

Abstract
The Aristaless-related homeobox (ARX) gene is implicated in intellectual disability with the most frequent pathogenic mutations leading to expansions of the first two polyalanine tracts. Here, we describe analysis of the ARX gene outlining the approaches in the Australian and Portuguese setting, using an integrated clinical and molecular strategy. We report variants in the ARX gene detected in 19 patients belonging to 17 families. Seven pathogenic variants, being expansion mutations in both polyalanine tract 1 and tract 2, were identifyed, including a novel mutation in polyalanine tract 1 that expands the first tract to 20 alanines. This precise number of alanines is sufficient to cause pathogenicity when expanded in polyalanine tract 2. Five cases presented a probably non-pathogenic variant, including the novel HGVS: c.441_455del, classified as unlikely disease causing, consistent with reports that suggest that in frame deletions in polyalanine stretches of ARX rarely cause intellectual disability. In addition, we identified five cases with a variant of unclear pathogenic significance. Owing to the inconsistent ARX variants description, publications were reviewed and ARX variant classifications were standardized and detailed unambiguously according to recommendations of the Human Genome Variation Society. In the absence of a pathognomonic clinical feature, we propose that molecular analysis of the ARX gene should be included in routine diagnostic practice in individuals with either nonsyndromic or syndromic intellectual disability. A definitive diagnosis of ARX-related disorders is crucial for an adequate clinical follow-up and accurate genetic counseling of at-risk family members.
Keywords: Aristaless-related homeobox gene, ARX, expanded polyalanine tract, intellectual disability, pathogenic variant
Introduction
Since its discovery in 2002, Aristaless-related homeobox gene (ARX; MIM# 300382; GenBank: NM_139058.2) has been implicated in X-linked intellectual disability (XLID) (Bienvenu et al. 2002; Shoubridge et al. 2010). The ARX gene, cytogenetically located on Xp21.3, encompasses 12.5 kb of genomic DNA and includes five coding exons encoding a 562 amino acid protein. This gene is a paired-type homeodomain transcription factor expressed predominately in the fetal and adult brain, testis, skeletal muscle, and pancreas. ARX has a critical role in brain development, particularly in GABAergic interneuron migration during cortical development (Miura et al. 1997; Kitamura et al. 2002; Stromme et al. 2002a). Hence, it is not surprising that epilepsy and structural brain malformations including lissencephaly and agenesis of corpus callosum are frequently observed in patients with ARX pathogenic mutations. Although patients may present with intellectual disability (ID) without additional clinical features (nonsyndromic ID), the concomitant observation of neurological deficits, with or without brain and/or genital anomalies suggests a number of recognizable syndromes. The ARX-associated spectrum of disorders includes Partington syndrome (OMIM 309510) (Partington et al. 2004), Early Infantile Epileptic Encephalopathy (OMIM 308350) (Kato et al. 2007), Agenesis of Corpus Callosum with Abnormal Genitalia (OMIM 300004) (Proud et al. 1992), and X-Linked Lissencephaly with Ambiguous Genitalia (OMIM 300215) (Kitamura et al. 2002). A genotype–phenotype relationship exists and has recently been discussed (Shoubridge et al. 2010). Despite a broad range of phenotypes associated with mutations in ARX, ID is a consistent clinical feature.
Although disease-causing variants occur across all five coding exons, most are located in the largest exon, exon 2. These include expansion of the first or second polyalanine tracts. In particular, duplication of 24 base pairs (bp), HGVS: c.441_464dup (referred to as dup24) in the second polyalanine tract is by far the most common pathogenic mutation in this gene. As most mutations are in this largest exon ARX mutation analysis is routinely confined to screening exon 2.
Report of ARX mutations in the literature is hampered by use of nonstandard nomenclature. Occasionally, publications describing “novel” sequence variants actually represent nomenclature errors, which are a cause of uncertainty and misinterpretation especially in medical diagnostic settings. For example, as many as seven descriptions can be identified for a single ARX gene variant expanding the first polyalanine tract (Stromme et al. 2002a; Wohlrab et al. 2005; Wallerstein et al. 2008; Kitamura et al. 2009; Fullston et al. 2011; Mirzaa et al. 2013). There is an obvious need for an effective molecular testing approach for ARX screening of ID patients and an unambiguous nomenclature for description of ARX variants. In this report, we have undertaken molecular analysis of the ARX gene using Portuguese and Australian cohorts of patients with syndromic and nonsyndromic ID. The continued characterization of ARX variants not only contributes to the identification of specific phenotypic features but also assists in unraveling the pathogenicity of additional rare variants. This type of information is required for accurate genetic counseling of ARX at-risk family members.
Materials and Methods
Patient recruitment
Clinical data and blood samples were collected after informed consent from the patients or their legal guardians. The National Committee for Data Protection authorized laboratory records. Relevant human research ethics committees have approved these studies. A total of 138 patients from two cohorts were enrolled for this study. Cohort 1 consisted of 101 Australian patients. Ninety patients were directly referred for polyalanine tract 1 (pA1) and tract 2 (pA2) ARX size-variant analysis. Thirteen patients were referred for sequencing of the entire open reading frame of ARX gene, including two cases that had already been tested negative for size variants in pA1 and pA2, but were included to rule out mutations elsewhere in the gene. Cohort 2 consisted of thirty-seven Portuguese patients. Twenty-four patients were referred for pA1 and pA2 ARX size-variant analysis. The remaining 13 patients were pulled out from a previous study consisting of the simultaneous analyses of fragile-X (FMR1), FRAXE (AFF2), and ARX (henceforward named multiplex screening) (Jorge et al. 2013). The selection of all subjects was based on clinical criteria assessed by the patients' geneticist or neurologist. Seventeen patients were subsequently sequenced for the entire open reading frame of ARX gene, based on clinical criteria (e.g. brain malformation) assessed by patient physicians. Consanguinity was excluded in the parents unless otherwise stated.
Molecular analysis
Cohort 1 – A PCR-based approach was used to screen for expansions in pA1 and pA2 in exon 2 of ARX. Any changes in migration of the amplicons detected by gel electrophoresis were subsequently analyzed by bidirectional cycle sequencing reactions. The PCR and electrophoresis conditions have been described in detail previously (Tan et al. 2013). Sequencing reactions were performed using ABI Big Dye Terminator chemistry version 3.1 and purified products subjected to an automated capillary sequencing on ABI 3100 sequencer (Applied Biosystems, Foster City, CA, USA) and analysis was aided by Seqscape V2.5 (Applied Biosystems). In 13 patients the entire open reading frame was screened. Each of the five exons of ARX was amplified by PCR using primers designed to amplify coding and flanking noncoding sequence (Table S1). The exception to this is exon 2, for which four overlapping amplicons were used to achieve robust amplification of GC-rich regions coding for three polyalanine tracts (Tan et al. 2013). Sequence of each amplicon was subsequently confirmed as outlined above.
