

Abstract
Malaria is one of the most devastating parasitic diseases worldwide. Plasmodium drug resistance remains a major challenge to malaria control and has led to the re-emergence of the disease. Chloroquine (CQ) and artemisinin (ART) are thought to exert their anti-malarial activity inducing cytotoxicity in the parasite by blocking heme degradation (for CQ) and increasing oxidative stress. Besides the contribution of the CQ resistance transporter (PfCRT) and the multidrug resistant gene (pfmdr), CQ resistance has also been associated with increased parasite glutathione (GSH) levels. ART resistance was recently shown to be associated with mutations in the K13-propeller protein. To analyze the role of GSH levels in CQ and ART resistance, we generated transgenic Plasmodium berghei parasites either deficient in or overexpressing the gamma-glutamylcysteine synthetase gene (pbggcs) encoding the rate-limiting enzyme in GSH biosynthesis. These lines produce either lower (pbggcs-ko) or higher (pbggcs-oe) levels of GSH than wild type parasites. In addition, GSH levels were determined in P. berghei parasites resistant to CQ and mefloquine (MQ). Increased GSH levels were detected in both, CQ and MQ resistant parasites, when compared to the parental sensitive clone. Sensitivity to CQ and ART remained unaltered in both pgggcs-ko and pbggcs-oe parasites when tested in a 4 days drug suppressive assay. However, recrudescence assays after the parasites have been exposed to a sub-lethal dose of ART showed that parasites with low levels of GSH are more sensitive to ART treatment. These results suggest that GSH levels influence Plasmodium berghei response to ART treatment.
Introduction
The development of drug resistance by Plasmodium parasites has become one of the major obstacles in the efforts to control malaria. Plasmodium falciparum, the deadliest and more severe malaria parasite, has developed resistance to the majority of the antimalarial drugs currently available [1]. Similarly, Plasmodium vivax, an important cause of malaria morbidity, has become resistant to chloroquine (CQ) and resistance has spread to almost all endemic countries [2]. Chloroquine (CQ), previously used as the first-line treatment and most cost-effective antimalarial, is currently ineffective in nearly all regions where malaria is endemic [1]. More alarming is the loss of sensitivity to artemisinin (ART), the current first-line treatment, which is emerging in the Thai-Cambodian border [3–5]. In addition, signs of P. falciparum ART resistance have been reported in Africa [6]. The development of drug resistance by malaria parasites poses a clear threat to recent efforts that have significantly reduced the burden of the disease.
Development of CQ resistance has been linked to the CQ resistance transporter (pfcrt) and the multidrug resistance analogue (pfmdr1) genes. However, glutathione (GSH)-mediated detoxification has been proposed to contribute to CQ resistance. A marked increase in GSH levels and the activity and expression of GSH-related enzymes has been reported in P. berghei and P. falciparum lines resistant to CQ [7–11]. In addition, a fraction of the toxic heme molecule produced during hemoglobin catabolism is detoxified by GSH, a process inhibited by CQ [8]. Therefore, increased GSH levels in the parasite might help overcome the CQ blockage of GSH-mediated heme degradation, resulting in an increased resistance to CQ [12].
The antimalarial activity of ART and its derivatives is proposed to be mediated by the iron-dependent generation of reactive oxygen species (ROS), which alters the redox balance of the parasite and consequently induces damage to cellular targets. ART reacts with hemin in vitro [13], and in vivo, the binding affinity to hemin correlates with the antiplasmodial activity of the drug [14]. In addition, the increased levels of intracellular ROS, and the antimalarial activity of ART require the uptake and degradation of hemoglobin by Plasmodium parasites [15]. Moreover, reduced GSH reacts and forms adducts with ART derived C-centered primary radicals [16], which might result in deprivation of GSH and consequently, an increase in intracellular ROS damage. As GSH is one of the parasite's main antioxidant systems, it is conceivable that increased levels of GSH could potentially detoxify the ROS-induced damage caused by ART treatment.
GSH is synthesized de novo by the sequential action of the rate-limiting enzyme gamma-glutamylcysteine synthetase (γ-GCS) and the GSH synthetase (GS) [17, 18]. Increased expression of the pbggcs mRNA was shown in P. berghei lines resistant to CQ and MQ [10]. Further evidence supporting a role for the pbggcs gene in CQ resistance comes from reports where the γ-GCS inhibitor L-buthionine sulfoximine (BSO) partially reverts the CQ resistance phenotype in P. berghei [7, 19]. In addition, CQ sensitive P. falciparum parasites are more susceptible to BSO treatment than CQ resistant parasites [9, 20]. These results support the association between increased GSH levels and CQ resistance in Plasmodium.
To further investigate the potential contribution of GSH to Plasmodium drug resistance, the development of genetically engineered P. berghei parasites overexpressing the pbggcs gene and displaying high levels of GSH is reported herein. We had previously disrupted the pbggcs gene, resulting in mutant parasites with significantly low levels of GSH [21]. Drug sensitivity responses were evaluated in mutants with the pbggcs silenced or overexpressed, as well as recrudescence and mice survival after treatment with an ART derivative. We report that altered GSH levels affect drug sensitivity to ART while the CQ response remains unchanged. This study provides new insights into the GSH involvement in the mechanism(s) of action of ART.
Materials and Methods
Mice and Parasites
Random-bred Swiss albino CD-1 female mice (Charles River Laboratories, Wilmington, MA, USA), 6–8 weeks old, weighting 20 to 35 g were used for the study. All mice procedures conducted at the AAALAC accredited UPR-School of Medicine were approved by the IACUC of the Medical Sciences Campus, University of Puerto Rico (Protocol numbers: 2480104; 2480106; 2480108, Animal Welfare Assurance # A3421-01). When an animal appeared to be in pain or disease the Veterinarian or Veterinary Technologist humanly euthanized the mouse by cervical dislocation or CO2 chamber following the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. All work was done in strict accordance with the “Guide for the Care and Use of Laboratory Animals” (National-Research-Council, Current Edition) and regulations of the PHS Policy on Humane Care and Use of Laboratory Animals. Mice were maintained and housed according to NIH and AAALAC regulations and guidelines and were allowed to acclimatize for 1 week prior to the beginning of studies.
