

Abstract
Local signals from tissue-specific extracellular matrix (ECM) microenvironments, including matrix adhesive ligand, mechanical elasticity and micro-scale geometry, are known to instruct a variety of stem cell differentiation processes. Likewise, these signals converge to provide multifaceted, mechanochemical cues for highly-specific tissue morphogenesis or regeneration. Despite accumulated knowledge about the individual and combined roles of various mechanochemical ECM signals in stem cell activities on 2-dimensional matrices, the understandings of morphogenetic or regenerative 3-dimenstional tissue microenvironments remain very limited. To that end, we established high-throughput platforms based on soft, fibrous matrices with various combinatorial ECM proteins meanwhile highly-tunable in elasticity and 3-dimensional geometry. To demonstrate the utility of our platform, we evaluated 64 unique combinations of 6 ECM proteins (collagen I, collagen III, collagen IV, laminin, fibronectin, and elastin) on the adhesion, spreading and fate commitment of mesenchymal stem cell (MSCs) under two substrate stiffness (4.6 kPa, 20 kPa). Using this technique, we identified several neotissue microenvironments supporting MSC adhesion, spreading and differentiation toward early vascular lineages. Manipulation of the matrix properties, such as elasticity and geometry, in concert with ECM proteins will permit the investigation of multiple and distinct MSC environments. This paper demonstrates the practical application of high through-put technology to facilitate the screening of a variety of engineered microenvironments with the aim to instruct stem cell differentiation.
Keywords: stem cell differentiation, 3-dimensional cell culture, high-through put screening, extracellular matrix
1. Introduction
There is abundant evidence suggesting that local signals from tissue-specific extracellular matrix microenvironments significantly affect cellular differentiation, phenotypic expression and maintenance [1–3]. Substrate biophysical signals, such as soluble factors [3], cell-ligand interactions [4], matrix elasticity [5, 6] and geometry [7] play critical roles in a diversity of biological events including cell adhesion, growth, differentiation, and apoptosis [7, 8]. Together these signals converge to provide a multifaceted, complex mechanochemical signaling environment for highly-specific tissue morphogenesis and regeneration. Despite accumulated knowledge regarding individual and combined roles of various mechanochemical ECM signals in stem cell activities, the intricacy exhibited by cellular microenvironments poses a considerable challenge in resolving the mechanisms ascribed to stem cell behavior and fate determination processes. This complexity mandates a systemic approach whereby integrative studies must be expanded to capture a more comprehensive understanding of the determinants which direct stem cell differentiation toward desired cell type and function. Conventional methods to elucidate these mechanisms have traditionally been executed in large scale, two-dimensional tissue culture platforms which are often limited by combinatorial brevity, substrate production, and reagent supply. Furthermore, these signals, matrix and biophysical, are often observed independently to differentiate cells on 2-dimensional substrates, an environment vastly different from the way cells are presented naturally in vivo, i.e. a 3-dimensional tissue context which elicits multiple signal inputs to regulate cell fate.
High through-put approaches have emerged in recent years to circumvent the limitations of traditional low through-put techniques (i.e. conventional cultureware), with the promise to develop complex platforms for combined biomolecule/substrate discovery. The salient features of microarray technology include the reproducibility and screening of multiple microenvironments with significantly less reagent and substrate requirements than traditional methods, while lending improved deconstruction of complex multivariable studies [9]. Several reports have demonstrated ECM protein microarrays [10], soluble factor screening [11], biomaterial chemistry screening [12, 13], and multiple signal integration arrays (i.e. elasticity and chemical factor) with encouraging results [14, 15]. However, despite the versatility afforded by current microarray technologies, the incorporation of multiple signals within engineered microarrays remain limited. Meanwhile the integration of current combinatorial microarray technologies in three-dimensions, coupled with other biophysical properties, such as tunable stiffness and geometry, have yet to reach fruition. Capturing complex, multifaceted 3-dimensional environments in high-throughput with combinatorial signaling will likely prove instrumental towards the design of future tissue regeneration biomaterial platforms.
To resolve the mechanisms associated with complex matrix signals and stem cell behavior and fate decisions, we established a high-throughput ECM platform based on soft, fibrous matrices meanwhile highly-tunable in elasticity and 3-dimensional geometry. The technology we demonstrate here is amenable to manipulation of several matrix properties, such as elasticity and geometry, in concert with customizable ECM protein micro-dot combination. Furthermore, selective cellular adhesion and isolation afforded by ECM microarrays permits the investigation of multiple and distinct cellular microenvironments in the presence of specific ECM signaling. Altogether, we demonstrate the practical adaptation of high-throughput technology to facilitate the screening of various tunable mechano-ligand microenvironments in three dimensions with the aim to optimize stem cell fate decisions.
2. Methods
2.1. Materials
Polyethylene glycol dimethacrylate (PEGDM) with a molecular weight of 750 and polyethylene oxide (PEO) (MW 400 kDa) were purchased from Sigma (St. Louis, MO). The photointiator Irgacure ® 2959 was purchased through Ciba Specialty Chemicals Corp. (Tarrytown, NY). (3-trimethoxysilyl)propyl methacrylate (TMPMA) was purchased through Sigma. Rhodamine-methacrylate was supplied by Polysciences, Inc. (Warrington, PA). Albumin-Cy3 and streptavidin-Cy5 protein conjugates we acquired through Life Technologies (Grand Island, NY). Microarray print buffer components, glycerol, triton X-100, were purchased through Sigma. Collagen I extracted from rat tail was supplied by Sigma. Collagen III and collagen IV were extracted by human placenta and provided by Sigma. Laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane was acquired through Sigma. Fibronectin purified from human plasma was obtained through EMD Millipore Corp. (Temecula, CA.). α-elastin extracted from bovine ligament was purchased from Elastin Products Co (Owensville, Missouri). Anti-collagen I, anti-collagen III, anti-collagen IV, anti-laminin, anti-fibronectin and anti-elastin primary antibodies were obtained from EMD Millipore Corp. Secondary antibody Cy3 conjugate was purchased through EMD Millipore Corp. Primary rat pulmonary arterial smooth muscle cells (PASMCs) were maintained in DME-F12 (Hyclone, Logan, UT), with 10% fetal bovine serum (FBS, Atlanta Biologicals, Flowery Branch, GA) and 1% Pen/Strep (Hyclone, Logan, Ut). Rat mesenchymal stem cells were maintained in DMEM (Corning, Corning, New York) with 10% defined FBS (Hyclone) for MSCs and 1% Penn/Strep (Hyclone). Bovine serum albumin (BSA) was obtained from Sigma. (4′, 6-diamidino-2-phenylindole) DAPI nuclear stain and Alexa488-phallodin cytoskeleton stain were purchased through Invitrogen, Inc. (Eugene, OR). Primary anti-PECAM antibody was supplied through Novus Biologicals (Littleton, CO). Secondary anti-rabbit IgG antibody conjugated with Alexa 555 was acquired through Invitrogen, Inc. Vectashield hard mount mounting media was obtained through Vector Laboratories, Inc. (Burlingame, CA).