Cohort 2 – A multiplex-PCR method designed to amplify a portion of the second exon of ARX gene that includes the first two of the three tracts of repetitive alanine coding triplets was previously published by the authors (Jorge et al. 2013). Further size estimation on capillary electrophoresis allows identification of size variants: a 380 bp amplicon matches the expected normal-sized allele and a 404 bp amplicon corresponds to the dup24. Any changes in migration of the amplicons detected by gel electrophoresis were subsequently analyzed by cycle sequencing as described above for the Australian cohort. M13-tailored primers were designed to amplify all exons and intron–exon borders (Table S1) and sequencing of each of the five ARX exons was done following standard procedures on ABI 3130xl (Applied Biosystems, Foster City, CA, USA).
Bioinformatics
Human Genome Variation Society (HGVS) nomenclature is stated for the ARX variants and refers to the GenBank mRNA entry NM_139058.2 (den Dunnen and Antonarakis 2000). The software Mutalyzer 2.0.beta-31 (www.mutalyzer.nl, HGVS nomenclature version 2.0) was used to check nomenclature of variants identified in the Portuguese and Australian cohorts. In addition, ARX-Locus Specific Database, LOVD (http://LOVD.nl/ARX; last accessed on March 2014) (Fokkema et al. 2011) and Human Gene Mutation Database, HGMD Professional (last accessed on April 2014) databases were crosschecked.
Statistical analyses
A chi-square (χ2) distribution test was performed, with a significance level (P) of 0.05 and a confidence of 95%, to verify differences between each cohort (cohort 1 Australian and cohort 2 Portuguese). Descriptive data are presented as median with interquartile range (IQR).
Results
Clinical summary of patients referred for diagnostic ARX study
Detailed clinical description and screening outcomes for family A to Q is described in Data S1. All 138 patients enrolled for ARX study had ID, with 127 (92.0%) being male and 11 (8.0%) female (Table1). Forty-two patients (30.4%) were referred exclusively due to ID. Approximately half of the patients were also affected by a movement disorder or epilepsy (67/138; 48.5%). A smaller proportion of patients presented with behavior abnormalities (17/138; 12%) or with brain malformations, with and without genital malformations (12/138; 8.6%). Interestingly, the cohorts differ in regards to the presence of a movement disorder or epilepsy (P < 0.007) and brain malformations, with and without genital malformations (P < 0.001) (Cohort 2 mainly without specific ARX-related referral). In total, 19 patients, four of whom are female, belonging to 17 families, had an ARX sequence change. Sequence variants were restricted to the polyalanine coding regions in ARX exon 2 and positioned in pA1 (n = 6) and pA2 (n = 13) and therefore all variants alter the ARX polyalanine content. Examples of partial electropherograms of ARX variants identified are shown in Figure S1. Inheritance was determined in eight male patients (maternal) and one female (paternal). In one male patient, a de novo variant was identified (one of six for which gDNA from the parents was available; 16.7%).
Table 1.
Clinical data of patients referred for diagnostic ARX study
| Cohort 1 (N = 101) [13 sequenced] | Cohort 2 (N = 37) [17 sequenced] | Total (%) | |
|---|---|---|---|
| Males: N; Age at diagnosis (years) (median; IQR) | 93; (5.5; 9.1) | 34; (11; 7) | 127 (92) |
| Females: N; Age at diagnosis (years) (median; IQR) | 8; (3.65; 38.15) | 3; (8; 7.5) | 11 (8) |
| Family history consistent with X-linked inheritance pattern: N | 13 | 3 | 16 (11) |
| Nonsyndromic ID: N (%) | 29 (28.7) | 13 (35) | 42 (30.4) |
| Syndromic ID: N (%) | |||
| With brain only or brain and genital malformations | 4 (4) | 8 (21) | 12 (8.6) |
| With movement disorders and/or seizures (without information on brain or genital malformations) | 56 (55.4) | 11 (30) | 67 (48.5) |
| With behavior abnormalities | 12 (11.9) | 5 (14) | 17 (12.3) |
ADHD, attention deficit hyperactivity disorder; ID, intellectual disability; IQR, interquartile range.
Standardization of nomenclature for the mutations in the polyalanine tracts of ARX
Publications were reviewed and ARX variants descriptions were standardized and detailed unambiguously according to recommendations of the HGVS (Table2). The HGVS recommends that duplications be designated by “dup” after an indication of the first and last nucleotide(s) duplicated. Moreover, the recommendations state that for all descriptions the most 3′ position possible is arbitrarily assigned to have been changed. This does not concur with what has been previously annotated for these mutations. For example, the most common mutation, c.429_452dup(24), should be annotated as c.441_464dup according to the HGVS. Given the sequence of this region we can see that the 24 bp that are duplicated (as identified from sequencing affected patients) can either be lined up from the position c.429 or c.441 as the most 3′ position. A similar 3′ shift in nomenclature is also observed when the longer 27 bp duplication (Demos et al. 2009) and the 33 bp duplication (Reish et al. 2009) in this pA2 tract are considered (Fig. S2).
Table 2.
Nomenclature standardization: comparison with the previously reported
| Polyalanine content variation | Nomenclature proposed according to HGVS | Present study | Also described as | References | |
|---|---|---|---|---|---|
| cDNA: NM_139058.2 | Protein-level: NP_620689.1 | ||||
| pA1 | |||||
| 16>23 | c.306GGC[17]/c.306_308[17]/c.315_335dup | p.(Ala109_Ala115dup)/p.(115Ala7) | dup/ins[GGC]7 | c.304ins(GCG)7(GCG)10+7c.333ins(GCG)7c.333_334(GCG)7c.333_334ins(GCG)7c.333_335dup(GGC)7c.335ins21 | Fullston et al. (2011)Stromme et al. (2002a)Stromme et al. (2002a)Wohlrab et al. (2005)Mirzaa et al. (2013)Mirzaa et al. (2013)Wallerstein et al. (2008) |
| 16>20 | c.306GGC[14]/c.306_308[14]/c.324_335dup | p.(Ala112_Ala115dup) p.(115Ala4) | dup/ins[GGC]4 | Novel | |
| 16>19 | c.306GGC[13]/c.306_308[13]/c.327_335dup | p.(Ala113_Ala115dup)/p.(115A3) | dup/ins[GGC]3 | c.304ins(GCG)3c.333_334(CGC)3 | Stromme et al. (2002b)Stromme et al. (2002b) |
| 16>17 | c.306GGC[11]/c.306_308[11]/c.333_335dup | p.(Ala115dup)/p.(115A) | dup/ins[GGC]1 | c.304ins(GCG)1c.333_334ins(GCG) | Fullston et al. (2011)Stromme et al. (2002b) |
| 16<12 | c.306GGC[6]/c.324_335del | p.(A112_A115del) | del12 | c.321-332del | Stromme et al. (2002b) |
| pA2 | |||||
| 12>23 (10A-G-12A) | c.426_458dup | p.(Gly143_Ala153dup) | Not reported in this study | c.423_455dup(33 bp) | Kuwaik et al. (2012) |
| 12>21 | c.435_461dup | p.(Ala147_Ala155dup)/p.(155A9) | Not reported in this study | c.430_456dup(27 bp) | Stromme et al. (2003) |
| 12>20 | c.441_464dup | p.(Ala148_Ala155dup)/p.(155A8) | dup24 | c.429_452dupc.428_451dupc.431_454dupc.464_465ins24 | Bienvenu et al. (2002)Stromme et al. (2002b)Gronskov et al. (2004) |
| 12<4 | c. 441_464del | p.(Ala148_Ala155del) | del24 | c.431_454delc.429del24 | Wallerstein et al. (2008)Bienvenu et al. (2002) |
| 12<7 | c. 441_455del;[=] | p.(Ala151_Ala155del);[=] | del15 | Novel | |
HGVS: Version 2.121101.
