

Abstract
The accompanying articles in this issue of the journal’s special collection describe attempts to improve on the dynamics of distribution and reduce side effects of analogs of etomidate and benzodiazepines. Both classes of drugs have their principal sites of action on gamma-aminobutyric acid type A receptors, although at very different binding sites and by different mechanisms of action. Herein, we review the structure of gamma-aminobutyric acid type A receptors and describe the location of the 2 likely binding sites. In addition, we describe how these drugs can interact with the nervous system at a systems level. We leave it to other reviewers to discuss whether these new drugs offer true clinical improvements.
Keywords: GABA, GABAaR, Etomidate
Recent bioinformatics studies and radiograph structural analysis have revealed that the highly conserved heteropentameric structure of the gamma-aminobutyric acid type A receptors (GABAaRs) has its roots in proton-gated ion channels found in prokaryotic bacteria.1,2 Herein, we review how this ancient GABAaR structure deals with new drugs; in particular, the new etomidate analogs3,4 and remimazolam5,6 presented in the accompanying articles, by examining what is already known about etomidate and benzodiazepines.
We consider these new drugs from 2 perspectives: (1) where they could bind in a molecular model of GABAaRs to affect the function of the receptor, and (2) where they could affect a systems-level representation of the many aspects of GABAaRs in the nervous system. Although both the etomidate analogs3,4 and remimazolam5,6 have their principal effects on the GABAaR, they bind in distinctly different sites (Fig. 1): between the alpha and beta subunits in the transmembrane domain in the case of etomidate and its analogs. In contrast, remimazolam and the remainder of the benzodiazepine family have a conserved binding site that is only found at the interface between alpha and gamma subunits and is in the ligand-binding domain. Moreover, it is important to note that the manuscripts in this collection on new GABAergic drugs were selected because of the drugs’ new properties of rapid kinetics. However, they are in fundamentally different drug families. That is, etomidate could be used as a general anesthetic whereas drugs in the benzodiazepine family are primarily used as sedatives and anxiolytics.3–6
Figure 1.
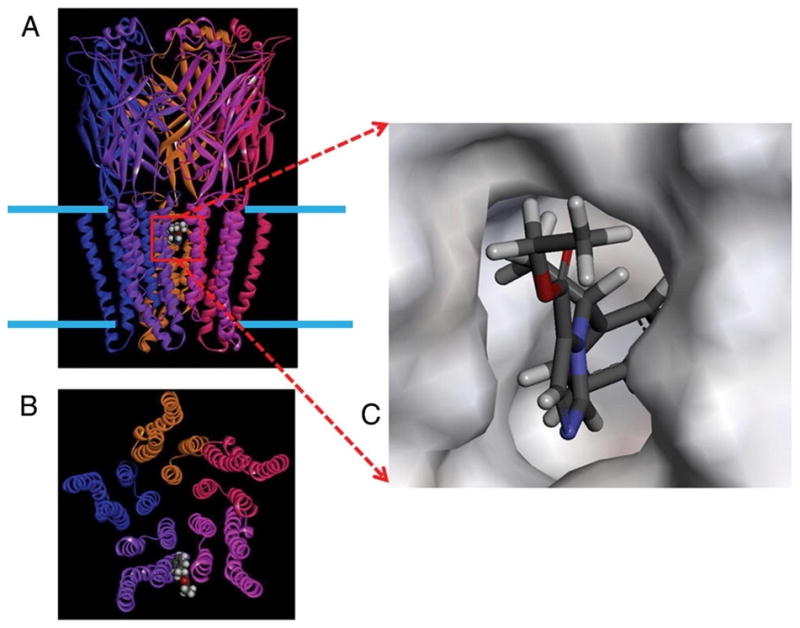
A homology model of the γ-aminobutyric acid type A receptor (GABAaR) built by threading the GABAaR sequence onto the radiograph structure of the homologous glutamate chloride channel template from Caenorhabditis elegans.18 A, The 5 subunits are viewed from the plane of the membrane (blue bars) and rendered as ribbons in distinct colors. A single molecule of etomidate (space filling surface) is docked to demonstrate the convergence of residues known to alter etomidate modulation of GABA-induced currents onto a common binding site. This site lies within the outer third of the transmembrane domain of the GABAaR and is located between subunits. B, The same model viewed down the axis of the ion pore from the extracellular side. C, The etomidate binding site with the etomidate molecule rendered as sticks (white, red, gray, and blue represent hydrogen, oxygen, carbon, and nitrogen). The “walls” of the binding cavity are rendered as a solid surface representing the van der Waals surface of the surrounding amino acid side chains.