2.1. Fabrication of PEGDM Soft Matrices
An electrospinning solution composed of 3.2% wt PEGDM 750, 3.4% wt PEO, 0.4 % wt of Irgacure 2959 and 93% DI H2O was mixed for 30 minutes with magnetic stir bar. PEGDM 750 photopolymerizable soft matrices were fabricated by electrospinning on a custom setup comprised of a high voltage power supply (Gamma High Voltage Research, Ormond Beach, FL), grounded collecting surface, motorized syringe pump (NE-300 New Era Pump Systems, Farmingdale, NY), and a 14mm syringe. The solution (2 ml) was spun at a distance of 26 cm from the stationary collecting surface, at the voltage of 30 kV, and a flow rate of 1.10 ml/hr. Electronspun matrices were deposited onto standard glass slides (25mm×75mm, Fischer Scientific Inc.) that were pretreated with TMPMA to present methacrylate groups that can bond the matrices to the glass. PEGDM matrices were subsequently introduced into an inert argon environment to remove oxygen, and then were stabilized with polymerization under UV exposure (352 nm light) with an average intensity of 5 mW/cm2 for predetermined time durations.
2.2. Characterization of PEGDM Soft Matrices
FTIR Analysis
PEGDM electrospun samples were first loaded into a sealed liquid-cell (Sigma), in the presence of an inert argon environment to prevent oxygen contamination during IR acquisition. The double bond conversion in PEGDM was evaluated using a real-time mid-range Fourier transform infrared spectroscopy (FTIR) (Nicolet 4700, Thermo Fisher Scientific, Waltham, MA) by examining the disappearance of the C=C peak within the methacrylate group (at ~1635nm) over time during polymerization under UV light (5 mW/cm2). To account for sample and background variation, data were normalized with the C=O peak located in the range from 1650 to 1726 cm−1.
Scanning Electron Microscopy Imaging
Scanning electron microscopy (FESEM, JSM-7401F, Jeol Ltd, Tokyo, Japan) was used to examine the microstructure of the electrospun PEGDM substrates in both dry and hydrated states. For hydrated samples, substrates were photopolymerized for 15 min and rinsed in DI H2O for 24 hr. To prepare for imaging, rinsed samples were shock frozen in liquid nitrogen (−195°C), and lyophilized for approximately 24 hr. ImageJ was used to analyze changes in fiber diameter and porosity.
Fluorescent Imaging
To image the structure of PEGDM soft matrices in their hydrated state, rhodamine-methacrylate was introduced into the electrospun fibers and subsequently stabilized with UV exposure to provide fluorescence of the fibrous structure. Matrices with PEGDM-rhodamine conjugates were then visualized using either a fluorescent microscope or a confocal laser scanning microscope.
Rheology
Changes in the storage modulus (G′) of PEGDM substrates with respect to photopolymerization time were characterized using a rheometer, (ARES TA rheometer, TA Instruments, New Castle, DE). PEGDM matrices, with approximately 0.3mm in thickness, were deposited onto TMPMA-modified circular coverslips (18mm in diameter) and photopolymerized for 2, 5, 10, or 15 min, and then rinsed in DI H2O for 24 hr. PEGDM soft matrices were tested with a parallel plate configuration. A vertical load of 5 grams was applied to all samples to prevent slippage. A strain sweep at a frequency of 1 rad/s and a frequency sweep at a strain of 5% were run on each sample. Specimen were inspected for slippage or tearing after shearing, and data collected from the linear viscoelastic region in the strain sweep were used to determine the storage modulus G′. Elastic modulus was calculated using the following relationship: E = G′(1+ν) where E is elastic modulus, G′ is storage modulus measured in shear and ν is the Poisson’s ratio taken as approximately zero [16].
2.3. ECM Protein Array Preparation
Protein printing efficiency and optimization was developed using control proteins albumin-Cy3 and streptavidin-Cy5 conjugates. A printing buffer consisting of 1% glycerol and 0.2% Triton X-100 was utilized for all protein depositions. To prepare ECM arrays, stock solutions of collagen I, collagen III, collagen IV, fibronectin, laminin, and elastin were suspended at a concentration of 250 μg/μl in printing buffer. For combinatorial arrays, collagen I is denoted as C1, collagen III as C3, collagen IV as C4, fibronectin as Fn, laminin as L, and elastin as E. Samples were deposited on the fibrous PEGDM matrix using Aushon 2470 arrayer with 185 micron pins (Aushon BioSystems, Billerica, MA), to achieve dots with a nominal diameter of 250 microns. Individual spots with 7 replicates (total of 8) of each protein combination were deposited with a 500μm pitch distance onto the PEGDM matrices. Between different sample depositions, the print needles were cleaned by sonication in cleaning solution before use. Approximately twenty ECM microarrays could be deposited simultaneously in this method within ~1hr. Prepared ECM microarrays were stored at 4°C in a humid environment for 24 h before use.