The recurrent pA1 expansion previously described as c.304insGCG is a duplicating insertion and should therefore be considered a duplication, and according to the 3′ rule, must be described as c.306GGC[11] or c.315_335dup. Concerning the identified pA1 deletion, there are two alternative descriptions: c.306GGC[6] and c.324_335del. If the repeat was not sequenced but instead the size was deduced from the length of a PCR fragment or while describing a predicted (not analyzed) protein change, curved brackets were used: p.(Ala151_Ala155del).
Clinical and molecular data of patients with an ARX variant
The clinical and molecular data for each of the 17 patients we are reporting with an ARX variant are detailed individually in Data S1 and summarized in Table3, including the age and reason for referral. Of the17 cases or variants identified, seven of these are known to be pathogenic, a detection rate of 5%. In cohort 1 there were two pathogenic mutations identified; (i) expansion of pA2 in Family G detected from the 93 patients referred and screened by ARX size-variation, and (ii) expansion of pA1 in Family A identified from the 13 patients referred and screened for sequencing of the entire open reading frame of ARX gene. In cohort 2, there were six males and one female from five families (Family H to L) identified with pathogenic expansions in pA2 from the 13 patients from the previous multiplex screening study.
Table 3.
Summary of available clinical and molecular data of patients with an ARX variant
| Family | Variant nomenclature | Change in ala | Age1 | Primary referral reason/index case | Number of cases and gender | Heredity | Pathogenicity |
|---|---|---|---|---|---|---|---|
| pA1 | |||||||
| A | c.306GGC[17]/c.306_308[17] | 16>23 | 2 months | Developmental delay and infantile seizures | 1M | Maternal | Pathogenic |
| B | c.306GGC[14]/c.306_308[14] | 16>20 | 2 | Developmental delay, microcephaly, spasticity, and dystonia | 1M | Maternal | VUS |
| C | c.306GGC[13]/c.306_308[13] | 16>19 | 4 | Developmental delay, intellectual, and speech delay | 1M | Unknown | VUS |
| D | c.306GGC[10];[11] | 16>17 | 4 | Developmental delay and autistic behavior | 1F | Unknown | VUS |
| E | c.306GGC[11]/c.306_308[11] | 16>17 | 4 | Mild developmental delay, attention deficit, and hyperactivity | 1M | Unknown | |
| F | c.306GGC[6]/c.324_335del | 16<12 | 10 | Intellectual disability | 1M | Unknown | VUS |
| pA2 | |||||||
| G | c.441_464dup | 12>20 | 6 months | Developmental delay, spasms | 1M | Maternal | Pathogenic |
| H | 12>20 | 2; 7 | Intellectual disability and congenital macrocephaly (in both) | 2M3 | Maternal | ||
| I | 12>20 | 4; 39 | Hyperactivity and developmental delay; Dysmorphisms and dystonia of the hands | 2M4 | Maternal | ||
| J | 12>20 | 8 | Intellectual disability, nonspecific cranio-facial dysmorphisms | 1M | De novo | ||
| K | 12>20 | 5 | Motor impairment, severe speech delay, and hyperactivity | 1M | Maternal | ||
| L | c.441_464dup;[=] | 12>20 | 10 | Intellectual diability and speech delay | 1F (heterozygote) | Unknown | |
| M | 12<4 | 4 | Developmental delay | 1M | Unknown | Unlikely | |
| N | c. 441_464del | 12<4 | 13 | Learning disability | 1M | Unknown | Unlikely |
| O | 12<4 | 8 | Learning disability, attention deficit, and hyperactivity | 1M | Maternal | Unlikely | |
| P | c.441_464del;[=] | 12<4 | 11 | Intellectual disability | 1F | Unknown | Unlikely |
| Q | c. 441_455del;[=] | 12<7 | 6 months | Developmental delay, nonspecific craniofacial dysmorphisms | 1F | Paternal | Notpathogenic2 |
VUS, variant of uncertain/unclassified significance; F, Female; M, Male.
In years unless otherwise specified.
Polymorphism absent in 200 control samples.
Brothers.
Nephew/uncle.
Polyalanine tract 1 variants
We identified six distinct families with a pA1 variant (Families A to F) (Table4). The pathogenic c.306GGC[17], p.(115Ala7) mutation in Family A leading to an increase in pA1 to 23 alanines was identified in a patient with infantile epileptic-dyskinetic encephalopathy. This variant has been reported previously and is not found in the control population (Stromme et al. 2002a; Poirier et al. 2006; Guerrini et al. 2007; Fullston et al. 2011). Table4 compares the clinical features of this case with other patients identified with this expansion. Overall, data in Table4 is compiled from 27 affected individuals and includes 8 de novo cases (Guerrini et al. 2007; Poirier et al. 2008; Shinozaki et al. 2008; Wallerstein et al. 2008; Absoud et al. 2009; Mirzaa et al. 2013), three cases of brother pairs (Guerrini et al. 2007; Cossee et al. 2011) two cases of families with maternal uncle–nephew pairs, one case of a pair of brothers and a maternal male cousin (Bruyere et al. 1999; Stromme et al. 2002a), and four cases without a family history (Mirzaa et al. 2013). There is only limited reporting of affected females in these families. The family in Cossee et al. (2011) lists a sister of the proband, with mild ID and epilepsy but no indication of age of onset or the type of seizure or movement disorder.
Table 4.
