

Abstract
Aarskog–Scott syndrome (AAS), also known as faciogenital dysplasia (FGD, OMIM # 305400), is an X-linked disorder of recessive inheritance, characterized by short stature and facial, skeletal, and urogenital abnormalities. AAS is caused by mutations in the FGD1 gene (Xp11.22), with over 56 different mutations identified to date. We present the clinical and molecular analysis of four unrelated families of Mexican origin with an AAS phenotype, in whom FGD1 sequencing was performed. This analysis identified two stop mutations not previously reported in the literature: p.Gln664* and p.Glu380*. Phenotypically, every male patient met the clinical criteria of the syndrome, whereas discrepancies were found between phenotypes in female patients. Our results identify two novel mutations in FGD1, broadening the spectrum of reported mutations; and provide further delineation of the phenotypic variability previously described in AAS.
Keywords: Aarskog–Scott syndrome, FGD1 gene, mental retardation, novel mutation, X-linked
Introduction
Aarskog–Scott syndrome (AAS, OMIM # 305400), also known as faciogenital dysplasia (FGD), is an X-linked syndrome with recessive inheritance, characterized by short stature, hypertelorism, short nose, brachydactyly, and shawl scrotum (Scott 1971; Orrico et al. 2004). Experience in Leuven (Belgium) and Manchester (United Kingdom) indicates population prevalence up to 1/25,000 (Orrico et al. 2014). The clinical abnormalities that can be used for diagnosis of AAS are varied; therefore, the criteria described by Teebi et al. (1993) are customarily utilized (Table1). Furthermore, it is difficult to establish carrier status in women, as they may be asymptomatic or show only partial expression of clinical manifestations due X inactivation (Pasteris et al. 1994).
Table 1.
Clinical description of the Teebi et al. (1993) criteria evaluated in patients and their mothers with AAS.
| Features | Patients evaluated | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Male | Female | ||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| Primary criteria | |||||||||
| Hypertelorism | + | + | + | + | + | + | + | + | − |
| Anteverted nostrils | + | − | + | − | + | + | − | + | − |
| Bottom lip Fold | + | + | + | + | + | + | + | − | + |
| Brachydactyly/wide fingers | + | − | + | + | + | + | − | + | − |
| Interdigital tracts | + | − | + | + | + | − | − | + | − |
| Shawl Scrotum | + | + | − | + | + | NA | NA | NA | NA |
| Syndactyly | − | − | + | + | − | − | − | − | − |
| Clinodactyly | + | − | − | − | + | + | − | − | − |
| Camptodactyly | − | − | − | + | + | + | − | − | − |
| Short stature | + | + | + | + | + | + | − | + | − |
| Secondary criteria | |||||||||
| Widow's peak | + | + | + | + | + | + | + | + | + |
| Ptosis | − | − | − | + | + | − | − | − | − |
| Downward slant palp | − | − | − | + | + | − | − | − | − |
| Joint hypermobility | + | + | − | + | + | − | + | + | − |
| Wide foot | + | − | − | + | + | − | − | − | + |
| Inguinal/umbilical hernia | − | + | + | − | + | − | − | − | − |
| Cryptorchidism | + | + | − | + | + | NA | NA | NA | NA |
| Dysplastic ears | + | + | − | + | + | − | − | − | − |
| Prominent umbilicus | − | − | + | − | − | − | − | + | − |
| Additional criteria | |||||||||
| Obesity | + | − | − | + | − | − | − | − | − |
| Long philtrum | + | + | − | + | + | − | − | + | − |
| Midface hypoplasia | + | − | + | + | + | + | + | + | − |
| Dental malocclusion | + | − | + | + | − | − | − | − | − |
| Transverse palmar crease | + | − | − | − | + | − | − | − | − |
| Hypospadias | − | + | − | − | − | NA | NA | NA | NA |
| Frontal bossing | + | + | + | − | − | − | − | + | − |
| Psychomotor retardation | − | − | − | + | − | − | − | + | − |
+, characteristic present; −, characteristic absent, NA, not applicable; ASS, Aarskog–Scott syndrome.