The P. berghei parasite lines used in this study were: ANKA 2.34 wild type reference line, P. berghei mutant clone pbggcs-ko [21], N clone (sensitive line) [22], RC line (selected under CQ pressure from the sensitive line) [22], and N/1100 (selected under MQ pressure) [23]. The Nclone, RC and N/1100 lines were derived from the K173 isolate while ANKA 2.34 clone was derived from the ANKA isolate [24].
For the RC or the N/1100 parasites, infections were started in a mouse by injecting an aliquot of parasites from liquid nitrogen stocks intraperitoneally. Mice were treated one day post-infection with CQ (for RC parasites, 60 mg/kg) or MFQ (for N/1100 parasites, 60 mg/kg) to maintain drug pressure. This treatment ensures that only resistant parasites will be further used in the study.
Generation and genotyping of pbggcs overexpression parasites
The pL1136 vector containing the Toxoplasma gondii dihydrofolate reductase—thymidylate synthase (tgdhfr/ts) selectable marker was used as a backbone for the creation of a pbggcs over-expressing plasmid (Fig 1). The complete pbggcs ORF, including 465 bp of the 3’UTR, was amplified from ANKA 2.34 genomic DNA using primers 2562 (5’-CATGCCATGGATGGGTTTTCTAAAAATTGGAACTCC-3’; KpnI site is underlined) and 2563 (5’- CGGGGTACCTGGTGTGTATATACCAAACCGTTTC-3’; KpnI site is underlined), cloned into the TOPO TA vector (Invitrogene) and sequenced. The pbggcs coding sequence containing the 3’UTR was excised from the pbggcs-TOPO plasmid using the NcoI and the KpnI restriction enzymes and subsequently cloned into the pL0017 after removing the GFP coding sequence from the plasmid. The resulting pL1136 plasmid was linearized using the SacII restriction enzyme and transfected into P. berghei (ANKA 2.34) purified schizonts. Transfection, selection of transformed parasites with pyrimethamine, and cloning of pbggcs-oe parasites were carried out as previously described [25]. Clonal parasites (pbggcs-oe1; pbggcs-oe2) obtained by limiting dilution were analyzed for correct integration of the pbggcs over-expression plasmid into the c/d-ssurrna on chromosome 5/6 by Southern analysis of chromosomes separated by Field Inverted Gel Electrophoresis (FIGE). Chromosome Southern blots were hybridized with the P. berghei 3’UTR dhfr-ts specific probe (chromosome 7, endogenous dhfr-ts and chromosome 5/6,dssurrna integration site). Additionally, integration into the dssurrna locus was confirmed by PCR analysis of genomic DNA from the two mutants clones using specific primers for pbggcs 213 (5’-TGGGAAAAAGTTGTATCAATTC-3’), pbdhfr/ts 214 (5’-AGTCGGGAAACGTGTCGTG-3’) and dssurrna genes 211 (5’-CTTGCCAGTAGTCATATGCTTGT-3’) and 212 (5’-CTTCCGCAGGTTCACCA-3’).
Fig 1. Generation of pbggcs overexpression mutants in P. berghei.

A. Diagram of the pbggcs overexpression plasmid (Top), the dssurrna genomic locus (Middle) and the predicted integration event of the pbggcs-oe vector at the dssurrna locus (chromosomes 5/6) via single cross-over recombination (Bottom). Open arrows, pbggcs coding region; light grey boxes, P. berghei elongation factor 1a promoter; white boxes, 3’UTR pbggcs; dark grey boxes, P. berghei dhfr-ts upstream and downstream regions; black arrows, T. gondii dhfr-ts coding region; hatched boxes, dssurrna DNA sequence; small arrows; positions of primer pairs for PCR analysis; dotted line, predicted size of PCR products; S, SacII; A, ApaI. B) Southern blot analysis of FIGE separated chromosomes of pbggcs-oe parasites. Correct integration of the pbggcs-oe plasmid to the dssurrna integration site was confirmed by incubating the chromosome blots with a P. berghei 3’UTR dhfr-ts specific probe that hybridizes to chromosomes 7 (endogenous dhfr-ts) and chromosome 5/6 (dssurrna integration site). C) PCR analysis confirming integration of the pbggcs over-expression constructs. Primers upstream and downstream from the endogenous dssurrna integration site (primers 211 and 212), to the T. gondii dhfr-ts selection cassette (primers 214) and to the integrated pbggcs sequence (primer 213) were used to verify integration into the dssurrna locus. Products of the predicted size (4.5 kb for primers 211/214 and 2.6kb for primers 212/213) were obtained for both clones. Lanes: m: 100 bp ladder (New England Biolabs), a: primers 211/214, b: primers 212/213.
Parasites with a disrupted pbggcs locus (pbggcs-ko1; pbggcs-ko2) were previously described in Vega-Rodríguez et al. (2009) [21].
RNase protection assay
Expression of the pbggcs gene was analyzed by RNase Protection Assay (RPA) as previously described [26]. Briefly, Alpha-32P UTP labeled riboprobes for the pbggcs and the β-tubulin genes were synthesized in vitro by antisense transcription using the T7 RNA polymerase (Maxiscript SP6/T7 Kit, Ambion). RPA’s were performed using the RPAIII system (Ambion, Austin, Texas) according to the manufacturer’s instructions. Riboprobes were hybridized with total RNA from the P. berghei pbggcs-oe or wild type parasites overnight at 42°C. The probe-RNA hybrids were resolved on denaturing 6% acrylamide gels, which were subsequently exposed to autoradiography films. Autoradiograms were scanned and analyzed using Quantity One 1-D Analysis Software (Bio-Rad, v. 4.4). The density of the pbggcs signal was normalized to the density of the β-tubulin signal. Density ratios of the normalized pbggcs signals were subsequently normalized to ANKA to estimate mRNA expression levels in the pbggcs-oe parasites.