The location of the benzodiazepine binding site and the clockwise gamma/alpha/beta/alpha/beta orientation (as viewed down the ion pore from the extracellular side) was finally confirmed by homology models7,8 based on the radiograph structure of the acetylcholine binding protein.9 Although the general location of the benzodiazepine binding site has been clear for a long time, the distinct binding site for etomidate has only recently been elucidated by a combination of mutagenesis and photolabeling.10,11 Benzodiazepines have differential effects depending on different affinity/efficacy to GABAaR subtypes. Although these differences in subunit composition will allow for advances in design of specific new drugs, they result in a complex pharmacology. This pharmacology and the development of new analogs for action at GABAaRs were recently reviewed.12
The somewhat complex pharmacology of etomidate and its analogs has been recently reviewed.13 The most notable demand from etomidate analogs is avoidance of prolonged adrenocortical suppression.13 This need has been addressed with the 2 new etomidate analogs described in this issue of the journal.3,4
A readily titratable state characterized by various levels of amnesia and loss of consciousness can be produced by many current medications such as benzodiazepines, barbiturates, volatile anesthetics, propofol, and etomidate. A significant proportion of their mechanism of action is mediated via effects at ligand-gated ion channels, notably the GABAaR. Although etomidate may have multiple effects at synapses containing GABAaRs, current work has focused on the enhancing effect that etomidate binding has on the modulation of GABA-induced chloride currents. Specifically, Belelli et al.10 have demonstrated that the mutation of a single amino acid residue within the transmembrane region of the beta3 subunit potently affects the ability of etomidate to enhance such currents. They, as well as Tomlin et al.,14 have demonstrated that this effect is also stereospecific at the GABAaR, a phenomenon that closely parallels the stereospecific clinical effects of the drug.15 Jurd et al.16,17 have taken this one step further to demonstrate that the single point mutation introduced into a knock-in mouse produces a nearly normal animal whose responses to propofol and etomidate are virtually obliterated. Molecular modeling of the GABAaR, based on the recent homologous GluCl template from Caenorhabditis elegans,18 in a manner similar to our previous work,19,20 clearly demonstrates the convergence of residues known to alter etomidate modulation of GABA-induced currents onto a common binding site. This site lies within the outer third of the transmembrane domain of the GABAaR (Fig. 1) and is located between subunits. This enhancing site is distinct from the inhibitory modulating site, which has been shown to exist within the transmembrane helical bundle of an individual subunit within the homologous prokaryotic GLIC protein. Molecular docking calculations clearly show the ways in which etomidate may bind to the GABAaR and lead us closer to a molecular understanding of the mechanism of anesthetic action.20
Etomidate Acts at Multiple Sites on GABAaR Synapses
Etomidate’s overall effect at GABAaR synapses is to enhance inhibition of postsynaptic neurons, depressing the neuron’s ability to discharge action potentials, leading to central nervous system depression associated with sedation and anesthesia. Similar to other general anesthetics, etomidate seems to act on several proteins located on both pre- and postsynaptic membranes. Thus, its mechanism(s) of action will be target protein–specific. GABAaRs located on postsynaptic membranes are major etomidate target proteins.21 Through a combination of actions produced by etomidate, these receptors respond more strongly to stimulation by GABA, resulting in a larger amount of chloride flux through the channels these receptors form through the membrane. Etomidate also seems capable of directly activating some GABAaRs, in the absence of GABA, to increase chloride currents in postsynaptic neurons.22 Increased chloride currents inhibit postsynaptic neurons by shunting excitatory currents and hyperpolarizing the membrane potential. Enhanced GABAaR/channel function comes about by at least 4 actions of etomidate: (1) enhanced affinity at the GABA binding site, (2) enhanced channel opening, (3) enhanced conductance, and (4) enhanced modulation (Fig. 2). It is possible that all of these actions come about when etomidate docks to a single binding site on the GABAaR/channel complex or additional sites may also be involved. For example, etomidate could bind to a single site and propagate actions through allosteric changes in protein 3-dimensional structure in multiple directions. It remains to be determined whether the etomidate binding sites involve protein pockets, or interfacial pockets that also include membrane lipid, and/or water. In any case, it seems that other protein targets also bind etomidate, and actions on these proteins contribute to neuronal depression. For example, etomidate and other general anesthetics can inhibit GABA uptake into neurons and glia, thus increasing GABA concentrations that are able to act more strongly on postsynaptic GABAaRs.23 This may be particularly important for extrasynaptic GABAaRs that mediate tonic inhibition through long-lasting chloride conductances.24 Also, by acting on presynaptic GABAaRs, etomidate seems to limit its own effects, because this presynaptic action results in a decreased release of GABA from interneurons. Interneurons appear to have at least 2 kinds of GABAaRs, because etomidate inhibits both action potential–dependent GABA release, by inhibiting discharge activity at the cell body, and also by directly depressing release at inhibitory interneuron presynaptic nerve terminals, that also express GABAaRs.