Since the identification in 1994 of mutations in FGD1 gene in patients with a phenotype consistent with AAS, about 56 different mutations across the 18 exons of the gene have been characterized worldwide (Orrico et al. 2011). The FGD1 locus is located on the short arm of the X chromosome (Xq11.22) and is essential for normal embryonic development in mammals (Estrada et al. 2001). The FGD1 protein product is a member of the DBL family, and acts as guanine nucleotide exchange factor of the Rho GTPase Cdc42, inducing a conformational change in the protein that allows interaction with effector proteins that handle a variety of biological processes (Genot et al. 2012). FGD1 mutations have been identified in approximately 20% of the known cases of AAS (Orrico et al. 2010; Verhoeven et al. 2012). Some studies have found that patients with confirmed molecular evidence have relatively consistent clinical presentations, including hypertelorism, short nose, short and broad hands, shawl scrotum, and a mild to moderate short stature with an acromelic prevalence (Scott 1971; Glover et al. 1993; Orrico et al. 2010).
Previous reports of molecularly proven cases have all corresponded to Caucasian patients, and the majority of mutations identified to date are unique within each family. Furthermore, no hotspot or common mutations exist that have a demonstrated genotype–phenotype correlation. Therefore, the purpose of this study was to clinically and genetically characterize four nonconsanguineous Mexican (non-Caucasian) families with an AAS syndrome phenotype. This led to the identification of two novel mutations in FGD1, representing 60% of the patients in these families (Table1).
Materials and Methods
Patients and sample collection
A clinical and dysmorphologic evaluation was performed on patients by applying the Teebi criteria (Teebi et al. 1993) to duos (mother/son) in four nonconsanguineous families with an AAS syndrome phenotype who received care at the Department of Genetics, “Dr. José Eleuterio González” University Hospital, the Autonomous University of Nuevo León. After written informed consent was obtained from patients, peripheral blood samples were collected, and total genomic DNA was extracted using the QIAamp® DNA Mini and Blood Mini extraction kits (Qiagen, Valencia, CA) according to the manufacturer's instructions. The study was approved by the ethics committee of the same institution.
Sequencing and mutation detection
PCR amplification for all FGD1 exons (NM_004463.2) was performed using the primers previously described by Orrico et al. (2010). The PCR products were sequenced bidirectionally using the same primers and BigDye version 3.1 (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. After purification, products were run on an ABI Prism 3130 Genetic Analyzer (Applied Biosystems). The results were analyzed by visual inspection using Geneious 4.7.6 software (Biomatters, Auckland, NZ).
Results
Four families were recruited with an AAS phenotype, from which nine samples were obtained from affected individuals: five males and four females, with mean age of 6 and 31 years, respectively. The clinical criteria utilized for diagnosing patients with AAS Syndrome are shown in Table1. During clinical evaluation, the following features were more prevalent in men: short stature, hypertelorism, and a fold of the lower lip were observed in 100% of patients; brachydactyly, interdigital tracts, and shawl scrotum were also seen in 80% of patients. Secondary criteria included a widow's peak, present in 100% of patients, followed by cryptorchidism, dysplastic ears, and joint hypermobility in 80% of patients. Finally, among the additional features described, long philtrum and mid-facial hypoplasia were present in 80% of patients, followed by poor dental occlusion and frontal bossing, present in 60% of patients. Photographs of some patients with the observed clinical characteristics are shown in Figure1.
Figure 1.
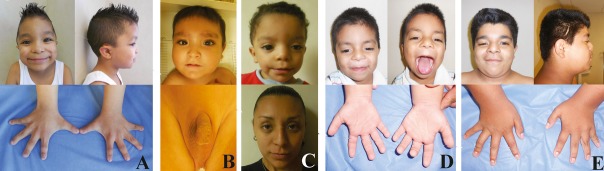
Patients with clinical features of Aarskog syndrome. (A) Patient 1; note distinctive facial characteristics and interdigital tracts in both hands. (B) Patient 2; discrete facial features and the shawl scrotum can be appreciated. (C) Patient 3 and his mother (patient 8); note prominent forehead, widow's peak, hypertelorism, and fold under the lower lip. (D) Patient 4 with widow's peak, midface hypoplasia, ptosis, clinodactyly, and brachydactyly. (E) Patient 5, brother of patient 4, with distinctive facial features, clinodactyly, and brachydactyly.
We showed two nonsense mutations (p.Gln664* and p.Glu380*) both inherited by each mother. The results of molecular testing are presented in Table2. In summary, a recognizable FGD1 mutation was identified in only 60% of individual who were analyzed.