Determination of GSH levels
Parasite GSH levels were determined by high-performance liquid chromatography (HPLC) as previously described [21, 27, 28]. Briefly, P. berghei infected blood was harvested from the donor mice with parasitemias between 5% and 15%. White blood cells were removed using a Whatman CF11 cellulose column [29]. The red blood cells (RBCs) were removed by lysis with saponin (0.15%) on ice, and free parasites resuspended at a concentration of 50X106/100 ml in HPLC buffer (3.5 mM MgCl2, 110 mM KCl, 40 mM NaCl, 20 mM Hepes, 6 mM EDTA, pH 7.4) with protease inhibitors [30]. Parasites were lysed by three freeze/thaw cycles and parasite extracts were treated with an optimal concentration of dithioerythritol (12.5 mM) to reduce all the GSH derivatives [31]. Samples were resolved on a Hewlett Packard HP ODS Hypersil column and analyzed as Monobromobimane (MBrB) fluorescence derivatization in a Hewlett Packard 1050 Series HPLC.
In vivo drug suppressive test
The Peters “4 day suppressive test” [32, 33] was carried out in P. berghei ANKA 2.34 wild type and the mutant parasites lacking (pbggcs-ko1; pbggcs-ko2) or overexpressing (pbggcs-oe1; pbggcs-oe2) the pbggcs gene. Two independent experiments were conducted for each parasite clone analyzed. CQ diphosphate salt and ART were obtained from Sigma-Aldrich. A 10 mg/ml stock solution was prepared in PBS for CQ and in 100% dimethyl sulfoxide (DMSO) for ART. Drugs were subsequently diluted in PBS to the appropriate dose for the drug assay. Five groups of mice (5 mice per group) were infected intravenously with 10X106 parasites from each line and treated with CQ (intraperitoneal) and ART (subcutaneous) 1 hr post infection and then daily for three consecutive days with different drug doses (10 mg/kg, 3 mg/kg, 2 mg/kg, 1 mg/kg). Mice in the control group received vehicle alone (PBS or DMSO). On day 4 post-infection (5th day of assay), blood was collected and parasitemias determined from Diff Quick stained blood smears. A minimum of 350 RBCs were counted. Dose response curves and ED50 values were calculated after analysis with GraphPad Prism, version 4.03.
ART (DHA) recrudescence assay
The 4 day suppressive test [32, 33] was modified to ascertain recrudescence after treatment with a 20 mg/kg dose of ART for four consecutive days. Mice (5 mice/group) were infected intravenously with either 10X106 P. berghei ANKA 2.34 wild type, pbggcs-ko or the pbggcs-oe parasites. Mice were treated with intramuscular doses of 20 mg/kg of dihydroartemisinin (DHA) beginning 1 hr post infection, and daily for four consecutive days. Parasitemia was monitored daily after the fifth day of infection for up to 28 days by Diff Quick stained blood smears. Animal health was closely monitored and strict defined endpoints were followed. When an animal appeared to be in pain or disease the Veterinarian or Veterinary Technologist humanly euthanized the mouse by cervical dislocation or CO2 chamber following the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. Euthanized mice are reported as mortality during recrudescence experiments.
Results
Overexpression of the pbggcs gene
To assess the potential contribution of Plasmodium GSH levels to CQ and ART resistance, the single copy gene encoding the P. berghei γ-GCS was disrupted or overexpressed using standard genetic modification techniques. To overexpress the pbggcs gene, P. berghei parasites (ANKA 2.34) were transfected in two independent experiments with a DNA-construct designed to express the pbggcs gene driven by the P. berghei eukaryotic elongation factor 1a promoter (Fig 1A). Two parasite clones (pbggcs-oe1; pbggcs-oe2) from the two independent transfection experiments were isolated for further analysis. Integration of the construct into the genome of pbggcs-oe1 and pbggcs-oe2 parasites was confirmed by chromosome blots on field inverted gel electrophoresis (FIGE) separated chromosomes (Fig 1B) and by PCR analysis (Fig 1C). Densitometric analysis of the hybridization intensity of the P. berghei 3’UTR dhfr-ts specific probe on chromosome 5/6 (transgene insertion site) compared to the hybridization intensity on chromosome 7 (endogenous pbdhfr/ts locus, single copy gene) shows insertion of 3 and 2 copies of the pbggcs transgene on the pbggcs-oe1 and the pbggcs-oe2 parasites respectively (Fig 1B). Overexpression of pbggcs mRNA in blood stages of the pbggcs-oe parasites was demonstrated by RNase Protection Assay (Fig 2A and 2B). The pbggcs mRNA levels in pbggcs-oe1 and pbggcs-oe2 parasites were 5.3 (P<0.001) and 4.3 (P<0.01) times higher respectively relative to wild type parasites. These results demonstrate the successful over-expression of the pbggcs gene in the two mutant lines.
Fig 2. Overexpression of the pbggcs gene.

A) Representative RPA showing pbggcs mRNA expression in wild type and pbggcs-oe parasites. Radiolabeled riboprobes for the pbggcs gene (top panel) and the P. berghei -tubulin gene (internal control, bottom panel) were used. B) Densitometric analysis of pbggcs expression by RPA. Densities of the pbggcs signals from each line were normalized to the density of the -tubulin signal. Normalized pbggcs signals from pbggcs-oe parasites were subsequently normalized to the signal of wild type. The horizontal line represents the mean relative expression of triplicate measurements from three independent experiments. Asterisks denote significant changes in pbggcs mRNA expression of pbggcs-oe parasites to wild type parasites as determined by a One-way ANOVA with Tukey’s Multiple Comparison Test (* = P<0.01, ** = P<0.001).
