085175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krentz S, Hof J, Mendioroz A, et al. Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblasticleukemia. Leukemia. 2013;27:295–304. doi: 10.1038/leu.2012.155. [DOI] [PubMed] [Google Scholar]
- 36.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape ofhypodiploidacute lymphoblasticleukemia. Nat Genet. 2013;45:242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guilloton F, Jean C, de Thonel A, Laurent G, Quillet-Mary A. Granzyme B induction signalling pathway in acute myeloid leukemia cell lines stimulated by tumor necrosis factor alpha and Fas ligand. Cell Signal. 2007;19(6):1132–40. doi: 10.1016/j.cellsig.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 38.Sharma V, Delgado M, Ganea D, Granzyme B. a new player in activation-induced cell death, is down-regulated by vasoactive intestinal peptide in Th2 but not Th1 effectors. J Immunol. 2006;176(1):97–110. doi: 10.4049/jimmunol.176.1.97. [DOI] [PubMed] [Google Scholar]
- 39.Puri KD, Di Paolo JA, Gold MR. B-cell receptor signaling inhibitors for treatment of autoimmune inflammatory diseases and B-cellmalignancies. Int Rev Immunol. 2013;32(4):397–427. doi: 10.3109/08830185.2013.818140. [DOI] [PubMed] [Google Scholar]
- 40.Mraz M, Kipps TJ. MicroRNAs and B cell receptor signaling in chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54(8):1836–9. doi: 10.3109/10428194.2013.796055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou FS, Mulloy JC. The thrombopoietin/MPL pathway in hematopoiesis and leukemogenesis. J Cell Biochem. 2011;112(6):1491–8. doi: 10.1002/jcb.23089. [DOI] [PubMed] [Google Scholar]
- 42.Pauls SD, Lafarge ST, Landego I, Zhang T, Marshall AJ. The phosphoinositide 3-kinase signaling pathway in normal and malignant B cells: activation mechanisms, regulation and impact on cellular functions. Front Immunol. 2012;3:224. doi: 10.3389/fimmu.2012.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrett D, Brown VI, Grupp SA, Teachey DT. Targeting the PI3K/AKT/mTOR signaling axis in children with hematologic malignancies. Paediatr Drugs. 2012;14(5):299–316. doi: 10.2165/11594740-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vora A, Goulden N, Mitchell C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukemia (UKALL 2003): a randomized controlled trial. Lancet Oncol. 2014;15(8):809–18. doi: 10.1016/S1470-2045(14)70243-8. [DOI] [PubMed] [Google Scholar]
- 45.Orgel E, Tucci J, Alhushki W, et al. Obesity is associated with residual leukemia following induction therapy for childhood B-precursor acute lymphoblastic leukemia. Blood. 2014;124(26):3932–8. doi: 10.1182/blood-2014-08-595389. [DOI] [PubMed] [Google Scholar]
- 46.Attarbaschi A, Panzer-Grümayer R, Mann G, et al. Austrian and German ALL-BFM (Berlin-Frankfurt-Münster) Study Group Minimal residual disease-based treatment is adequate for relapse-prone childhood acute lymphoblastic leukemia with an intrachromosomal amplification of chromosome 21: the experience of the ALL-BFM 2000 trial. Klin Padiatr. 2014;226(6/07):338–43. doi: 10.1055/s-0034-1387795. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan EM, Jeha S, Kang G, Cheng C. NK cell genotype and phenotype at diagnosis of acute lymphoblastic leukemia correlate with postinduction residual disease. Clin Cancer Res. 2014;20(23):5986–94. doi: 10.1158/1078-0432.CCR-14-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paganin M, Fabbri G, Conter V, et al. Postinduction minimal residual disease monitoring by polymerase chain reaction in children with acute lymphoblastic leukemia. J Clin Oncol. 2014;32(31):3553–8. doi: 10.1200/JCO.2014.56.0698. [DOI] [PubMed] [Google Scholar]
- 49.Margolin JF, Rabin KR, Steuber CP, et al. Acute lymphoblastic leukemia. In: Pizzo AP, Poplack DG, editors. Principles and Practice of Pediatric Oncology. Philadelphia: Lippincott Williams & Wilkins, Inc; 2011. pp. 518–65. [Google Scholar]
- 50.Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13 A, 13B and 14 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:371–82. doi: 10.1038/leu.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pui CH, Sandlund JT, Pei D, et al. Total Therapy Study XIIIB at St Jude Children’s Research Hospital Improved outcome for children with a cute lymphoblasticleukemia: results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood. 2004;104:2690–96. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 52.Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14:18–24. doi: 10.1200/JCO.1996.14.1.18. [DOI] [PubMed] [Google Scholar]
- 53.Greaves MF, Colman SM, Beard ME, et al. Geographical distribution of acute lymphoblastic leukaemia subtypes: second report of the collaborative group study. Leukemia. 1993;7(1):27–34. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. List of the 691 significantly (P < 0.005) differentially expressed genes and their estimates of P-values between positive day 46 MRD and negative MRD patients in each subtypes of B-ALL and their direction of expression values.
Supplementary Table 2. Complete list of the 270 genes used in network analysis, their predicted scores for correctly assigning them to the appropriate molecular networks and information on the molecular functions in which the genes are involved.