Table 2.
Changes found in cDNA and its effect on the protein.
| Case | Gender | Clinical criteria | gDNA | cDNA | Protein | Type of mutation |
|---|---|---|---|---|---|---|
| 1 | M | 19 | g.54495273C>A | c.1138G>T | p.Glu380* | Novel |
| 2 | M | 12 | – | – | NC | – |
| 3 | M | 13 | – | – | NC | – |
| 4 | M | 20 | g.54481906G>A | c.1990C>T | p.Gln664* | Novel |
| 5 | M | 20 | g.54481906G>A | c.1990C>T | p.Gln664* | Novel |
| 6 | F | 9 | g.54495273C>A | c.1138G>T | p.Glu380* | Novel |
| 7 | F | 7 | – | – | NC | – |
| 8 | F | 11 | – | – | NC | – |
| 9 | F | 3 | g.54481906G>A | c.1990C>T | p.Gln664* | Novel |
gDNA, genomic DNA; cDNA, complementary DNA; M, male; F, female; NC, no change.
Discussion
Clinically, patients with AAS present with various abnormalities (Teebi et al. 1993). In this study, several primary characteristics were observed, namely, short stature, brachydactyly, shawl scrotum, hypertelorism, and a fold below the lower lip (Scott 1971; Orrico et al. 2004). Mutations of FGD1 gene represent the etiology of AAS (Orrico et al. 2010; Verhoeven et al. 2012). Various types of FGD1 mutations have been identified, ranging from point mutations to large deletions distributed throughout the gene; most result in a truncated protein (Orrico et al. 2000; Hou et al. 2003; Orrico et al. 2011; Volter et al. 2014). In this study, FGD1 mutation detection was conducted in four Mexican families; two novel mutations were identified in two no Caucasian families with AAS (c.1138G>T and c.1990C>T); in each case, these produce a stop codon leading to a truncated protein. These two mutations arose in different domain types of the FGD1 protein: Dbl homology/RhoGEF activity (DH); and pleckstrin homology (PH).
The c.1138G>T (p.Glu380*) variant, a previously unreported mutation identified in exon 5 that produces a stop codon, is located within the FGD1 DH domain (Fig.2), which has been shown to play an important role in regulating cell growth and differentiation (Pasteris et al. 1994). Interestingly, the residue 380 was already detected as involved in mutational events, although the type of mutation is missense (p.E380A) (Orrico et al. 2004). This mutation was identified in the mother and child (patients 1 and 6). Patient 1, a 6-year-old boy evaluated due to short stature at 2 years of age, had eight primary, clinical criteria identified (Fig.1A), while his mother, patient 6, had attenuated features of this syndrome.
Figure 2.
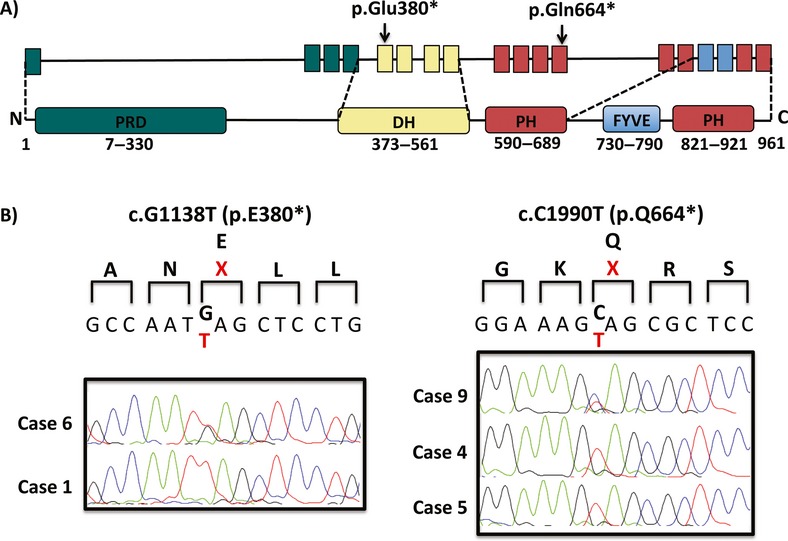
Detection of FGD1 mutations. (A) Schematic representation of the domains of the FGD1 protein showing mutations (p.Glu380* and p.Gln664*) identified in patients with AAS. Arrows indicate the positions of the mutated nucleotides in FGD1. (B) sequencing results (p.Glu380* and p.Gln664*) detected in exon 5 and 12, respectively. The altered amino acids are shown in red.