Total GSH levels are increased in pbggcs-oe and in CQ and MQ P. berghei resistant parasites
To investigate whether or not overexpression of the pbggcs gene results in increased parasite GSH levels, total GSH was determined in pbggcs-oe and wild type parasites by HPLC. Total GSH levels were significantly higher in pbggcs-oe1 (17.5 nmol/109 parasites, SD ±15.2, P<0.05) and pbggcs-oe2 (22.3 nmol/109 parasites, SD ±19.3, P<0.001) parasites when compared to wild type (7.4 nmol/109 parasites, SD ±1.7) (Fig 3A). These results show that overexpression of the pbggcs gene in P. berghei results in augmented GSH levels.
Fig 3. Total GSH levels in blood stages of P. berghei parasites.
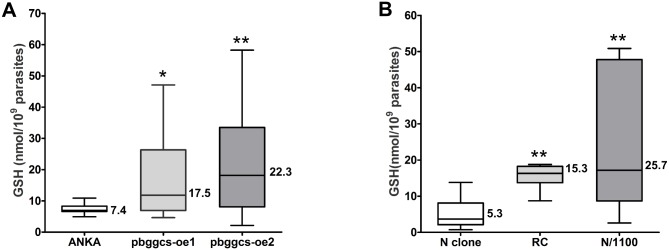
GSH levels were determined by HPLC in extracts of purified blood stage P. berghei parasites obtained from asynchronous infections of (A) ANKA wild type, pbggcs-oe1 and pbggcs-oe2, (B) N clone (drug sensitive), RC (selected for CLQ resistant) and N/1100 (selected for MFQ resistant) parasites. GSH concentration was significantly increased in the pbggcs-oe parasites as compared to ANKA wild type. Similarly, the drug resistant lines RC and N/1100 displayed significantly higher GSH levels as compared to the sensitive one (N clone). Numbers on boxes represent the mean concentration of GSH. Asterisks denote significant changes in GSH concentration as determined by a One-way ANOVA with Tukey’s Multiple Comparison Test (* = P<0.05, ** = P<0.001).
Increased levels of GSH were reported in CQ resistant P. falciparum (9)] and P. berghei [7, 11, 19]. Total GSH content was determined in the sensitive P. berghei N clone and the CQ-resistant RC [22] and MQ-resistant N/1100 [23] derived lines (Fig 3B). Significantly higher GSH levels were determined in the P. berghei CQ resistant RC (15.3 nmol/109 parasites, SD ±3.1, P<0.001) and the MQ resistant N/1100 (25.7 nmol/109 parasites, SD ±18.3, P<0.001) as compared to the sensitive N clone (5.3 nmol/109 parasites, SD ±3.8). These results confirm previous reports which establish that in P. berghei, resistance to CQ is accompanied by an increase in total GSH levels [7–11].
GSH levels do not alter P. berghei CQ or ART response in a 4-day suppressive assay
CQ and ART responses of P. berghei parasites displaying significantly high (pbggcs-oe) or significantly low (pbggcs-ko) [21] GSH levels were investigated. As determined by the 4-day suppressive assay, no major differences in CQ ED50 values were detected in the dose-response curves from parasites with low or high GSH levels when compared to wild type (Fig 4A and 4B). The CQ ED50 values for the pbggcs-ko1 and pbggcs-ko2 parasites were 1.7 mg/kg and 0.19 mg/kg respectively, while in the pbggcs-oe1 and pbggcs-oe2 were 1.01 mg/kg and 1.80 mg/kg respectively. The CQ ED50 value of wild type parasites was 2.21 mg/kg. Similarly, no significant differences in ART ED50 values were observed for pbggcs-ko and pbggcs-oe parasites when compared to wild type (Fig 4C and 4D). The ART ED50 values for the pbggcs-ko1 and pbggcs-ko2 parasites were 0.25 mg/kg and 0.015 mg/kg respectively, while in the pbggcs-oe1 and pbggcs-oe2 were 0.008 mg/kg and 3.58 mg/kg respectively. The ART ED50 value of wild type parasites was 0.016 mg/kg. These results show that either reducing or increasing the GSH levels in the P. berghei ANKA strain do not alter the response to CQ or ART in a 4-day drug suppressive test.
Fig 4. Drug response curves to CLQ and ART.

Dose response curves show percent growth for each parasite clone relative to the untreated control on day 4 of the assay (5th day post infection) versus drug concentration. The 4-day suppressive test was carried out for CLQ (A and C) and ART (B and D) on two independent pbggcs-ko (pbggcs-ko1; pbggcs-ko2) and pbggcs-oe (pbggcs-oe1; pbggcs-oe2) clones. To compare the drug responses from the mutant parasites, the parasitemia from each parasite line was normalized to the parasitemia of the untreated control. No significant differences in the EC50 values were observed between parasites with the pbggcs gene silenced or overexpressed as compared to wild type control. Bars represent standard deviation of the mean.
P. berghei pbggcs-ko parasites failed to recover after ART treatment
ART resistance is defined as an increase of parasite clearance time after ART treatment, or the presence of parasites on day 3 after treatment with recrudescence of the disease within 28 to 42 days [1]. The design of the 4 day test does not allows for detection of small changes in drug sensitivity as the one reported for ART resistance in field isolates [4, 5, 34]. A modification of the 4 day sensitivity test was employed in order to detect recrudescence after treatment with DHA, the active compound of ART and its derivatives. First, we determined that treatment of P. berghei-wild type infected mice with doses of 20 mg/kg DHA in a 4 day drug sensitivity assay results in microscopically undetectable levels of parasites at day 4 of the assay with reappearance of parasites circulating in the mouse peripheral blood within 2–3 days after treatment (Fig 5A). The pbggcs-ko and the pbggcs-oe parasites were subsequently analyzed for infection recrudescence after receiving a daily dose of 20 mg/kg DHA for four consecutive days. Parasites were undetectable after the fourth day of DHA treatment in all the parasite lines used (Fig 5). Mice infected with wild type parasites showed recrudescence between 2–3 days after treatment. In addition, nine out of the ten mice infected with pbggcs-oe parasites showed recrudescence (Fig 5A and 5B). Surprisingly, only two of the ten mice infected with the pbggcs-ko parasites showed recrudescence after ART treatment, one of which eventually cleared the infection (Fig 5B). The survival curves show that after treatment with DHA, survival of mice infected with pbggcs-ko was significantly higher (90%, P < 0.0001) than that of mice infected with wild type (0% survival) or pbggcs-oe (10% survival, P < 0.8021) parasites (Fig 6).