2.4. Cell Seeding and Cell Culture
The microarray slides of fibrous PEGDM matrix containing ECM proteins were rinsed in DI H2O for 1 h, followed by sterilization with 70% ethanol for 1hr prior to cell seeding. Matrix microarray slides were equipped with 16mm×16mm silicone multiwall chamber (Grace Bio-Labs) to partition individual microarray replicates. Cell seeding protocols were optimized using rat mesenchymal stem cells (MSCs) and primary cell rat pulmonary arterial smooth muscle cells (PASMCs) obtained from rat vascular pulmonary arteries (Figure S1, Supplement Information). Cells with passages of 3–8 were used for all experiments. PASMCs were detached from culture flask and suspended at a concentration of 106 cells per ml in serum free media. The cell suspension was dispensed onto the 3-dimensional matrix microarray within the gasket region at a cell density of 105 cells per array and incubated for 2 hours. The arrays were then gently aspirated by submerging into a large chamber filled with pre-warmed media. Culture media was changed daily. Rat MSCs were extracted from femurs of 10 week old Sprague-Dawley rats weighing approximately 200g each. Metaphyseal heads of the femurs were removed and marrow was flushed out with ice-cold MSC culture media using 25g needles. Clumps in the marrow were dissociated by repeated aspiration with 18g needles and marrow suspension was filtered through 40μm nylon strainer. After a brief centrifugation, cells were re-suspended in warm culture media and seeded. Media was completely replaced after 24h to remove any unattached cells. For neotissue cell seeding, a cell suspension of passages 2–5 with concentration of 106 cells per ml in serum free media was prepared. The cell suspension was dispensed onto the 3-dimensional matrix microarray within the gasket region at a cell density of 105 cells per neotissue array and incubated for 4 hours. The arrays were then gently aspirated into a large chamber filled with prewarmed media. Following aspiration, culture media (10% serum) was introduced into the microarray wells. For cell culture lasting longer than 24 h, the culture media was changed daily.
2.4. Immunofluorescent Staining
Following cell culture, neotissue arrays were samples were fixed with 3.7% formaldehyde at room temperature, permeated with 0.1% Triton X-100 and blocked with 3% BSA. Immunofluorescent staining of cells for cell nuclei (DAPI) and cellular cytoskeleton (Alexa488-phallodin) were utilized to observe cell adhesion and spreading respectively. Platelet endothelial cell adhesion molecule (PECAM-1) antigenic staining was performed to characterize vascular differentiation. For vascular marker immunostaining, samples were first incubated with primary anti-PECAM in 1% BSA overnight at 4°C. Following primary antibody coupling, samples were washed 3X in PBS and incubated with secondary antibody anti-rabbit IgG antibody conjugated with Alexa 555 for 2 h at room temperature. All samples were finally mounted with Vectashield Hard Set mounting media and stored at 4°C for imaging.
2.5. Confocal imaging
Confocal images were acquired using a Nikon A1R laser scanning confocal microscope piloted by NIS-Elements 4.0 and equipped with 405 nm, 488 nm, 561 nm, and 640 nm laser lines. Typically, the relative z position of the focal plane was ensured by using an equipped Nikon Perfect Focus System. Unless otherwise stated, a 10×0.5NA objective with the pinhole set to 1.2 Airy Units (AU) was used. Neotissue array large images were obtained using a motorized XY stage with piezo Z-Drive insert for rapid multidimensional (XYZ) imaging. The image overlap was set to 5% and the resulting digital montage was generated using the same NIS-Elements 4.0 software. When needed, multiple z planes were acquired in order to capture all of the cells within each micro printed well. A maximum intensity projection image was then generated using the piloting software.
2.5. Image & Statistical Analysis
Neotissue image intensities were obtained using National Institute of Health (NIH) ImageJ software (v.1.4). For each neotissue array condition, a minimum of 3 replicate arrays were imaged for statistical significance. Replicate dots per a single array (8 per condition) were averaged and reported with standard error for each intensity evaluated respectively. Nuclear intensities were normalized to the max adhesion values on each array. F-actin and PECAM intensities were further normalized to their respective nuclear intensities to observe cellular spreading and vascular differentiation independent to cell number. To evaluate significant differences between the different protein and stiffness conditions, a 27 (6 proteins + elasticity) full factorial design was performed on all normalized 24 h data using Minitab statistical software (Minitab, State College, PA) to determine the magnitudes of main and interaction effects and their statistical significance respectively. A minimum of 3 arrays for each experimental parameter investigated were used for all factorial analysis.
3. Results
3.1 Characterization of 3-dimensional Fibrous Soft Hydrogel Matrix
Fibrous hydrogels were prepared by electrospinning a photopolymerizable polymer (PEGDM) onto a TMSPSA-functionalized glass surface, followed by UV stabilization (Figure 1). The presence of methacrylate groups on the glass surface allows firm attachment of fibrous hydrogels for substrate stability and longevity after multiple rinses in aqueous solution, facilitating unabridged function for extended biological assays. PEGDM (MW 750) was selected for its biocompatibility, ease of manipulation, elasticity, anti-fouling, and commercial availability [17]. The stabilization of PEGDM substrates is achieved via radical chain photopolymerization between the methacrylate groups in the presence of a photoinitiator and UV light (352nm). We employed mid-range FTIR to characterize the degree of PEGDM conversion by monitoring the disappearance of the reactive methacrylate peak at 1637 cm−1 for samples over the course of 15 minutes UV exposure (Figure 2A). Results showed the attenuation of the methacrylate peak with up to 46% reduction after 15 minutes of UV exposure. The lack of efficient methacrylate conversion is likely due to the occurrence of polymerization in the dry state, reducing chain mobility and active crosslinking domains for polymerization.
Figure 1.
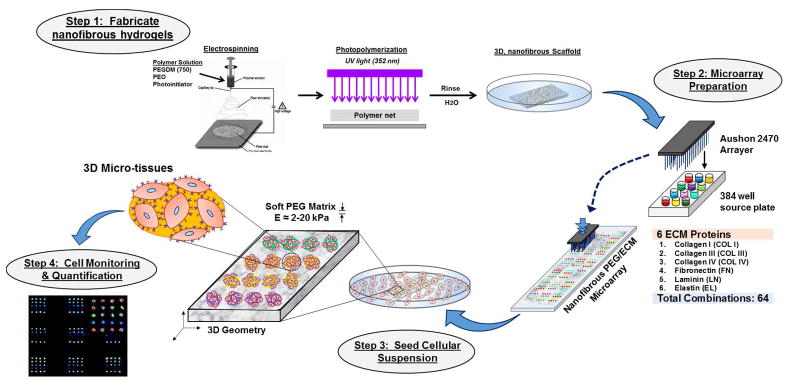
Illustration of ECM Neotissue Fabrication and Utility for Multivariate Cell Culture Platforms.