Clinical features of patients with an ARX mutation leading to expansion of polyalanine tract 1 to 23 alanines
| Clinical data | Previous studies (frequency) families (27 males)# | This study family A (male, 2.5 years) | Total % (frequency) |
|---|---|---|---|
| DD/ID | 100% (27/27) | + | 100% (28/28) |
| Age at diagnosis (median, IQR) | Onset 0–7 (median 3; range 2.25 m) (data from 9/27) | 8 weeks | Onset 0–7 (median 2; range 2 m) (data from 10/28) |
| Formal development evaluation (IQ, median age of evaluation in years) | 1/27 mild, 15/27 severe, 2/27 Profound, DD (median 3 Y) | Profound DD | 1/28 mild, 15/28 severe, 3/28 Profound (median 2.5 Y) |
| DD onset prior to seizure onset (age at diagnosis) | 22% (6/27) (median age 2 m) | + | 25% (7/28) |
| Developmental regression with seizure onset | 22% (6/27) | + | 25% (7/28) |
| Evaluation of MRI | Data for 14/27 patients | Data for 15/28 patients | |
| MRI abnormalities | |||
| Basal ganglia2 (age at diagnosis) | 14% (2/14) (median age 5.5 Y) | 13% (2/15) | |
| Lateral ventricles3 (age at diagnosis) | 21% (3/14) (median age 5 Y) | 20% (3/15) | |
| Brain atrophy (age at diagnosis) | 21% (3/14) (median age 7 m) | 20% (3/15) | |
| Delayed myelination (age at diagnosis) | 7% (1/14) (4 m) | 7% (1/15) | |
| No abnormalities (age at MRI) | 50% (7/14) (age not supplied) | 5 m – Neuroimage normal | 53% (8/15) |
| Epilepsy | 100% (27/27) | + | 100% (28/28) |
| Age at diagnosis (median, IQR) | Median 3.75 m (Onset 0–18 m:2 m) | 4 m | Median 4 m |
| Initial phenotype | |||
| IS (age at diagnosis) | 85% (23/27) (median 4, 1.95; m) | + | 85% (24/28) |
| Ohtahara (age at diagnosis) | 4% (1/27) | 4% (1/28) | |
| Other – tonic clonic (age at diagnosis) | 15% (4/27) (median 4.5, 8; m) | 14% (4/28) | |
| Later phenotype(s) | No ongoing seizures at 2.5Y | ||
| Focal seizures | 7% (2/27) | 7% (2/28) | |
| Myoclonic jerks (age at diagnosis) | 15% (4/27) (median 2.5, 3.25 Y) | 14% (4/28) | |
| Tonic spasms, tonic clonic, and frontal lobe epilepsy (age at diagnosis) | 22% (6/27) (median 4, 3.3 Y) | 21% (6/28) | |
| Treatments reported to be of benefit | ACTH, vigabatrin, phenytonin, phentobarbitol, felbamate, zonisamide, sodium valporate | Topiramate | |
| Movement disorders | 93% (25/27)* | + | 93% (26/28) |
| Age at diagnosis (median, IQR) | 15/27 reported age of onset 2 to 11 months (5 m, 1.5 m) | 7 m | |
| Type (age at diagnosis often not reported) | |||
| Dystonia | 52% (13/25) | + (report details in the text) | 53% (14/26) |
| Chorea | 28% (7/25) | + | 30% (8/26) |
| Dysphagia | 4% (1/25) | 4% (1/26) | |
| Episodes of status dystonicus | 12% (3/25) | + | 15% (4/26) |
| Abnormalities of tone | 64% (16/25) | 61% (16/26) | |
| Hypotonia | 88% (14/16) | + | 88% (15/17) |
| Spasticity | 43% (7/16) | − | 41% 7/17) |
| Family history | Maternally inherited | ||
Basal ganglia: cavitated, fragmented, indistinct, small, or normal (Kato et al, 2004).
Lateral ventricles: mildly to moderately enlarged lateral ventricles, sometimes in continuity with an interhemispheric fluid space (Kato et al, 2004).
Remaining two patients are deceased and complete clinical assessments were not undertaken or reported.
8 de novo cases.
Variants in Families B, C, D, E, and F are classified as variants of uncertain clinical significance (VUS). There are reports of triplet expansions increasing the pA1 tract by one, two, and three residues (Bienvenu et al. 2002; Gronskov et al. 2004; Oegema et al. 2012). The pathogenicity of these shorter expansions is not well established. In this study, there are four cases of diminishing size of duplicating/insertion GGC triplet repeats, with a novel four GCG triplet repeat: c.306GGC[14], leading to a pA1 tract of 20 alanines, one case of a three GGC triplet repeat: c.306GGC[13], and two cases of a single addition of a GCG triplet repeat: c.306GGC[10];[11] and: c.306GGC[11]. The last case describes a 12 bp deletion: c.306GGC[6] leading to a shorter pA1 tract of 12 alanines.
Polyalanine tract 2 variants
The most prevalent ARX mutation, a 24 bp duplication in exon 2 leading to expansion of pA2 to 20 alanines, c.441_464dup (herein named as dup24), was identified in eight patients, including one female, belonging to six distinct families (Families G to L) (Table S2). Interestingly, in the two brothers of Family H a recognizable episodic dystonic movement of the hands was identified in a retrospective revaluation (at 16 and 11 years old) allowing the diagnosis of the Partington-like syndrome (Fig. S3). Variants that are unlikely to be, or are certainly not pathogenic were identified in the remaining families with a pA2 variant (Families M to Q) (as shown in Table3). In cases of families M to P the same nucleotide region within pA2 was deleted. This deletion is classified as a variant that is unlikely to negatively influence ARX function. In Family Q we report a novel pA2 deletion of 15 base pairs, c.441_455del;[=] resulting in a contraction of five alanines in tract pA2 of ARX. Segregation studies in this family revealed that two healthy male relatives inherit the same variant. Analyses of 200 control chromosomes of healthy Portuguese controls did not detect this novel variant indicating that this contraction represents a very rare, benign polymorphism, and is not pathogenic.
Discussion
Molecular analysis of ARX identified sequence variants in 17 families. There were seven families harboring disease-causing mutations, four families had unlikely pathogenic variants and in the remaining five families a variant of unclear/unclassified pathogenic significance (VUS) was identified, including one novel variant. Expansions of the first two polyalanine tracts in ARX account for more than half (56%) of all mutations previously reported in this gene (Shoubridge et al. 2010). Our strategy in the main (127/138 patients) was based on screening exon 2 by size-variant analysis (PCR and gel electrophoresis or multiplex analysis) followed by sequence analysis where indicated. Thirty of the 138 patients underwent sequencing of the entire ARX coding regions, many after a negative outcome by size-variant analysis. Of the 8 variants identified, 5 ARX variants were detected using multiplex screening that enables the simultaneous investigation of FMR1, AFF2, and ARX genes (Jorge et al. 2013) and three ARX variants were detected by direct sequencing of the gene. Despite these different approaches, all ARX variants identified in this study were located in exon 2 of the gene, specifically in pA1 and pA2. In our previous study, of the eight pathogenic variants identified only three were outside the pA tracts in a cohort of 613 mostly isolated cases, 500 of which had the entire open reading frame sequenced (Fullston et al. 2011). Hence, we predict the likelihood of missing a mutation elsewhere in the gene in the patients screened only by size-variant analysis would be quite low. From 138 patients we identified 7 known pathogenic variants, an overall detection rate of 5%. It remains challenging to assess the true prevalence of ARX mutations given the potential ascertainment bias. Here, we report a small number of mostly isolated cases and some brother pairs with a rate higher than the 1.3–1.5% from isolated cases (de Brouwer et al. 2007; Fullston et al. 2011), and 2.2% from brother pairs (Poirier et al. 2006) but substantially less than the 9.5% reported when screening large XLID families (Poirier et al. 2006).