Fig 5. Recrudescence of P. berghei pbggcs deficient or overexpressing mutants after treatment with DHA.

Mice infected with wild type (A), pbggcs-ko (B) or pbggcs-oe (C) parasites were treated with intramuscular doses of 20 mg/kg of DHA beginning 1 hr post infection, and daily for four consecutive days. After the 4th DHA dose, every mouse on each group has undetectable parasite levels by microscopic examination. To determine recrudescence, the parasitemia was monitored in each mouse after the 4th day of DHA treatment (day 0). 100% of the mice infected with wild type parasites (n = 15) and 90% of the mice infected with pbggcs-oe parasites (n = 10) presented recrudescence of the disease showing that these parasites were able to recover from the DHA treatment. Importantly, 80% of the mice (n = 10) infected with pbggcs-ko parasites remained parasite free throughout the duration of the assay suggesting that the knockout parasites did not recover from the DHA treatment. †, euthanized mouse due to malaria disease; ‡, euthanized parasite-free mouse. Each graph represents a biological replicate.
Fig 6. Kaplan-Meir survival curve of mice infected with P. berghei parasites lacking or overexpressing the pbggcs gene.

Mice infected with wild type (green, n = 15), pbggcs-ko (red, n = 10) or pbggcs-oe parasites (blue, n = 10) were treated with intramuscular doses of 20 mg/kg of DHA beginning 1 hr post infection, and daily for four consecutive days. Mice survival was monitored after the 4th day of DHA treatment (day 0). Mice mortality is defined as mice humanly euthanized due to distress caused by severe malaria. Survival of mice infected with wild type parasites was comparable to the survival of mice infected with pbggcs-oe parasites and rapidly declined after treatment with DHA. In contrast, 90% of the mice infected with pbggcs-ko parasites survived after the DHA treatment. Asterisk denote significant changes in survival as determined by a Log-rank (Mantel-Cox) Test (* = P<0.0001).
Discussion
In this study, we demonstrate that overexpression of the pbggcs gene resulted in significantly increased GSH levels in blood stages. More importantly, we show that contrary to previous findings, low or high total GSH levels do not affect sensitivity to QC or ART in a 4-days drug suppressive test. Interestingly, recrudescence of parasites with low GSH levels (pbggcs-ko) after treatment with DHA is highly impaired, sustaining its role in the parasite’s response to ART.
Plasmodium resistance to CQ is mainly attributed to the acquisition of mutations in the pfcrt gene [35, 36]. In addition, mutations in the pfmdr1 gene, encoding an ABC transporter, modulate levels of resistance. These mutations are associated with increased CQ efflux from the parasite’s digestive vacuole where CQ interferes with heme detoxification [35–38]. However, additional genes might be involved in conferring Plasmodium resistance to CQ [36, 39, 40]. Previous studies report that increased GSH levels are associated with P. falciparum and P. berghei resistance to CQ [7, 9, 19, 20, 41]. It is proposed that CQ can interfere with hemozoin polymerization by interacting with the m-oxo dimer form of oxidized heme [42–47]. Inhibition of hemozoin polymerization increases the parasites heme levels, which in turn increases oxidative stress resulting in damage to membranes and proteins [48, 49].
In this study, we analyzed the contribution of the antioxidant GSH on Plasmodium CQ resistance by using genetically transformed P. berghei lines with decreased (pbggcs knockout) or increased (pbggcs overexpression) levels of GSH. The response to CQ in both mutant lines was not affected when compared to wild type parasites suggesting that altered GSH levels do not modulate CQ drug resistance in P. berghei. The increased GSH levels previously reported in CQ resistant parasites could be the result of a parasite response to an oxidative stressed environment induced by CQ, including an increase in GSH production and/or changes in GSH transport. One of the most conclusive reports relating GSH to CQ resistance in Plasmodium is the reversion of CQ resistance by using BSO to deplete the GSH levels [7]. However, a study in Trypanosoma brucei suggested that BSO may have additional targets in the parasite besides the inhibition of the γ-GCS enzyme [50]. Supplementation of T. brucei with GSH rescued the lethal phenotype induced by the depletion of GSH after ggcs RNAi knockdown. However, supplementation with GSH did not complement the lethal phenotype seen by BSO treatment.
Alternatively, the increased GSH levels detected in CQ resistant Plasmodium parasites could be part of a resistance phenotype which in conjunction with other genes contributes to maintain the resistant phenotype [7, 9, 19, 41]. The genetic modifications (pbggcs knockout and knockin) resulting in altered GSH levels in P. berghei were done in the drug sensitive ANKA 2.34 strain of P. berghei which allows the analysis of drug resistance in parasites with high or low GSH levels under a similar genetic background. It is plausible that this strain does not possess the CQ resistant genetic background containing any of the CQ-resistance associated mutations present in the pbcrt gene. In support of this hypothesis, Patzewitz et al. (2013) reported that the transporter encoded by the P. falciparum CQ resistant pfcrt allele was able to transport GSH more efficiently than the CQ sensitive allele [20]. They suggested that an increase of GSH import into the digestive vacuole, presumably mediated by the CQ resistant PfCRT, and not the augmented levels of total parasite GSH, could cause an increase in parasite resistance to CQ [20]. It is conceivable that the sensitivity of the pbggcs-ko (low GSH) and the pbggcs-oe (high GSH) parasites did not change to that of wild type parasites because of the absence of the CQ resistance genotype, such as crt mutations. In this report significantly increased GSH levels were demonstrated in the P. berghei CQ resistant RC line and in the MQ and CQ resistant N/1100 line when compared to the sensitive N clone. Disruption of the pbggcs gene in the CQ resistant RC line to reduce the GSH levels could help to prove the above hypothesis, as this line may possess mutated crt and/or mdr genes.