Figure 2.

Electrospun PEGDM fibrous hydrogel characterization. (A) Mid-range IR identifies methacrylate conversion with UV exposure. (B) Fibrous architecture was investigated using confocal laser microscopy and scanning electron microscopy in both wet and dry states. (C) Shear stress vs. shear strain relationships for several PEGDM substrates prepared under different UV exposures. (D) Translation of shear-strain relationships into elastic modulus using a Poisson ratio ν ~ 0 [16].
The fibrous architecture of electrospun PEGDM substrates was examined using different microscopy techniques (Figure 2B) in both hydrated and dry states. Copolymerizing the PEGDM fibrous hydrogels with rhodamine-methacrylate permitted the visualization of individual fiber diameter and geometry under confocal microscopy. Employing scanning electron microscopy, higher magnification images were obtained of the fibrous substrates in dry and wet states. Both imaging methods demonstrate the fiber diameter increased after hydration to approximately 0.5–1μm. Lack of beading or webbing of the electrospun matrices indicates optimal spinning parameters with minimal artifacts.
To evaluate our capability to regulate the elastic properties of these fibrous substrates, mechanical properties were evaluated under shear using a parallel plate rheometer for PEGDM specimens prepared under different UV exposures at 2, 5, 10 or 15 minutes. Results are presented in Figures 2C–2D. The storage modulus increased with the UV exposure time from 400 Pa to 10kPa after 2- and 15- minute UV exposure, respectively. The elastic modulus determined with the shear modulus measured here (Figure 2D) are in good agreement with compressive modulus determined in our previous work [8].
3.2 Design and Optimization of 3-dimensional Protein Microarray
The deposition of protein microdots is illustrated in Figure 3. Array deposition produces repeatable distinct microdots of 240 ± 11.4 μm in diameter and 580 ± 16.8 μm in pitch to pitch distance (Figure 3A). To optimize the presentation, homogeneity and longevity of protein dots in the 3-dimensional fibrous PEGDM substrates, we have performed iterations with a number of printing buffers using a quality control protein, albumin (Figure 3B). The glycerol content in the buffer was found to influence printing parameters significantly. With increasing glycerol content, protein dot circularity increased whereas the fluorescent intensity of dots decreased. The glycerol content of 1% (v/v) was sufficient in retaining dot circularity without markedly reducing protein intensity after incubation, and thus was used for all studies here. Serial dilutions of quality control proteins, cy3-albumin and cy5-streptavidin, revealed strong protein uptake by the fibrous PEGDM hydrogels, with proteins detected at a deposition concentration as low as 15 μg/ml (Figure 3C). The fluorescent intensity of microdots is correlated well with the deposition concentration. Figure 3D demonstrates 3-dimensional presentation of protein microdots with approximately 200μm in diameter and 50μm in depth penetration of PEGDM fibrous substrates. Finally, optimized printing conditions are applied to produce 3-dimensional protein array with global deposition over a large area (10mm×20mm); results show minimal perturbations occur in the array organization and layout (Figure 3E).
Figure 3.

Protein Microdot Optimization. (A) Array layout depicted through color dye control. (B) Optimization of buffer glycerol content achieves ideal spotting. Inset images scale bar 50 μm. (C) Serial dilution of two model proteins (Albumin-Cy3, Streptavidin-Cy5) demonstrating distinct dot deposition and periodicity. Scale bar 500 μm. (D) Confocal microscopy rendering of albumin-Cy3 deposition illustrating 3-dimensional dot presentation. Scale bar 50 μm. (E) Printing optimization techniques allow for global array deposition onto PEGDM substrates. Scale bar 1 mm. Inset image scale bar 500 μm.
To further assess printing efficiency, we used six types of ECM proteins and their combinations. Imaging results demonstrate that collagen I microdots could be detected at the same resolution – concentrations similar to the quality control proteins (i.e. 15–250 μg/ml), which was stable after several days of continuous rinsing in PBS (Figure 4A). Figure 4B represents the design of a combinatorial ECM protein microarray comprised of 6 ECM proteins, resulting in a total of 64 protein conditions (rows) and replicates of eight (columns) for each condition. To ensure the protein retention for all ECM proteins, antigenic immunostaining was performed, with immunofluorescence results on collagen I and collagen IV shown in Figures 4C and 4D (note: all other protein data not shown for brevity). For all proteins investigated, we found that the immunofluorescence intensity correlated well with expected protein distribution and density. Nevertheless, it should be noted that some detectable fluorescence was observed on the protein combinations absent of the immunostained protein, for example, as shown on the slide stained with collagen IV antibody (Figure 4D). We attributed this phenomenon to the cross reactivity (<10% for collagen I and III) of the collagen IV primary antibody used, as reported by the manufacturer (EMD Millipore). To prevent potential for protein carryover between depositions, we performed consecutive rinsing and sonication of the array tips between each deposition, ameliorating deposition artifacts between replicate dots. Therefore, distinct immunofluorescent detection of specific ECM proteins demonstrate successful deposition of combinatorial designs without condition carryover or contamination.
Figure 4.
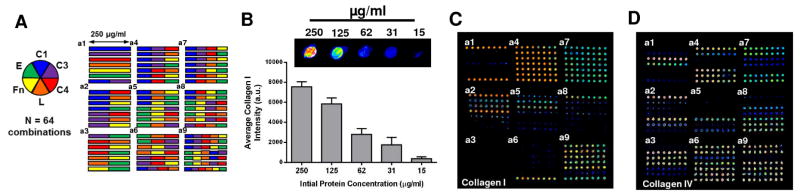
Design and Characterization of Combinatorial Protein Neotissue Array. (A) Design of a combinatorial ECM matrix with 6 proteins yielding 64 unique spotting combinations: C1 (collagen I), C3 (collagen III), C4 (collagen IV), L (Laminin), Fn (Fibronectin), E (Elastin). (B) Serial dilutions of Collagen I are retained after several rinsing stages and detectable at concentrations as low as 15 μg/ml (n=8). Immunostaining of combinatorial ECM matrix for collagen I (C) and collagen IV (D) after deposition and rinsing. Scale bar 1 mm.