The variant c.1990C>T (p.Gln664*), identified in exon 12 of FGD1, is also a novel mutation that produces a stop codon. This mutation disrupts the coding sequence of the PH domain (amino acids, aa, 590–689; Fig.2). It is widely accepted that the majority of proteins with domains in the PH family are involved in the phosphorylation of inositol phospholipids. They play a central role in a number of cell processes, ranging from intracellular signal transduction in the plasma membrane, actin cytoskeleton organization, and prevention of apoptosis, to regulation of vesicular endocytosis (Orrico et al. 2000). The mutation was identified in two siblings (patients 4 and 5) and their mother (patient 9). Patient 4, at 12 years of age had the most phenotypic characteristics of AAS of all the patients reported in this study, as defined by Teebi et al. (1993), and shared the vast majority of clinical features with his younger brother of 4 years of age (patient 5). Interestingly, patient 4 also displayed delayed learning, a finding that may not yet have been evident in his brother. Delayed learning has been observed in other cases (Lebel et al. 2002; Orrico et al. 2004), although apparently has not reported genotype–phenotype correlation that can predict whether a patient will have delayed learning, even this feature is considered rare in AAS. The boys’ mother, who was heterozygous for the mutation, phenotypically only, displayed a “widow's peak” and wide feet as unique features compatible with AAS.
The failure to identify pathogenic mutations in FGD1 in the other two families (patient 2 and mother, patient 7; patient 3 and mother, patient 8) can be attributed largely to the clinical heterogeneity of AAS being as a clinical and not a molecular diagnosis, meaning that clinical heterogeneity and the lack of identifiable mutation do not exclude the diagnosis. Both patient 3 and his mother had sufficient clinical findings to support the diagnosis; however, a molecular basis for the disorder in this family cannot be ruled out. A recent publication documents, for example, the occurrence of a mutation in the branch point of exon 13 by exome sequencing, that conditions premature termination of translation due to a skipped exon (Aten et al. 2013); making it important to consider additional molecular diagnostic technologies. However, the differential diagnosis must also consider other disease such as Noonan syndrome, SHORT syndrome, and Robinow syndrome, as their pathologies overlap with many clinical features of AAS as short stature, long philtrum, micrognathia, hypertelorism, broad nasal bridge, anteverted nostrils, cryptorchidism, brachydactyly, clinodactyly, small hands, ptosis, skeletal and genitourinary abnormalities (Orrico et al. 2011; Tartaglia et al. 2011).
Whereas the best characterized form of AAS is associated with mutations in FGD1, only about 20% of disease prevalence can be explained by this mechanism, and the genetic cause has not yet been identified in most families. Therefore, it is believed that other genes may be involved in the etiology of this disorder. GEFs comprise a large family of regulatory proteins with over 21 having been identified across the human genome to date. GEFs control various intracellular processes such as gene expression, rearrangements of the cytoskeleton, intracellular trafficking, and metabolism (Hall 2005; Jaffe and Hall 2005; Gupta et al. 2013). For example, in vitro studies have shown that FGD4 and Vav1 activate signaling downstream from Cdc42 as does FGD1; and that these proteins play critical roles in the formation of the actin cytoskeleton and embryonic development (Olson et al. 1996). Therefore, a form of autosomal recessive inheritance, with similar underlying mechanism to the X-linked variety, cannot be ruled out for this disorder.
Clinical examination of the mothers of the probands showed that four women between 28 and 37 years old found phenotypic heterogeneity in most cases. Dramatic similarity was observed between patient 6 and her son, patient 1. Patient 6 was the mother with the most primary clinical features, sharing the same, albeit attenuated, phenotypic traits with her child, with the exception of interdigital tracts; but not sharing the secondary and additional characteristics, of which she shared only the “widow's peak” and midface hypoplasia. Molecularly, the same mutation was found in both patients. As has been described elsewhere, the carriers of a mutation may exhibit an attenuated phenotype AAS (Mikelsaar and Lurie 1992; Pasteris et al. 1994); therefore, behaves as an X-linked recessive disorder, with phenotypic attenuation in females likely due to wild-type FGD1 expression from the second X chromosome (Park et al. 2010). It is further possible that other genes or regulatory factors may modulate FGD1 expression in females, moderating the phenotype (Gao et al. 2011; Zou et al. 2011).