When pbggcs-ko or pbggcs-oe parasites were tested for ART response in a 4 day sensitivity assay, both mutant parasites presented a drug response similar to the wild type control. However, when analyzed in a recrudescence assay, the pbggcs-ko parasites did not recover from the treatment as evidenced by the lack of infection after day five. These results show that reduced levels of the antioxidant GSH renders P. berghei parasites more sensitive to ART treatment. This is compatible with the ART resistance phenotype detected in P. falciparum from Southeast Asia which is characterized by the presence of parasites on day 3 after ART treatment with a concomitant recrudescence of the disease [1].
The antimalarial activity of ART is thought to result from an altered redox balance in the parasite caused by this endoperoxide-containing drug. ART induces the autoxidation of flavin cofactors, including FADH2, which is required by the GSH reductase enzyme for the reduction of GSSG to GSH [51]. GSH can also form adducts with an ART derived C-centered primary radical [16]. In addition, hemoglobin degradation by the parasite is required for both the increased levels of ROS and the antimalarial activity of ART [15]. Malaria parasites are rich in hemin, which results from hemoglobin degradation. ART interacts with hemin to produce ROS, resulting in cellular damage [13]. In support of these findings, Paitayatat et al. (1997) reported that the ART binding affinity to hemin correlates with the ART anti-plasmodial activity [14]. The above mentioned reactions will reduce the intracellular pools of reduced GSH resulting in an increase of ROS-induced damage. Our previous report show that P. berghei blood stages survive with very low GSH levels [21]. However, this reduced pool of GSH might render the parasite even more sensitive to the oxidative stress induced by ART. In addition, ART can react with GSH in vitro and it is proposed that glutathione S-transferase might be involved in the metabolism of ART [52].
Recently, mutations in the parasites PF3D7_1343700 kelch propeller domain (K13-propeller) protein were associated with resistance to artemisinin in P. falciparum laboratory strains and field isolates [6, 53, 54]. Some of these mutations are highly prevalent among P. falciparum isolates from patients that show a delayed clearance of parasites after ART treatment [6, 53]. Removal of these mutations from ART resistant isolates reduced the ART survival rate of the parasite [54]. In addition, introduction of the K13-propeller mutations into the ART sensitive allele increased the ART resistance levels to those observed in P. falciparum field isolates carrying this mutation [54, 55]. The P. falciparum K13-propeller has homology with the human KEAP1, a kelch domain-containing protein [53]. KEAP1 is a repressor of the transcription factor Nrf2, which in turn induces the expression of cytoprotective and antioxidant enzymes, including some of the enzymes of the GSH metabolism like γ-GCS and GSH S-transferase [56]. Based on homology to other kelch domain-containing proteins, it is possible that the mutations associated with ART resistance in the P. falciparum K13-propeller could destabilize the kelch domain scaffold and alter the protein function [53].
In summary, we report here that altered GSH levels do not change P. berghei sensitivity to CQ. However, reduction of GSH levels renders P. berghei parasites more sensitive to clearance by ART treatment. These results suggest that ART resistant parasites could be using antioxidant molecules like GSH to reduce the pro-oxidant effects of ART resulting in an increase of tolerance to the drug. A better understanding of the potential contribution of GSH to the modulation of ART resistance could help improve current strategies using ART or ART-based combination therapies to control malaria.
Acknowledgments
The authors thank Dr. Blandine Fanke-Fayard, Dr. Chris Janse and Dr. Andrew P. Waters for their contributions and discussions on the generation of the pbggcs-oe parasites during the early stages of the project. The authors also want to thank Dr. Dennis E. Kyle for helpful discussions on artemisinin resistance.
Data Availability
All relevant data are within the paper.
Funding Statement
This study was partially supported by the following awards of the National Institute of Health (AES): National Institute of General Medicine S06GM008224, National Institute on Minority Health and Health Disparities G12-MD 007600, The Fogarty International Center and National Center on Minority Health and Health Disparities T37 MD001477, and the National Institute of Allergy and Infectious Diseases F31A1056662 (Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship award to JVR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.WHO. World Malaria Report 2013. World Health Organization. 2013. http://www.who.int/malaria/publications/world_malaria_report_2013/en/
- 2. Price RN, von Seidlein L, Valecha N, Nosten F, Baird JK, White NJ. Global extent of chloroquine-resistant Plasmodium vivax: A systematic review and meta-analysis. Lancet Infect Dis. 2014; 14: 982–991. 10.1016/S1473-3099(14)70855-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. Status report on artemisinin resistance. 2014. Available: http://www.who.int/malaria/publications/atoz/update-artemisinin-resistance-jan2014/en/.