3.3. Stem Cell Adhesion and Spreading within 3-dimensional Neotissue Arrays
To demonstrate the feasibility of our matrix hydrogel arrays to support cell adhesion and spreading for the formation of 3-dimensional engineered microenvironments, we developed seeding protocols for MSCs (progenitor/stem cells) and PASMCs (primary cells). Images of PASMC arrays were shown in Figure S1 (supplemental information). MSCs were seeded onto the soft hydrogel arrays (E = 4.6 kPa) in serum-free media for 4 hours at 37°C, followed by gentle aspiration and extended culture in serum-containing media for an additional 20 hr. The MSCs attached preferentially to the protein deposited regions with little to no cell attachment observed on the neat PEGDM fibrous matrix (Figures 5A–5B). Upon closer inspection, distinct cellular islands formed 3-dimensional engineered neotissue microdomains within specialized matrix microenvironments, showing a tissue thickness of approximately 50–100 μm (Figures 5C–5D). MSC attachment and spreading are analyzed with fluorescent microscopy imaging, after stained with DAPI (nucleus) and phallodin-488 (F-actin). Staining for cell nuclei identified distinct cell populations associated with the ECM protein depositions (Figure 5E). Results on quantification of MSC adhesion are illustrated in Figures 5F–5G, which show preferential matrix conditions for 3-dimensional cell adhesion. For instance, a mixture of collagen I and collagen III yielded approximately 3-fold increase in DAPI intensity compared to elastin alone. High-resolution micrographs of the cells in relevant protein matrix environments taken from the DAPI intensity images, confirmed the affinity of certain protein conditions that support cell adhesion over others. To evaluate matrix effects on cellular spreading and morphology, fluorescently labeling of cell F-actin was performed and analyzed with normalization to the respective DAPI intensity measurement for each protein condition. Figure 5H demonstrates the strong dependence of MSC spreading on protein environments of the matrix. Several protein conditions were identified to significantly influence MSC spreading in the neotissue array. The spreading for MSCs cultured on collagen I microdots showed nearly 2-fold increase when compared to MSCs cultured on elastin microdots. Notably, eight combinations of a total of 63 combinatorial matrix environments exhibited F-actin intensity of less than 1 relative to the DAPI intensity, which indicates minimal spreading. Overall, these results highlight the potential of using our fabricated neotissue array platform to capture cellular phenomenon in precisely-defined microenvironments and to determine cause-effect relationship of matrix environments on cells.
Figure 5.

Rat mesenchymal stem cells (MSCs) adhered on neotissue slides (E = 4.6 kPa substrate). (A) Confocal montage image of neotissue array after 24 h cell culture with distinct cellular islands visible for all protein spotting conditions (scale bar 1 mm)(green, f-actin; blue, DAPI). (B) Magnification of a4 subarray depicting cellular dot circular geometry and periodicity (scale bar 500 μm). (C, D) Confocal 3-dimensional rendering of cell loaded subarray (C)(scale bar 250 μm) and of a single cellular dot (D). (E) Nuclear staining of MSCs seeded on the neotissue arrays for image analysis and quantification. (F) 3-dimensional bar graph representing the average nuclear pixel intensities for all protein combinations for image (A). (G, H) Sorted average pixel intensities for adhesion (G) and spreading (H) for all protein combinations after 24 h cell culture (n = 3 neotissue arrays); Insets depict cellular dot images for adhesion and spreading of representative protein conditions (scale bars 100 μm).
3.4. Effect of Protein Environment and Matrix Elasticity on MSC Adhesion and Spreading
To investigate the effects of matrix elasticity and ECM protein environment on MSC adhesion and spreading, we evaluated MSCs on the 3-dimensional fibrous matrix arrays prepared with two UV exposure times, 5 and 15 minutes, which produce substrates with different elasticity, 4.6 and 20 kPa, respectively. Figure 6 shows the distinct patterns of 3-dimesional adhesion and spreading of MSCs on soft (4.6 kPa) and stiff (20 kPa) matrix arrays after 24-hour cell culture. Cell attachment and spreading on both arrays were confirmed, as illustrated in Figure 6A. Statistical analysis of the MSC adhesion and spreading profiles highlighted a diverse set of protein combinations that are correlated to either positive or negative influence with respect to different matrix elasticity conditions (Figure 6B). In an effort to delineate the regulatory mechanisms underlying the effects of elasticity, protein environment, and their interaction on cell activity, we compared the variations of average MSC adhesion and spreading for each protein environment on the 4.6 kPa array with those on the 20 kPa array (Figures 6C–6D). Comparison of the average MSC adhesion profiles on 4.6 kPa and 20 kPa matrices revealed a nearly linear relationship with the exception of few outliers, suggesting cell adhesion in most protein environments is independent of matrix elasticity. Among the few outliers, MSC adhesion on C1·C4·L was favored in the 20 kPa matrix, whereas C3·C4·Fn was favored in the 4.6 kPa matrix. These trends are further illustrated with corresponding images which depict distinct adhesion profiles in various elasticity and protein environments. MSC spreading was found to be strongly influenced by the matrix elasticity (Figure 6D). Compared to the 20 kPa array, MSC spreading on the 4.6 kPa array significantly increased. Compared to cell adhesion, MSC spreading was more synergistically regulated by matrix elasticity and adhesive protein environments. Cell spreading in several protein environments (i.e. C1·C4·Fn·E and L) favored the 4.6 kPa array and others (i.e. C3·E, Fn·E) favored 20 kPa array. Several distinct cell spreading conditions existential to elasticity and protein combinations are also illustrated with corresponding images (Figures 6D–6F).
Figure 6.