The finding in this study of two separate mutations within FGD1 is consistent with previous studies, wherein each identified mutation was unique to each family (Orrico et al. 2010), with the exception of mutations c.528insC, p.R656X, and p.R443, which were reported in several unrelated families (Al-Semari et al. 2013). However, these families do not share general clinical characteristics, nor is there any evidence suggesting that the mutation influences the clinical phenotype of the disease.
In conclusion, this study represents the first molecular analysis of the FGD1 gene in Mexican patients with characteristic AAS phenotypes, and presents the discovery of two novel mutations not previously reported in the literature. The main features observed in our probands were short stature, hypertelorism, a lower lip fold, and a widow's peak, confirming the variability of phenotypic expression described in previous publications. Our data demonstrate that even when the clinical criteria dictated by Teebi et al. (1993) are met, conventional sequencing techniques are only able to identify recognizable mutations in FGD1 in a few of families. Numerous other possible causes for AAS, such as deletions or intronic mutations of FGD1, are not detectable by this technique. Finally, the results obtained in this study allowed us to provide more accurate genetic counseling for the women identified as mutation carriers, as opposed to relying upon the general familial recurrence risk.
Acknowledgments
We thank all the patients and families who participated in the study, as well as the clinicians who provided patient blood samples and clinical information.
Conflict of Interest
None declared.
References
- Al-Semari A, Wakil SM, Al-Muhaizea MA, Dababo M, Al-Amr R, Alkuraya F, et al. Novel FGD1 mutation underlying Aarskog-Scott syndrome with myopathy and distal arthropathy. Clin. Dysmorphol. 2013;22:13–17. doi: 10.1097/MCD.0b013e32835b6dc4. [DOI] [PubMed] [Google Scholar]
- Aten E, Sun Y, Almomani R, Santen GW, Messemaker T, Maas SM, et al. Exome sequencing identifies a branch point variant in Aarskog-Scott syndrome. Hum. Mutat. 2013;34:430–434. doi: 10.1002/humu.22252. [DOI] [PubMed] [Google Scholar]
- Estrada L, Caron E. Gorski JL. Fgd1, the Cdc42 guanine nucleotide exchange factor responsible for faciogenital dysplasia, is localized to the subcortical actin cytoskeleton and Golgi membrane. Hum. Mol. Genet. 2001;10:485–495. doi: 10.1093/hmg/10.5.485. [DOI] [PubMed] [Google Scholar]
- Gao L, Gorski JL. Chen CS. The Cdc42 guanine nucleotide exchange factor FGD1 regulates osteogenesis in human mesenchymal stem cells. Am. J. Pathol. 2011;178:969–974. doi: 10.1016/j.ajpath.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genot E, Daubon T, Sorrentino V. Buccione R. FGD1 as a central regulator of extracellular matrix remodelling–lessons from faciogenital dysplasia. J. Cell Sci. 2012;125:3265–3270. doi: 10.1242/jcs.093419. [DOI] [PubMed] [Google Scholar]
- Glover TW, Verga V, Rafael J, Barcroft C, Gorski JL, Bawle EV, et al. Translocation breakpoint in Aarskog syndrome maps to Xp11.21 between ALAS2 and DXS323. Hum. Mol. Genet. 1993;2:1717–1718. doi: 10.1093/hmg/2.10.1717. [DOI] [PubMed] [Google Scholar]
- Gupta M, Kamynina E, Morley S, Chung S, Muakkassa N, Wang H, et al. Plekhg4 is a novel Dbl family guanine nucleotide exchange factor protein for rho family GTPases. J. Biol. Chem. 2013;288:14522–14530. doi: 10.1074/jbc.M112.430371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005;33:891–895. doi: 10.1042/BST20050891. [DOI] [PubMed] [Google Scholar]
- Hou P, Estrada L, Kinley AW, Parsons JT, Vojtek AB. Gorski JL. Fgd1, the Cdc42 GEF responsible for faciogenital dysplasia, directly interacts with cortactin and mAbp1 to modulate cell shape. Hum. Mol. Genet. 2003;12:1981–1993. doi: 10.1093/hmg/ddg209. [DOI] [PubMed] [Google Scholar]