- 4. Noedl H, Se Y, Sriwichai S, Schaecher K, Teja-Isavadharm P, Smith B, et al. Artemisinin resistance in Cambodia: A clinical trial designed to address an emerging problem in Southeast Asia. Clin Infect Dis. 2010; 51: e82–89. 10.1086/657120 [DOI] [PubMed] [Google Scholar]
- 5. Amaratunga C, Sreng S, Suon S, Phelps ES, Stepniewska K, Lim P, et al. Artemisinin-resistant Plasmodium falciparum in Pursat province, Western Cambodia: A parasite clearance rate study. Lancet Infect Dis. 2012; 12: 851–858. 10.1016/S1473-3099(12)70181-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014; 371:411–423. 10.1056/NEJMoa1314981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubois VL, Platel DF, Pauly G, Tribouley-Duret J. Plasmodium berghei: Implication of intracellular glutathione and its related enzyme in chloroquine resistance in vivo. Exp Parasitol. 1995; 81: 117–124. [DOI] [PubMed] [Google Scholar]
- 8. Ginsburg H, Famin O, Zhang J, Krugliak M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem Pharmacol. 1998; 56: 1305–1313. [DOI] [PubMed] [Google Scholar]
- 9. Meierjohann S, Walter RD, Muller S. Regulation of intracellular glutathione levels in erythrocytes infected with chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum . Biochem J. 2002; 368: 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perez-Rosado J, Gervais GW, Ferrer-Rodriguez I, Peters W, Serrano AE. Plasmodium berghei: Analysis of the gamma-glutamylcysteine synthetase gene in drug-resistant lines. Exp Parasitol. 2002; 101: 175–182. [DOI] [PubMed] [Google Scholar]
- 11. Safeukui I, Mangou F, Malvy D, Vincendeau P, Mossalayi D, Haumont G, et al. Plasmodium berghei: Dehydroepiandrosterone sulfate reverses chloroquino-resistance in experimental malaria infection; correlation with glucose 6-phosphate dehydrogenase and glutathione synthesis pathway. Biochem Pharmacol. 2004; 68: 1903–1910. [DOI] [PubMed] [Google Scholar]
- 12. Lehane AM, McDevitt CA, Kirk K, Fidock DA. Degrees of chloroquine resistance in Plasmodium—is the redox system involved? Int J Parasitol Drugs Drug Resist. 2012; 2: 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meshnick SR, Thomas A, Ranz A, Xu CM, Pan HZ. Artemisinin (qinghaosu): The role of intracellular hemin in its mechanism of antimalarial action. Mol Biochem Parasitol. 1991; 49: 181–189. [DOI] [PubMed] [Google Scholar]
- 14. Paitayatat S, Tarnchompoo B, Thebtaranonth Y, Yuthavong Y. Correlation of antimalarial activity of artemisinin derivatives with binding affinity with ferroprotoporphyrin IX. J Med Chem. 1997; 40: 633–638. [DOI] [PubMed] [Google Scholar]
- 15. Klonis N, Crespo-Ortiz MP, Bottova I, Abu-Bakar N, Kenny S, Rosenthal PJ, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A. 2011; 108: 11405–11410. 10.1073/pnas.1104063108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang DY Wu YL. A possible antimalarial action mode of qinghaosu (artemisinin) series compounds. Alkylation of reduced glutathione by C-centered primary radicals produced from antimalarial compound qinghaosu and 12-(2,4-dimethoxyphenyl)-12-deoxoqinghaosu. Chemical Communications. 2000; 5: 2193–2194. [Google Scholar]
- 17. Seelig GF, Meister A. Glutathione biosynthesis; gamma-glutamylcysteine synthetase from rat kidney. Methods Enzymol. 1985; 113: 379–3. [DOI] [PubMed] [Google Scholar]
- 18. Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994; 54: 4313–4320. [PubMed] [Google Scholar]
- 19. Platel DF, Mangou F, Tribouley-Duret J. Role of glutathione in the detoxification of ferriprotoporphyrin IX in chloroquine resistant Plasmodium berghei . Mol Biochem Parasitol. 1999; 98: 215–223. [DOI] [PubMed] [Google Scholar]
- 20. Patzewitz EM, Salcedo-Sora JE, Wong EH, Sethia S, Stocks PA, Maughan SC, et al. Glutathione transport: A new role for PfCRT in chloroquine resistance. Antioxid Redox Signal. 2013; 19: 683–695. 10.1089/ars.2012.4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vega-Rodriguez J, Franke-Fayard B, Dinglasan RR, Janse CJ, Pastrana-Mena R, Waters AP, et al. The glutathione biosynthetic pathway of Plasmodium is essential for mosquito transmission. PLoS Pathog. 2009; 5: e1000302 10.1371/journal.ppat.1000302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peters W. Drug resistance in Plasmodium berghei vincke and lips, 1948. I. Chloroquine resistance. Exp Parasitol. 1965; 17: 80–89. [DOI] [PubMed] [Google Scholar]
- 23. Peters W, Portus J, Robinson BL. The chemotherapy of rodent malaria, XXVIII. The development of resistance to mefloquine (WR 142,490). Ann Trop Med Parasitol. 1977; 71: 419–427. [DOI] [PubMed] [Google Scholar]
- 24. Killick-Kendrick R. Parasitic protozoa of the blood of rodents: a revision of Plasmodium berghei . Parasitology. 1974; 69(2):225–237. [DOI] [PubMed] [Google Scholar]
- 25. Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei . Nat Protoc. 2006; 1: 346–356. [DOI] [PubMed] [Google Scholar]
- 26. Gonzalez-Pons M, Szeto AC, Gonzalez-Mendez R, Serrano AE. Identification and bioinformatic characterization of a multidrug resistance associated protein (ABCC) gene in Plasmodium berghei . Malar J. 2009; 8: 1–13. 10.1186/1475-2875-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mansoor MA, Svardal AM, Ueland PM. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal Biochem. 1992; 200: 218–229. [DOI] [PubMed] [Google Scholar]
- 28. Rodriguez JF, Cordero J, Chantry C, Gonzalez S, Rivera C, Febo I, et al. Plasma glutathione concentrations in children infected with human immunodeficiency virus. Pediatr Infect Dis J. 1998; 17: 236–241. [DOI] [PubMed] [Google Scholar]