Cultured MSCs display differential adhesion and spreading characteristics in response to protein combination and matrix elasticity. (A, B) Cellular structures, nuclei and f-actin, are stained, imaged (A) and quantified using software to produce an average intensity map (B) for distinct elastic environments (4.6 kPa; 20 kPa). (C, D) Comparison of average adhesion (C) and spreading (D) of MSCs on each protein condition for soft (4.6 kPa) and stiff (20 kPa) neotissue substrates. Conditions denoted by blue or red significantly favor adhesion or spreading for soft (4.6 kPa) or stiff (20 kPa) matrix elasticity respectively, purple denotes both. Insets depict adhesion and spreading of representative protein conditions screened from the arrays in (A) (scale bars 100 μm). (E, F) Results of 27 full factorial ANOVA of significant (p < 0.01) main and interaction effects for DAPI (E) and f-actin (F) intensities supporting either soft (4.6 kPa) or stiff (20 kPa) matrix conditions (n = 3 neotissue arrays). Blue and red dots represent prominent protein conditions supporting significant effects only on soft (4.6 kPa) or stiff (20 kPa) or both (purple dot) substrates respectively.
The equivocal activity of several protein combinations and their effects on MSCs under different elastic substrates inspired us to perform full factorial design and subsequent analysis of variance (ANOVA) for main (1-factor) and interaction (27-factor) effects with a significance level of p<0.05. Results are shown in Figures 6E–6F, which respectively demonstrate the effect magnitude for matrix elasticity on cell adhesion (Figure 6E) and cell spreading (Figure 6F) in all protein environments (see Supplementary Figure S2 for enumeration of all effects). Our results revealed that the protein environment exhibiting the most significant upregulation on cell adhesion was C4 for both stiffness environments (20 kPa and 4.6 kPa), while few protein environments were found to decrease (e.g. C3·C4) cell adhesion on both arrays, with protein-regulated variations independent of elasticity. Finally, cell adhesion variations for other protein environments were differentially regulated by the matrix elasticity. For example, C1 promoted cell adhesion on the 20 kPa array but weakened adhesion on 4.6 kPa. Interestingly, we revealed an inverse adhesive profile for C1·C3·E condition, whereby enhanced attachment was observed for soft substrate when compared to stiff substrate which had reduced adhesion. Similar to adhesion, we estimated the contribution of matrix elasticity, protein environment and their interaction on MSC spreading variation using factorial ANOVA (Figure 6F). Cell spreading was markedly enhanced by C4 but reduced by Fn·E on both 20 kPa and 4.6 kPa arrays. Several protein environments, however, significantly influence cell spreading only under one matrix elasticity: C3·L·Fn enhanced spreading on 4.6 kPa alone, while C1·C3·C4·L·Fn·E enhanced spreading on 20 kPa alone. In contrast, reduced spreading conditions were revealed to be C1·C3·C4·Fn and C1·C3·L·Fn·E for 4.6 kPa and 20 kPa, respectively. Overall, performing linear regression (Figures 6E–6F) revealed positive trends for MSC adhesion and spreading on softer substrates (4.6 kPa) compared to stiff (20 kPa). Furthermore, both adhesion and spreading array analyses revealed a tendency to support similar significant effects on attachment and spreading potential (positive slope) of all protein environments on these matrices.
3.5. Effects of Matrix Elasticity and Adhesive Protein Environment on MSC Fate Commitment in 3-dimensional Engineered Neotissues
To explore effects of neotissue microenvironments on stem cell differentiation, we cultured MSCs within our neotissue arrays for 24 h under two different elasticities and then stained cells for the vascular marker PECAM. Representative MSC cultures within neotissue arrays are depicted in Figure 7A, stained for cell nuclei (blue), F-actin (green), and PECAM (red). Immunostaining for the differentiation marker PECAM resulted in detectable levels after 24 h culture on both elastic arrays (Figure 7B). Cell images on several relevant protein conditions and as per elasticity are illustrated in Figure 7C. Interestingly, PECAM expression intensity was notably greater on stiff (20 kPa) vs. soft (4.6 kPa) matrix (Figure 7D). On each elastic array, several protein environments were identified to significantly influence PECAM expression. Notably, C1·C4·Fn·E and C1 provided the greatest difference in PECAM intensity for 4.6 kPa matrix when compared to 20 kPa matrix. Likewise, C3·E, C3·L·E, and C4·L·Fn resulted in upregulated PECAM expression on 20 kPa matrix and much lower expression on 4.6 kPa. Of the top ten protein environments that enhanced PECAM expression on 4.6 kPa matrix, 2 conditions contained laminin; however, comparison to the bottom ten environments, 7 conditions contained laminin, while all other types of proteins see nominal fluctuation between the top ten and bottom ten protein environments for PECAM. Further, of those 7 conditions, 5 represented combinations of laminin and fibronectin. This suggests laminin reduces PECAM expression on 4.6 kPa substrate. Comparably, the ten highest PECAM intensities for 20 kPa matrices all lacked collagen I, with laminin and elastin constituting the greatest frequency at 6 and 5 conditions out of 10 respectively. Interestingly, the ten lowest PECAM intensities for 20 kPa matrices showed a considerable increase in collagen I content (5 out of 10) and a significant decrease (2 of 10) in elastin frequency. Altogether, these results suggest that elastin and collagen I play important but opposite roles in determining overall PECAM expression on stiff matrices, while laminin, in particular laminin mixed with fibronectin, play a role in reducing PECAM on soft matrices.
Figure 7.

Cultured MSCs differentiate in response to protein combination and matrix elasticity on neotissue arrays. (A) Confocal montage images of neotissue arrays stained for nuclei, f-actin and PECAM for both elasticities investigated. Scale bar 1mm. (B) Differentiation marker PECAM average intensities obtained from neotissue arrays (A) and rendered into intensity maps for distinct elastic environments (4.6 kPa; 20 kPa). (C) Confocal images of cell nuclei (DAPI), f-actin (green) and PECAM (red) of MSCs cultured on specific protein conditions and matrix elasticity. Scale bar 50 μm. (D) Average PECAM intensity of MSCs after 24 h for each protein condition compared against soft (4.6 kPa) and stiff (20 kPa) neotissue substrates. Conditions denoted by blue or red significantly favor PECAM expression for soft (4.6 kPa) or stiff (20 kPa) or both (purple dot) matrix elasticity respectively. (E–G) Results of 27 full factorial ANOVA of significant (p < 0.01) main and interaction effects for PECAM intensities (G) and compared against either soft (4.6 kPa) or stiff (20 kPa) matrix conditions (E)(n = 3 neotissue arrays). Blue and red dots represent prominent protein conditions supporting significant effects only on soft (4.6 kPa) or stiff (20 kPa) or both (purple dot) substrates respectively. (F) Significant effects of matrix elasticity on all cellular phenomenon investigated (adhesion, spreading, PECAM) as reported from 27 full factorial ANOVA.