ARX polyalanine tract expansion mutations of the same size and composition can cause a range of overlapping, but distinct clinical phenotypes (Fig.1). Each reported case is depicted as a single circle on the graph against the length of the tract involved. Some circles represent a single individual, whereas others represent multiple affected individuals within a family. For familial cases, the prevailing phenotype reported was used to designate the position on the axis of Figure1. This unusual breadth of variation in the clinical presentations, intra and interfamilial variability, associated with the frequent expansion mutations in pA1 and pA2 in particular has been noticed, but not adequately explained (Turner et al. 2002; Shoubridge et al. 2010). The clinical features of each of these cases are summarized in Table4 and Table S2, respectively.
Figure 1.
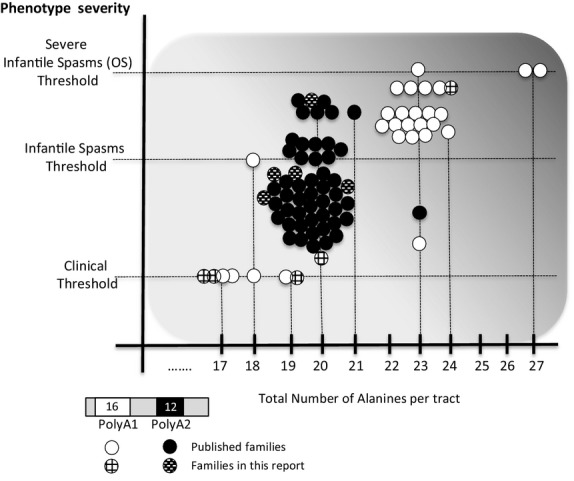
Clinical variability associated with expanded in polyalanine tract mutations in ARX. The panel below the graph shows the normal length of the pA1 as 16 residues and pA2 as 12 residues. The graph shows the relationship between the number of alanines and the phenotype severity in published families with pA1 (white circles) and pA2 families (black circles). Each circle represents a separate published case; some cases being a single affected individual whereas other cases are comprised of multiple affected individuals within a family. The black circle with horizontal line indicates a polyalanine tract expansion interspersed with a glycine residue (10A-G-12A) (Demos et al. 2009). The cases we are reporting in this study are shown as hatched circles. The same mutation can lead to different clinical outcomes, whereas different mutations can lead to consistent outcomes. With increasing length of residues in the pA tracts, the clinical presentation becomes more severe, with early onset seizures being a frequent but not consistent finding. Modified with permission from (Shoubridge et al. 2014).
The most frequent mutation in pA1 expands the 16 reside tract to 23 alanines. To date there are 18 families reported (across 27 individuals), increasing to 19 families (and 28 individuals) with our report of Family A. This particular mutation is associated with severe clinical outcomes ranging from generalized dystonia in the absence of infantile spasms (Shinozaki et al. 2008), to the more common X-linked infantile spasms (Stromme et al. 2002a; Wohlrab et al. 2005; Guerrini et al. 2007; Poirier et al. 2008; Cossee et al. 2011; Mirzaa et al. 2013), through to infantile epileptic-dyskinetic encephalopathy (Reish et al. 2009) and Ohtahara syndrome (Demos et al. 2009) (Table4). The age of onset and severity of the phenotype also varies; with reports of rapid neurodegeneration and death within the first year of life (Absoud et al. 2009). An added complexity is the overlapping clinical spectrum with mutations in other regions of ARX, with a number of patients reported with the severe early infantile encephalopathy of Ohtahara syndrome due to mutations outside of expansions to pA1 and pA2. These mutations include early truncating mutations (suggested to re-initiate translation at a subsequent methionine) (Fullston et al. 2009) and missense and nonsense mutations in the Aristaless domain (Giordano et al. 2010; Kato et al. 2010; Eksioglu et al. 2011; Sartori et al. 2011; Bettella et al. 2012). This highlights the need for detailed case reports of patients with expanded polyalanine tract mutations (and other mutations) in ARX to better identify the key clinical features associated with these infantile spasms.
The most frequent pA2 mutation expands the 12 reside tract to 20 alanines. To date there are 46 cases/families reported with this mutation, increasing to 52 cases with our report of Families G to L. Clinical outcomes always include mild to moderate ID either as the only consistent clinical feature, or in association with dystonia, particularly of the hands known as Partington syndrome, though to infantile spasms or West syndrome (Shoubridge et al. 2014). No syndromic features were recognized at the first referral in 13 of 17 patients, identified with ARX mutations. A retrospective detailed evaluation of some cases with the dup24 mutation revealed features of an “ARX-syndrome.” Cognitive profile evaluation in three patients with ID, and subsequent clinical assessment of a maternal male relative also with ID, enabled identification of hand dystonia in each male patient. Thus, identification of the dup24 pathogenic mutation was essential for the diagnosis of Partington syndrome and for appropriate genetic counseling.
In addition to the well-established pathogenic expansions to both tracts there are several examples of triplet expansions increasing the pA1 tract by one, two, and three residues (Bienvenu et al. 2002; Gronskov et al. 2004; Oegema et al. 2012). The pathogenicity of these shorter expansions is not well established. In this study, we identified triplet repeat expansion increasing pA1 by one (Family D and E), three (Family C), and a novel 4 residues (Family B). In this last case, the novel mutation in pA1 expands the first tract to 20 alanines. This precise number of alanines is sufficient to cause pathogenicity when expanded in pA2. Hence, it is difficult to predict the pathogenic impact of this expansion in pA1. In the absence of functional studies to determine the in vivo pathogenic status, haplotype analysis, in-silico and population studies are of particular importance. When we examine the genotype–phenotype spectrum associated with all cases of expansions to pA1 and pA2 of ARX we see that our patients generally cluster quite well with previously reported patients with the same mutations (Fig.1). However, there are outliers that do not fit with the genotype–phenotype correlation of a more severe outcome with a longer length of expansion. The novel mutation increasing the pA1 from 16 to 20 alanines in Family B gives rise to a phenotype more severe than seen in many patients with pA2 mutations increasing pA2 to 20 alanines in length. Although the authors have listed this mutation as a variant of unknown significance, additional cases may add weight to the potential pathogenicity of this mutation. Differential diagnoses are being considered for this patient. In the case of Family D, the variant was reported in a female patient. The variant in pA1 was of unknown significance, with the alanine tract increasing from 16 to 17 alanines (Family D). The female proband in Family D was also diagnosed with classic galactosemia, which can explain a mild intellectual impairment even in patients who avoid galactose. There is one other report of this change (Gronskov et al. 2004). Pathogenicity should not be ruled out in the variant c.331_333dupGCG, nor in others that cause a small increase in alanine residues of pA1 with further evidence needed to clarify the pathogenicity of small increases to alanine content in this tract.