- Jaffe AB. Hall A. Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Lebel RR, May M, Pouls S, Lubs HA, Stevenson RE. Schwartz CE. Non-syndromic X-linked mental retardation associated with a missense mutation (P312L) in the FGD1 gene. Clin. Genet. 2002;61:139–145. doi: 10.1034/j.1399-0004.2002.610209.x. [DOI] [PubMed] [Google Scholar]
- Mikelsaar RV. Lurie IW. Atypical case of Aarskog syndrome. J. Med. Genet. 1992;29:349–350. doi: 10.1136/jmg.29.5.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Pasteris NG, Gorski JL. Hall A. Faciogenital dysplasia protein (FGD1) and Vav, two related proteins required for normal embryonic development, are upstream regulators of Rho GTPases. Curr. Biol. 1996;6:1628–1633. doi: 10.1016/s0960-9822(02)70786-0. [DOI] [PubMed] [Google Scholar]
- Orrico A, Galli L, Falciani M, Bracci M, Cavaliere ML, Rinaldi MM, et al. A mutation in the pleckstrin homology (PH) domain of the FGD1 gene in an Italian family with faciogenital dysplasia (Aarskog-Scott syndrome) FEBS Lett. 2000;478:216–220. doi: 10.1016/s0014-5793(00)01857-3. [DOI] [PubMed] [Google Scholar]
- Orrico A, Galli L, Cavaliere ML, Garavelli L, Fryns JP, Crushell E, et al. Phenotypic and molecular characterisation of the Aarskog-Scott syndrome: a survey of the clinical variability in light of FGD1 mutation analysis in 46 patients. Eur. J. Hum. Genet. 2004;12:16–23. doi: 10.1038/sj.ejhg.5201081. [DOI] [PubMed] [Google Scholar]
- Orrico A, Galli L, Faivre L, Clayton-Smith J, Azzarello-Burri SM, Hertz JM, et al. Aarskog-Scott syndrome: clinical update and report of nine novel mutations of the FGD1 gene. Am. J. Med. Genet. A. 2010;152A:313–318. doi: 10.1002/ajmg.a.33199. [DOI] [PubMed] [Google Scholar]
- Orrico A, Galli L, et al. Clinical utility gene card for: Aarskog-Scott Syndrome (faciogenital dysplasia) 2014. . European journal of human genetics: EJHG. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Carrel L. Makova KD. Strong purifying selection at genes escaping X chromosome inactivation. Mol. Biol. Evol. 2010;27:2446–2450. doi: 10.1093/molbev/msq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasteris NG, Cadle A, Logie LJ, Porteous ME, Schwartz CE, Stevenson RE, et al. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: a putative Rho/Rac guanine nucleotide exchange factor. Cell. 1994;79:669–678. doi: 10.1016/0092-8674(94)90552-5. [DOI] [PubMed] [Google Scholar]
- Scott CI. Unusual facies, joint hypermobility, genital anomaly and short stature: a new dysmorphic syndrome. Birth Defects Orig. Artic. Ser. 1971;7:240–246. [PubMed] [Google Scholar]
- Tartaglia M, Gelb BD. Zenker M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2011;25:161–179. doi: 10.1016/j.beem.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teebi AS, Rucquoi JK. Meyn MS. Aarskog syndrome: report of a family with review and discussion of nosology. Am. J. Med. Genet. 1993;46:501–509. doi: 10.1002/ajmg.1320460508. [DOI] [PubMed] [Google Scholar]
- Verhoeven WM, Egger JI. Hoogeboom AJ. X-linked Aarskog syndrome: report on a novel FGD1 gene mutation. Executive dysfunction as part of the behavioural phenotype. Genet. Couns. 2012;23:157–167. [PubMed] [Google Scholar]
- Volter C, Martinez R, Hagen R. Kress W. Aarskog-Scott syndrome: a novel mutation in the FGD1 gene associated with severe craniofacial dysplasia. Eur. J. Pediatr. 2014;173:1373–1376. doi: 10.1007/s00431-014-2317-3. [DOI] [PubMed] [Google Scholar]
- Zou W, Greenblatt MB, Shim JH, Kant S, Zhai B, Lotinun S, et al. MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J. Clin. Invest. 2011;121:4383–4392. doi: 10.1172/JCI59041. [DOI] [PMC free article] [PubMed] [Google Scholar]