Figure 2.

Etomidate acts at several sites to enhance γ-aminobutyric acid type A receptor (GABAaR) synapse function, resulting in stronger neuronal inhibition. A GABAaR synapse is represented as a stylized set of gears, all of which work together to inhibit postsynaptic neurons. Etomidate acts at some sites to enhance function (represented by an oil can) or to inhibit function (represented by a monkey wrench). By inhibiting interneuron excitability and GABA release, etomidate has a self-limiting effect on GABAaR synapses, because less GABA is available to bind with receptors. However, etomidate also blocks uptake of GABA, so any GABA that is released can act longer and stronger; etomidate also enhances GABA binding by increasing receptor affinity for its agonist. Several additional actions combine to enhance inhibition at GABAaR synapses. Etomidate enhances the ability of other channel modulators to increase chloride flux, such as the benzodiazepines and steroids. In addition, both channel opening and channel conductance can be increased by etomidate, whereas the efficacy of channel blockers is reduced. The net effect produced by etomidate is a 2- to 4-fold increase of inhibition at GABAaR synapses.
Etomidate does not treat all GABAaRs equally and receptors associated with different kinds of GABAaR synapses seem to contribute to different therapeutic end points for this anesthetic. For example, etomidate acts at beta3 subunit–containing receptors located at GABAAslow synapses much more strongly than on GABAAfast receptors.25 This result could account for some of etomidate’s effects on lower-frequency electroencephalogram rhythms (i.e., delta and theta oscillations) versus higher-frequency activity (i.e., gamma oscillations). Similarly, etomidate acts on alpha5–containing receptors forming “tonic” GABAaRs that contribute to the amnesic effects produced by etomidate, but not to the sedative or immobilizing effects produced by this anesthetic.26 The extent to which etomidate enhances GABA inhibition to produce immobility remains to be determined. Clearly, etomidate (and other general anesthetics) act at GABAaRs to produce some of the important clinical effects of this sedative, but there is also evidence for etomidate actions on other ion channels and signaling pathways that will likely also prove important for loss of recall, sedation, loss of consciousness, and surgical immobility.22
Of course, in the future, we will have to incorporate any new findings about the etomidate analogs3,4 and remimazolam5,6 into both the molecular models and the system level models. There might be some surprises.