There were three other cases in which females were identified with an ARX variant, this time in pA2 (Family L, P and Q). The female in Family L heterozygote for dup24 was lost to follow-up. The remaining two female cases had deletions in pA2. Deletions in pA2 are difficult to interpret in terms of pathogenicity (Shoubridge et al. 2010). The heterozygous c. 441_464del in one case from our cohort (Family P) was considered as a causative mutation in a male patient with nonsyndromic mental retardation previously published (Troester et al. 2007). Troester et al. interpreted the heterozygous deletion in the patient's asymptomatic mother and sister as a result of favorable X-inactivation in the brain. Evidence from several other publications places in frame deletions in exon 2, leading to retraction of pA1 and pA2, in an unlikely pathogenic group, based in part on some of these deletions also being reported in healthy, unaffected males (Bienvenu et al. 2002; Gronskov et al. 2004; Conti et al. 2010; Fullston et al. 2011). In agreement, the c.44a_455del;[=] variant identified in the female proband of Family Q was paternally inherited and did not segregate with cognitive impairment in the family, suggesting a nonpathogenic variant classification. The last case identified a previously unreported, presumed rare polymorphism (HGVS: c.441_455del; [=]) of paternal origin was identified in a heterozygous state in one family (Family Q).
Due to the overlap of phenotypic features in ARX-related and Fragile-X syndromes and family history of X-linked inheritance pattern, patients previously classified as having a normal FMR1 profile by Southern blot irrespective of their phenotypic presentation were subsequently screened for an ARX-related disorder (exon 2 size-variant). Clinical details given by the referring physicians requesting screening for fragile-X syndrome often only relates specifically to fragile-X diagnosis. Consequently, in many cases there is little or no available clinical information specifically referable to ARX-related phenotypes. Given the outcomes of our screening strategy, we recommend that all patients referred for fragile-X (after confirming a normal karyotype), particularly those with a priori nonsyndromic ID or XLID, but also including cases with isolated cognitive impairment, be routinely screened for exon 2 size variants in ARX.
This study aims to establish standard nomenclature for ARX sequence variation description based on the published guidelines of the HGVS. One of the pitfalls in standardization of description of genetic variants concerns the absence of electropherogram visualization, hindering an effective variant comparison. However, variable description of ARX variants, combined with the low prevalence of familial cases and in many cases a lack of functional studies, hampers the establishment of pathogenicity and leads to confusion and misinterpretation, particularly in medical diagnostic settings. Hence, we have collated and compared the nomenclature of the variants to clarify this process and provide a useful resource. We encourage clinical researchers and clinical laboratory specialists to use Locus Specific Databases (LSDBs) (such as LOVD for ARX gene) to continue the process of collecting and describing identified sequence variants (http://databases.lovd.nl/shared/variants/ARX). This can ultimately be utilized to suggest and discuss accurate variant terminology and ensure clarity of variant descriptions essential for proper genetic counseling.
Conclusion
The screening of mutations in the exon 2 of the ARX gene, followed by sequencing of the entire ARX gene, enabled the diagnosis of ARX-related disorders in eight patients from six families, presenting with different clinical phenotypes. Mutations were described using standardized and consistent nomenclature. In addition, several other pA1 expansions including the novel mutation expanding the first pA tract by four alanines in one family remain of unknown clinical significance. Further studies are required to characterize the impact of these variants on the functionality of ARX. Molecular screening of ARX gene is frequent in patients with XLID, ID presenting with infantile epilepsy or movement disorder or brain and genital malformations. Data from this and other studies led us to recommend that molecular screening of exon 2 of the ARX gene be done in all patients referred for fragile-X (after a normal karyotype), particularly those with apparent nonsyndromic ID of unknown cause, or in which the “Partington” hand dystonia is recognized.
Acknowledgments
We thank the patients, their families, and physicians for their participation in this study, also the psychologist Carla Carmona, for enabling and providing the photos of the younger brother from Family H (Fig. S3). Unit for Multidisciplinary Research in Biomedicine, UMIB, ICBAS-UP, Porto, Portugal was funded by FEDER funds of the Operational Program for Competitiveness Factors – COMPETE through FCT – Foundation for Science and Technology under the project: Fcomp-01-0124-FEDER-015896. The Neurogenetics research program in the Department of Paediatrics, University of Adelaide, Australia was funded by the Australian National Health and Medical Research Council (Grant No. 1063025). C. S. is supported Australian Research Council (Future Fellowship FT120100086).
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Supplementary note.
Figure S1.ARX mutations identified.
Figure S2. Differences in regions designated as duplicated in pA2 expansion mutations of ARX. The sequence spanning c.421 and c.471 is shown encompassing pA2 of ARX. The region duplicated is indicated for the existing published nomenclature (underlined and in bold) as compared to the HGVS nomenclature (dotted box) for the 24dup (top panel), 27 bp (middle panel), and the 33 bp duplication (bottom panel).
Figure S3. Block design subtests of WISC-III (Wechsler Scale), performed by the younger brother from Family H, demonstrate his inability to make purposeful hand movements due to lack of manipulative skills, thus suggesting hand dystonia.
Table S1.PCR primers designed to amplify pA1 and pA2 or ARX ORFs.
Table S2. Clinical features of patients with an ARX mutation leading to expansion of polyalanine tract 2 to 20 alanines (dup 24).