We estimated the contribution of matrix elasticity, protein environment and their interaction on PECAM expression variation using factorial ANOVA for each of 128 neotissues assayed Figures 7E–G. Detecting significant effects of those factors on PECAM expression level represent PECAM variation for elasticity regulation (E), protein environmental effect (P), and elasticity-regulated variation for protein environmental effect (E × P, elasticity-by-environment interaction). A total of 67 conditions varied in PECAM expression level between two elasticity matrices independently of the protein environment (pE<0.05, pE × P>0.05). This is a set of conditions whose PECAM level is regulated by the matrix elasticity but this variation is not protein-dependent. The PECAM level of 20 conditions was affected by the protein environments independently of the matrix elasticity (pP<0.05, pE×P>0.05), which represent roughly 15% of the conditions surveyed. Finally, 27 conditions showed significant interaction between the stiffness and the environments on PECAM expression (pE × P<0.05). This latter group represents MSC expression of PECAM that responds differently to the protein environments examined, depending on the matrix elasticity, and therefore represent engineered neotissues with MSC differentiation synergistically regulated by matrix elasticity and protein environments. Notably, C4·L·Fn·E significantly enhanced PECAM expression on stiff matrices (20 kPa) alone, while C3·L·Fn on soft matrices (4.6 kPa) alone. Further, C1·C3·L·Fn·E significantly reduced PECAM expression on stiff matrices alone, while C3·Fn on soft matrices alone. Unlike analysis results for cell adhesion, no protein condition resulted in inverse PECAM expression on the different elastic matrices. Some of the prominent protein conditions that significantly influenced PECAM expression positively and negatively, irrespective of elasticity, include C3·L·Fn·E and C3·C4·L·Fn, respectively. Linear regression of all significant PECAM effects revealed a positive regression slope suggesting significant PECAM expression conditions generally followed similar trends between both elastic matrices.
To further decouple the stiffness effect on cell differentiation from protein effects, comparison of the general effects obtained through 27 full factorial ANOVA of substrate elasticity over all cellular phenomenon is presented in Figure 7F. In agreement with our previous findings, MSC adhesion and spreading significantly (p < 0.001) correlated negatively for all protein conditions on stiff (20 kPa) matrix compared to soft (4.6 kPa). Interestingly, despite negative influences on adhesion and spreading, stiff matrices significantly (p < 0.05) upregulated PECAM expression irrespective of protein condition, when compared to soft matrices. Of the significant (p < 0.05) effects leading to reduced PECAM expression, thirteen conditions were found on 20 kPa substrate versus eighteen conditions on 4.6 kPa substrate. We note that of the eighteen significantly negative effects on PECAM expression for soft substrate, eight were not shared with the stiff substrate (20 kPa). Investigating the average PECAM intensities for both substrates we find the maximum intensity reported for 20 kPa to be approximately 15% greater than that measured on 4.6 kPa (data not shown). Collectively, these trends suggest that matrix elasticity may have a more significant role in PECAM expression of MSCs cultured on our neotissue arrays than any of the protein environments presented.
4. Discussion
The need for engineered stem cell niches integrating several extrinsic stimuli has become a significant challenge within the research community. Recent evidence suggests that cells react to a complex mechanosensing apparatus whereby the interaction of ligand tethering and ECM stiffness can impart differential cellular functions [18], including differentiation [19]. Indeed, emerging reports have highlighted the significance of protein conformational status which is influenced by the underlying matrix stiffness [20], or chemistry [21] effectively modulating the presentation of binding sites for cell receptors and/or growth factors [22], the entirety of which can significantly influence cellular processes [23]. Due to the lack of existing methods to accurately and efficiently capture these complex microenvironments, we created a high throughput method whereby 3-dimensional matrix physical properties and biological ligand could be modulated. The design of a multivariate protein array for screening of stem cell microenvironments required the fabrication of an appropriate platform incorporating 3-dimensional substrate with fiber architecture and tunable elasticity and finally the integration of a combinatorial ECM protein library upon our engineered substrates.
The choice of neotissue substrate was an important requisite of our design which necessitated: 1) tunable elasticity, 2) 3-dimensional architecture, 3) reproducible fabrication, and 4) ease of sample production. For this work, we chose a soft, fibrous hydrogel platform prepared from an electrospinning method as previously described [8, 15]. Recent studies have highlighted the importance of 3-dimensional, fibrous matrices to optimize stem cell niche environments [24, 25], as well as their candidacy as platforms for MSC differentiation into vascular lineages [8, 15]. We find electrospinning, followed by UV polymerization to represent a highly versatile technique whereby stiffness and microstructure can be reproduced with high fidelity. The electrospun matrices produced here require facile production techniques, meanwhile providing reproducible elastic moduli of ~3–20 kPa which represent biologically relevant stiffness range for native vasculature [8], while avoiding disparity in substrate architecture and geometry.
We chose a contact style arrayer for the protein deposition which is capable of depositing up to 3200 distinct protein dots, of ~240μm diameter and pitch 500μm, onto a standard 25mm×75mm microscope slide. Protein deposition volume is on the order of picoliters [26], which represents several orders of magnitude less reagent required for traditional-scale cultures. Tailoring of the printing buffer for ECM proteins was found to significantly affect doting efficiency upon our unique substrates. Others have reported buffer reformulations for 2-dimensional microarray technologies [10, 27]. We found significantly less glycerol content was necessary to retain our protein deposits on our 3-dimensional substrates. Our methodology requires printing upon a dry electrospun surface, whereby the lack of substrate moisture likely assists in dot uptake and retention, removing the necessity for increased glycerol concentration.