Footnotes
Disclosures: None
References
- 1.Bocquet N, Prado de Carvalho L, Cartaud J, Neyton J, Le Poupon C, Taly A, Grutter T, Changeux JP, Corringer PJ. A prokaryotic proton-gated ion channel from the nicotinic acetylcholine receptor family. Nature. 2007;445:116–9. doi: 10.1038/nature05371. [DOI] [PubMed] [Google Scholar]
- 2.Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–31. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 3.Pejo E, Cotten JF, Kelly EW, Ge RL, Cuny GD, Laha JK, Liu J, Lin XJ, Raines DE. In vivo and in vitro pharmacological studies of methoxycarbonyl-carboetomidate. Anesth Analg. 2012;115:297–304. doi: 10.1213/ANE.0b013e3182320559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ge RL, Pejo E, Haburcak M, Husain SS, Forman SA, Raines DE. Pharmacological studies of methoxycarbonyl etomidate’s carboxylic acid metabolite. Anesth Analg. 2012;115:305–308. doi: 10.1213/ANE.0b013e318239c6ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiltshire HR, Kilpatrick GJ, Tilbrook GS, Borkett KM. A placebo- and midazolam-controlled phase I single ascending dose study evaluating the safety, pharmacokinetics and pharmacodynamics of remimazolam (CNS 7056): Part II. Population pharmacokinetic and pharmacodynamic modeling and simulation. Anesth Analg. 2012;115:284–96. doi: 10.1213/ANE.0b013e318241f68a. [DOI] [PubMed] [Google Scholar]
- 6.Antonik LJ, Goldwater DR, Kilpatrick GJ, Tilbrook GS, Borkett KM. A placebo- and midazolam-controlled, phase I, single ascending dose study evaluating the safety, pharmacokinetics and pharmacodynamics of remimazolam (CNS 7056): Part I. Safety, efficacy and basic pharmacokinetics. Anesth Analg. 2012;115:274–83. doi: 10.1213/ANE.0b013e31823f0c28. [DOI] [PubMed] [Google Scholar]
- 7.Trudell JR. Unique assignment of inter-subunit association in GABAa alpha1 beta3 gamma2 receptors determined by molecular modeling. Biochim Biophys Acta. 2002;1565:91–6. doi: 10.1016/s0005-2736(02)00512-6. [DOI] [PubMed] [Google Scholar]
- 8.Cromer BA, Morton CJ, Parker MW. Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–7. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- 9.Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der OJ, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–76. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 10.Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci U S A. 1997;94:11031–6. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov. 2011;10:685–97. doi: 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forman SA. Clinical and molecular pharmacology of etomidate. Anesthesiology. 2011;114:695–707. doi: 10.1097/ALN.0b013e3181ff72b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomlin SL, Jenkins A, Lieb WR, Franks NP. Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A receptors and animals. Anesthesiology. 1998;88:708–17. doi: 10.1097/00000542-199803000-00022. [DOI] [PubMed] [Google Scholar]
- 15.Belelli D, Muntoni AL, Merrywest SD, Gentet LJ, Casula A, Callachan H, Madau P, Gemmell DK, Hamilton NM, Lambert JJ, Sillar KT, Peters JA. The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology. 2003;45:57–71. doi: 10.1016/s0028-3908(03)00144-8. [DOI] [PubMed] [Google Scholar]
- 16.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–2. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- 17.Siegwart R, Jurd R, Rudolph U. Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3) gamma(2)gamma-aminobutyric acid(A) receptors. J Neurochem. 2002;80:140–8. doi: 10.1046/j.0022-3042.2001.00682.x. [DOI] [PubMed] [Google Scholar]
- 18.Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murail S, Wallner B, Trudell JR, Bertaccini E, Lindahl E. Microsecond simulations indicate that ethanol binds between subunits and could stabilize an open-state model of a glycine receptor. Biophys J. 2011;100:1642–50. doi: 10.1016/j.bpj.2011.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertaccini EJ, Wallner B, Trudell JR, Lindahl E. Modeling anesthetic binding sites within the glycine alpha one receptor based on prokaryotic ion channel templates: the problem with TM4. J Chem Inf Model. 2010;50:2248–55. doi: 10.1021/ci100266c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanelian DL, Kosek P, Mody I, MacIver MB. The role of the GABAA receptor/chloride channel complex in anesthesia. Anesthesiology. 1993;78:757–76. doi: 10.1097/00000542-199304000-00020. [DOI] [PubMed] [Google Scholar]
- 22.Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–10. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Mantz J, Lecharny JB, Laudenbach V, Henzel D, Peytavin G, Desmonts JM. Anesthetics affect the uptake but not the depolarization-evoked release of GABA in rat striatal synaptosomes. Anesthesiology. 1995;82:502–11. doi: 10.1097/00000542-199502000-00020. [DOI] [PubMed] [Google Scholar]
- 24.Bieda MC, Su H, Maciver MB. Anesthetics discriminate between tonic and phasic gamma-aminobutyric acid receptors on hippocampal CA1 neurons. Anesth Analg. 2009;108:484–90. doi: 10.1213/ane.0b013e3181904571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai S, Perouansky M, Pearce RA. Amnestic concentrations of etomidate modulate GABAA,slow synaptic inhibition in hippocampus. Anesthesiology. 2009;111:766–73. doi: 10.1097/ALN.0b013e3181b4392d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HT, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, Wafford KA, Orser BA. Alpha5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. J Neurosci. 2006;26:3713–20. doi: 10.1523/JNEUROSCI.5024-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]