References
- Absoud M, Parr JR, Halliday D, Pretorius P, Zaiwalla Z. Jayawant S. A novel ARX phenotype: rapid neurodegeneration with Ohtahara syndrome and a dyskinetic movement disorder. Dev. Med. Child Neurol. 2009;52(3):305–7. doi: 10.1111/j.1469-8749.2009.03470.x. doi: 10.1111/j.1469-8749.2009.03470.x. [DOI] [PubMed] [Google Scholar]
- Bettella E, Di Rosa G, Polli R, Leonardi E, Tortorella G, Sartori S, et al. Early-onset epileptic encephalopathy in a girl carrying a truncating mutation of the ARX gene: rethinking the ARX phenotype in females. Clini. Genet. 2012;84(1):82–5. doi: 10.1111/cge.12034. . doi: 10.1111/cge.12034. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Poirier K, Friocourt G, Bahi N, Beaumont D, Fauchereau F, et al. ARX, a novel prd-class-homeobox gene highly expressed in the telencephalon, is mutated in X-linked mental retardation. Hum. Mol. Genet. 2002;11:981–991. doi: 10.1093/hmg/11.8.981. [DOI] [PubMed] [Google Scholar]
- de Brouwer AP, Yntema HG, Kleefstra T, Lugtenberg D, Oudakker AR, de Vries BB, et al. Mutation frequencies of X-linked mental retardation genes in families from the EuroMRX consortium. Hum. Mutat. 2007;28:207–208. doi: 10.1002/humu.9482. . doi: 10.1002/humu.9482. [DOI] [PubMed] [Google Scholar]
- Bruyere H, Lewis S, Wood S, MacLeod PJ. Langlois S. Confirmation of linkage in X-linked infantile spasms (West syndrome) and refinement of the disease locus to Xp21.3-Xp22.1. Clini. Genet. 1999;55:173–181. doi: 10.1034/j.1399-0004.1999.550305.x. [DOI] [PubMed] [Google Scholar]
- Conti V, Marini C, Mei D, Falchi M, Ferrari AR. Guerrini R. Contractions in the second polyA tract of ARX are rare, non-pathogenic polymorphisms. Am. J. Med. Genet. A. 2010;155A(1):164–7. doi: 10.1002/ajmg.a.33753. , and Jan;. doi: 10.1002/ajmg.a.33753. [DOI] [PubMed] [Google Scholar]
- Cossee M, Faivre L, Philippe C, Hichri H, de Saint-Martin A, Laugel V, et al. ARX polyalanine expansions are highly implicated in familial cases of mental retardation with infantile epilepsy and/or hand dystonia. Am. J. Med. Genet. A. 2011;155A:98–105. doi: 10.1002/ajmg.a.33785. . doi: 10.1002/ajmg.a.33785. [DOI] [PubMed] [Google Scholar]
- Demos MK, Fullston T, Partington MW, Gecz J. Gibson WT. Clinical study of two brothers with a novel 33 bp duplication in the ARX gene. Am. J. Med. Genet. A. 2009;149A:1482–1486. doi: 10.1002/ajmg.a.32851. doi: 10.1002/ajmg.a.32851. [DOI] [PubMed] [Google Scholar]
- den Dunnen JT. Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum. Mutat. 2000;15:7–12. doi: 10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. doi: 10.1002/(SICI)1098-1004(200001)15:1<3c7:AID-HUMU4>3e3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Eksioglu YZ, Pong AW. Takeoka M. A novel mutation in the aristaless domain of the ARX gene leads to Ohtahara syndrome, global developmental delay, and ambiguous genitalia in males and neuropsychiatric disorders in females. Epilepsia. 2011;52:984–992. doi: 10.1111/j.1528-1167.2011.02980.x. doi: 10.1111/j.1528-1167.2011.02980.x. [DOI] [PubMed] [Google Scholar]
- Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF. den Dunnen JT. LOVD v. 2.0: the next generation in gene variant databases. Hum. Mutat. 2011;32:557–563. doi: 10.1002/humu.21438. doi: 10.1002/humu.21438. [DOI] [PubMed] [Google Scholar]
- Fullston T, Brueton L, Willis T, Philip S, Macpherson L, Finnis M, et al. Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X) Eur. J. Hum. Genet. 2009;18(2):157–62. doi: 10.1038/ejhg.2009.139. . doi: 10.1038/ejhg.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullston T, Finnis M, Hackett A, Hodgson B, Brueton L, Baynam G, et al. Screening and cell-based assessment of mutations in the Aristaless-related homeobox (ARX) gene. Clini. Genet. 2011;80(6):510–22. doi: 10.1111/j.1399-0004.2011.01685.x. Dec;. doi: 10.1111/j.1399-0004.2011.01685.x. [DOI] [PubMed] [Google Scholar]
- Giordano L, Sartori S, Russo S, Accorsi P, Galli J, Tiberti A, et al. Familial Ohtahara syndrome due to a novel ARX gene mutation. Am. J. Med. Genet. A. 2010;152A:3133–3137. doi: 10.1002/ajmg.a.33701. . doi: 10.1002/ajmg.a.33701. [DOI] [PubMed] [Google Scholar]
- Gronskov K, Hjalgrim H, Nielsen IM. Brondum-Nielsen K. Screening of the ARX gene in 682 retarded males. Eur. J. Hum. Genet. 2004;12:701–705. doi: 10.1038/sj.ejhg.5201222. doi: 10.1038/sj.ejhg.5201222. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Moro F, Kato M, Barkovich AJ, Shiihara T, McShane MA, et al. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology. 2007;69:427–433. doi: 10.1212/01.wnl.0000266594.16202.c1. [DOI] [PubMed] [Google Scholar]
- Jorge P, Oliveira B, Marques I. Santos R. Development and validation of a multiplex-PCR assay for X-linked intellectual disability. BMC Med. Genet. 2013;14:80. doi: 10.1186/1471-2350-14-80. doi: 10.1186/1471-2350-14-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, et al. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome) Am. J. Hum. Genet. 2007;81:361–366. doi: 10.1086/518903. . doi: 10.1111/j.1528-1167.2010.02559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Koyama N, Ohta M, Miura K. Hayasaka K. Frameshift mutations of the ARX gene in familial Ohtahara syndrome. Epilepsia. 2010;51:1679–1684. doi: 10.1111/j.1528-1167.2010.02559.x. doi: 10.1111/j.1528-1167.2010.02559.x. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 2002;32:359–369. doi: 10.1038/ng1009. . doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Itou Y, Yanazawa M, Ohsawa M, Suzuki-Migishima R, Umeki Y, et al. Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Hum. Mol. Genet. 2009;18:3708–3724. doi: 10.1093/hmg/ddp318. . doi: 10.1093/hmg/ddp318. [DOI] [PubMed] [Google Scholar]
- Kuwaik GA, Saldivar JS. Yoon G. Novel mutation in ARX associated with early hand preference and a mild phenotype. J. Dev. Behav. Pediatr. 2012;33:586–588. doi: 10.1097/DBP.0b013e31825e2310. doi: 10.1097/DBP.0b013e31825e2310. [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Paciorkowski AR, Marsh ED, Berry-Kravis EM, Medne L, Alkhateeb A, et al. CDKL5 and ARX mutations in males with early-onset epilepsy. Pediatr. Neurol. 2013;48:367–377. doi: 10.1016/j.pediatrneurol.2012.12.030. . doi: 10.1016/j.pediatrneurol.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Yanazawa M, Kato K. Kitamura K. Expression of a novel aristaless related homeobox gene ‘Arx’ in the vertebrate telencephalon, diencephalon and floor plate. Mech. Dev. 1997;65:99–109. doi: 10.1016/s0925-4773(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Oegema R, Maat-Kievit A, Lequin MH, Schot R, Nanninga-van den Neste VM, Doornbos ME, et al. Asymmetric polymicrogyria and periventricular nodular heterotopia due to mutation in ARX. Am. J. Med. Genet. A. 2012;158A:1472–1476. doi: 10.1002/ajmg.a.35365. . doi: 10.1002/ajmg.a.35365. [DOI] [PubMed] [Google Scholar]