- 29. Baggaley VC, Atkinson EM. Use of CF 12 columns for preparations of DNA from rodent malarias. Trans R Soc Trop Med Hyg. 1972; 66: 4–5. [DOI] [PubMed] [Google Scholar]
- 30. Birago C, Marchei E, Pennino R, Valvo L. Assay of gamma-glutamylcysteine synthetase activity in Plasmodium berghei by liquid chromatography with electrochemical detection. J Pharm Biomed Anal. 2001; 25: 759–765. [DOI] [PubMed] [Google Scholar]
- 31. Pastrana-Mena R, Dinglasan RR, Franke-Fayard B, Vega-Rodriguez J, Fuentes-Caraballo M, Baerga-Ortiz A, et al. Glutathione reductase-null malaria parasites have normal blood stage growth but arrest during development in the mosquito. J Biol Chem. 2010; 285: 27045–27056. 10.1074/jbc.M110.122275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peters W. Chemotherapy and drug resistance in malaria. 2nd ed Orlando: Academic Press; 1987. [Google Scholar]
- 33. Peters W. The chemotherapy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontocidal activity. Ann Trop Med Parasitol. 1975; 69: 155–171. [PubMed] [Google Scholar]
- 34. Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009: 361: 455–467. 10.1056/NEJMoa0808859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000; 6: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sidhu AB, Verdier-Pinard D, Fidock DA. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science. 2002; 298: 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martin RE, Marchetti RV, Cowan AI, Howitt SM, Broer S, Kirk K. Chloroquine transport via the malaria parasite's chloroquine resistance transporter. Science. 2009; 325: 1680–1682. 10.1126/science.1175667 [DOI] [PubMed] [Google Scholar]
- 38. Summers RL, Dave A, Dolstra TJ, Bellanca S, Marchetti RV, Nash MN, et al. Diverse mutational pathways converge on saturable chloroquine transport via the malaria parasite's chloroquine resistance transporter. Proc Natl Acad Sci U S A 111: E1759–1767. 10.1073/pnas.1322965111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jelinek T, Peyerl-Hoffmann G, Muhlberger N, Wichmann O, Wilhelm M, Schmider N, et al. Molecular surveillance of drug resistance through imported isolates of Plasmodium falciparum in Europe. Malar J. 2002; 1:11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lim P, Chy S, Ariey F, Incardona S, Chim P, Sem R, et al. Pfcrt polymorphism and chloroquine resistance in Plasmodium falciparum strains isolated in Cambodia. Antimicrob Agents Chemother. 2003; 47:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Srivastava P, Puri SK, Kamboj KK, Pandey VC. Glutathione-S-transferase activity in malarial parasites. Trop Med Int Health. 1999; 4: 251–254. [DOI] [PubMed] [Google Scholar]
- 42. Chou AC, Chevli R, Fitch CD. Ferriprotoporphyrin IX fulfills the criteria for identification as the chloroquine receptor of malaria parasites. Biochemistry. 1980; 19: 1543–1549. [DOI] [PubMed] [Google Scholar]
- 43. Moreau S, Perly B, Biguet J. Interaction of chloroquine with ferriprotophorphyrin IX. nuclear magnetic resonance study. Biochimie. 1982; 64: 1015–1025. [DOI] [PubMed] [Google Scholar]
- 44. Egan TJ, Mavuso WW, Ross DC, Marques HM. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J Inorg Biochem. 1997; 68: 137–145. [DOI] [PubMed] [Google Scholar]
- 45. Dorn A, Vippagunta SR, Matile H, Jaquet C, Vennerstrom JL, Ridley RG. An assessment of drug-haematin binding as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials. Biochem Pharmacol. 1998; 55: 727–736. [DOI] [PubMed] [Google Scholar]
- 46. Famin O, Krugliak M, Ginsburg H. Kinetics of inhibition of glutathione-mediated degradation of ferriprotoporphyrin IX by antimalarial drugs. Biochem Pharmacol. 1999; 58: 59–68. [DOI] [PubMed] [Google Scholar]
- 47. Leed A, DuBay K, Ursos LM, Sears D, De Dios AC, Roepe PD. Solution structures of antimalarial drug-heme complexes. Biochemistry. 2002; 41: 10245–10255. [DOI] [PubMed] [Google Scholar]
- 48. Pandey AV, Bisht H, Babbarwal VK, Srivastava J, Pandey KC, Chauhan VS. Mechanism of malarial haem detoxification inhibition by chloroquine. Biochem J. 2001; 355: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sullivan DJ. Theories on malarial pigment formation and quinoline action. Int J Parasitol. 2002; 32:1645–1653. [DOI] [PubMed] [Google Scholar]
- 50. Huynh TT, Huynh VT, Harmon MA, Phillips MA. Gene knockdown of gamma-glutamylcysteine synthetase by RNAi in the parasitic protozoa Trypanosoma brucei demonstrates that it is an essential enzyme. J Biol Chem. 2003; 278: 39794–39800. [DOI] [PubMed] [Google Scholar]
- 51. Haynes RK, Chan WC, Wong HN, Li KY, Wu WK, Fan KM, et al. Facile oxidation of leucomethylene blue and dihydroflavins by artemisinins: Relationship with flavoenzyme function and antimalarial mechanism of action. ChemMedChem. 2010; 5: 1282–1299. 10.1002/cmdc.201000225 [DOI] [PubMed] [Google Scholar]
- 52. Mukanganyama S, Naik YS, Widersten M, Mannervik B, Hasler JA. Proposed reductive metabolism of artemisinin by glutathione transferases in vitro . Free Radic Res. 2001; 35: 427–434. [DOI] [PubMed] [Google Scholar]
- 53. Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014; 505: 50–55. 10.1038/nature12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, et al. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science. 2014; 347: 428–431. 10.1126/science.1260867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ghorbal M, Gorman M, Macpherson CR, Martins RM, Scherf A, Lopez-Rubio JJ. Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol. 2014; 32: 819–821. 10.1038/nbt.2925 [DOI] [PubMed] [Google Scholar]
- 56. Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011; 85: 241–272. 10.1007/s00204-011-0674-5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.