- Partington MW, Turner G, Boyle J. Gecz J. Three new families with X-linked mental retardation caused by the 428-451dup(24 bp) mutation in ARX. Clin. Genet. 2004;66:39–45. doi: 10.1111/j.0009-9163.2004.00268.x. doi: 10.1111/j.0009-9163.2004.00268.x. [DOI] [PubMed] [Google Scholar]
- Poirier K, Lacombe D, Gilbert-Dussardier B, Raynaud M, Desportes V, de Brouwer AP, et al. Screening of ARX in mental retardation families: consequences for the strategy of molecular diagnosis. Neurogenetics. 2006;7:39–46. doi: 10.1007/s10048-005-0014-0. . doi: 10.1007/s10048-005-0014-0. [DOI] [PubMed] [Google Scholar]
- Poirier K, Eisermann M, Caubel I, Kaminska A, Peudonnier S, Boddaert N, et al. Combination of infantile spasms, non-epileptic seizures and complex movement disorder: a new case of ARX-related epilepsy. Epilepsy Res. 2008;80:224–228. doi: 10.1016/j.eplepsyres.2008.03.019. . doi: 10.1016/j.eplepsyres.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Proud VK, Levine C. Carpenter NJ. New X-linked syndrome with seizures, acquired micrencephaly, and agenesis of the corpus callosum. Am. J. Med. Genet. 1992;43:458–466. doi: 10.1002/ajmg.1320430169. [DOI] [PubMed] [Google Scholar]
- Reish O, Fullston T, Regev M, Heyman E. Gecz J. A novel de novo 27 bp duplication of the ARX gene, resulting from postzygotic mosaicism and leading to three severely affected males in two generations. Am. J. Med. Genet. A. 2009;149A:1655–1660. doi: 10.1002/ajmg.a.32842. doi: 10.1002/ajmg.a.32842. [DOI] [PubMed] [Google Scholar]
- Sartori S, Polli R, Bettella E, Rossato S, Andreoli W, Vecchi M, et al. Pathogenic role of the X-linked cyclin-dependent kinase-like 5 and aristaless-related homeobox genes in epileptic encephalopathy of unknown etiology with onset in the first year of life. J. Child Neurol. 2011;26:683–691. doi: 10.1177/0883073810387827. . doi: 10.1177/0883073810387827. [DOI] [PubMed] [Google Scholar]
- Shinozaki Y, Osawa M, Sakuma H, Komaki H, Nakagawa E, Sugai K, et al. Expansion of the first polyalanine tract of the ARX gene in a boy presenting with generalized dystonia in the absence of infantile spasms. Brain Dev. 2008;31(6):469–72. doi: 10.1016/j.braindev.2008.08.006. . doi: 10.1016/j.braindev.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Shoubridge C, Fullston T. Gecz J. ARX spectrum disorders: making inroads into the molecular pathology. Hum. Mutat. 2010;31:889–900. doi: 10.1002/humu.21288. doi: 10.1002/humu.21288. [DOI] [PubMed] [Google Scholar]
- Shoubridge CLR, Field M. Gecz J. Developmental abnormalities due to mutations in the Aristaless-related homeobox gene. In: H. Brunner IKrantz., editor; Epstein's inborn errors of development: the molecular basis of clinical disorders of morphogenesis (IED) 3rd ed. New York: Oxford University Press; 2014. [Google Scholar]
- Stromme P, Mangelsdorf ME, Shaw MA, Lower KM, Lewis SM, Bruyere H, et al. Mutations in the human ortholog of aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 2002a;30:441–445. doi: 10.1038/ng862. . doi: 10.1038/ng862. [DOI] [PubMed] [Google Scholar]
- Stromme P, Mangelsdorf ME, Scheffer IE. Gecz J. Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev. 2002b;24:266–268. doi: 10.1016/s0387-7604(02)00079-7. doi: 10.1016/S0387-7604(02)00079-7. [DOI] [PubMed] [Google Scholar]
- Stromme P, Bakke SJ, Dahl A. Gecz J. Brain cysts associated with mutation in the Aristaless related homeobox gene, ARX. J. Neurol. Neurosurg. Psychiatry. 2003;74:536–538. doi: 10.1136/jnnp.74.4.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Gecz J. Shoubridge C. PCR amplification and sequence analysis of GC-rich sequences: Aristaless-related homeobox example. Methods Mol. Biol. 2013;1017:105–120. doi: 10.1007/978-1-62703-438-8_8. doi: 10.1007/978-1-62703-438-8_8. [DOI] [PubMed] [Google Scholar]
- Troester MM, Trachtenberg T. Narayanan V. A novel mutation of the ARX gene in a male with nonsyndromic mental retardation. J. Child Neurol. 2007;22:744–748. doi: 10.1177/0883073807304000. [DOI] [PubMed] [Google Scholar]
- Turner G, Partington M, Kerr B, Mangelsdorf M. Gecz J. Variable expression of mental retardation, autism, seizures, and dystonic hand movements in two families with an identical ARX gene mutation. Am. J. Med. Genet. 2002;112:405–411. doi: 10.1002/ajmg.10714. doi: 10.1002/ajmg.10714. [DOI] [PubMed] [Google Scholar]
- Wallerstein R, Sugalski R, Cohn L, Jawetz R. Friez M. Expansion of the ARX spectrum. Clin. Neurol. Neurosurg. 2008;110:631–634. doi: 10.1016/j.clineuro.2008.03.007. doi: 10.1016/j.clineuro.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Wohlrab G, Uyanik G, Gross C, Hehr U, Winkler J, Schmitt B, et al. Familial West syndrome and dystonia caused by an Aristaless related homeobox gene mutation. Eur. J. Pediatry. 2005;164:326–328. doi: 10.1007/s00431-005-1622-2. . doi: 10.1007/s00431-005-1622-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary note.
Figure S1.ARX mutations identified.
Figure S2. Differences in regions designated as duplicated in pA2 expansion mutations of ARX. The sequence spanning c.421 and c.471 is shown encompassing pA2 of ARX. The region duplicated is indicated for the existing published nomenclature (underlined and in bold) as compared to the HGVS nomenclature (dotted box) for the 24dup (top panel), 27 bp (middle panel), and the 33 bp duplication (bottom panel).
Figure S3. Block design subtests of WISC-III (Wechsler Scale), performed by the younger brother from Family H, demonstrate his inability to make purposeful hand movements due to lack of manipulative skills, thus suggesting hand dystonia.
Table S1.PCR primers designed to amplify pA1 and pA2 or ARX ORFs.
Table S2. Clinical features of patients with an ARX mutation leading to expansion of polyalanine tract 2 to 20 alanines (dup 24).