The neotissue arrays we prepared here revealed a robust biological response of MSCs cultured within these substrates. MSCs were found to attach preferentially to protein deposited microdots, retaining the array structure and periodicity. In the absence of protein spotting, lack of cellular attachment and spreading was ostensible due to the inert and anti-fouling properties [17] of the neat PEGDM electrospun matrices. Furthermore, evidence of 3-dimensional cellular organization was detected over a diversity of protein and substrate conditions. Using both stem cell and matured cells in our study, we showed the method described here could be amenable to virtually any cell line of interest, with minor modification to current protocols. For this work, we only investigated the cellular response on MSCs over 24 h and 72hr (Figure S1, Supplemental Information); however, preliminary extended cell culture experiments demonstrated that these neotissue arrays retained their structure and functionality up to 7 days continuous culture. Therefore, we suspect these neotissue arrays can easily be adapted for longer cell culture regiments, with the potential to integrate spatiotemporal signals.
Our focus on vascular regeneration inspired the choice of proteins employed: collagen I, collagen III, collagen IV, laminin, fibronectin and elastin; all of which are amply represented in the native vasculature [28] and have distinct roles in development and vasculogenesis [28, 29]. However, we note that our developed technique is adaptable to virtually any protein combination or formulation of interest for the end user. The choice of protein concentration utilized for cell culture was another important design consideration for our neotissue arrays. We found cellular attachment with as little as 15 μg/ml spotting concentration of pure collagen I could be achieved on our PEGDM electrospun substrates (data not shown). Therefore, we chose a protein dot concentration of 250 μg/ml for our experiments in order to ensure sufficient representation of each protein mixture.
The ability to deposit complex protein combinations onto our engineered soft matrices with high reproducibility and accuracy encouraged us to adapt this technology towards a high-throughput neotissue platform. Current cellular microarray technologies lack the amenity to observe substrate parameters such as elasticity, geometry and biological ligand in synergy. Attempts to integrate multiple signals into microarray technologies have been reported previously. Gobaa et al. successfully coupled tunable elastic microwells with array spotting technology to investigate the effects of cell density, substrate elasticity and protein on the adipogenic or osteogenic differentiation of MSCs in 2-dimensions [14]. However, emerging evidence suggests a drastically different ensemble of biological signals exists for stem cells when cultured in 3-dimensions [7, 30]. Recent attempts at 3-dimensional microarray platforms have emerged with promising results [31, 32], but the integration and independent modulation of several mechano-chemical factors in 3-dimensional formats is still a considerable challenge in the microarray community [33]. A key aspect of our neotissue arrays is their ability to incorporate significant matrix parameters, including 3-dimensional geometry, elasticity and biological ligand, and investigate their effects in synergy or independently on cellular phenomenon in high-throughput fashion. However, one limitation of our neotissue arrays, compared to other micro-engineered niche environments [34], is that it’s comprised of a single medium chamber, thereby allowing cellular crosstalk events among different conditions. Adaption of our technology to a multiwell format [35] or proper spotting randomization [14] could obviate this problem.
Our interest in vascular regeneration prompted us to evaluate the potential of our neotissue arrays to instruct vascular commitment of MSCs. Several reports have observed the differentiation of MSCs into vascular lineages in vitro [36, 37]. The majority of literature regarding stem cell differentiation into vascular lineages involves precise soluble factor regiment [36,], application of shear [38], matrix rigidity [8, 15], composition of the ECM [39–41], or multiple factors [15, 42]. For example, it has been shown that the administration of vascular endothelial growth factor (VEGF) [36] or combination with shear stress [43] or matrix elasticity [15] instructs MSC differentiation into vascular lineages. Wang et al demonstrated the significance of ECM proteins in determining the vascular commitment of MSCs [41]. Others have reported on specific ECM environments for improved vascular differentiation of MSCs [44]. However, few studies have investigated the role of insoluble matrix factors combined with elasticity in modulating vascular commitment of MSCs. Abdeen et al demonstrated a positive correlation between 2-dimensional fibronectin-modified hydrogels and stiffness towards proangiogenic signaling of MSCs [45]. Despite significant progress in defining MSC to vascular differentiation protocols, a fundamental understanding of how matrix ligand, in concert with tunable elasticity in 3-dimensional environments, as presented in this study, is largely missing.
We postulated that the merging of biological ligand with appropriate elasticity in 3-dimensional environment could augment or repress the fate commitment of stem cells into specific lineages. Our work revealed a strong dependence of matrix protein composition and elasticity on MSC cellular processes including adhesion, spreading and differentiation toward early vascular lineages. Interestingly, we observed a negative correlation between MSC adhesion and spreading with substrate stiffness. Though stiffer 2D substrates generally increase receptor-ligand activity leading to increased F-actin expression and thus cell spreading, our results showed a different trend with 3D matrices which might be caused by different focal adhesion mechanisms employed by 3D cell-matrix interaction compared to 2D cell-substrate [46]. In fact, evidence suggests that the creation of cellular focal adhesions on compliant 3-dimensional substrates is abrogated preventing cell traction forces, whereas rigid substrate analogs prevent cells from exerting sufficient force to deform their matrix [7]; with these events directly influence cellular spreading upon substrates. In contrast, the inclusion of biological ligands at specific densities can result in differential spreading of cells regardless of substrate stiffness. Trappmann and colleagues observed similar spreading of cells on stiff substrates with a 5-fold reduction in active ligand binding sites compared to neat soft substrates [19]. The mechanism by which ECM protein and substrate elasticity effects MSC adhesion and spreading here is not fully understood. From the cited literature, it is likely protein-elasticity crosstalk influences MSC attachment and spreading observed here. Probing the ability to activate and deactivate certain integrin signaling events via protein interaction with substrate stiffness may help elucidate these complex phenomena [47].
We evaluated the efficacy of our neotissue arrays to instruct MSC differentiation towards vascular lineage. Our data revealed a strong dependence of matrix protein composition and elasticity on MSC differentiation toward vascular lineages. Indeed, several combinatorial environments were arrived at that significantly up- or down- regulate expression of PECAM. For instance, we observed a negative correlation for PECAM expression for laminin on soft matrix, when compared to stiff which was comparably positive. Likewise, we found an inverse relationship between collagen I and elastin protein to down and upregulate PECAM expression on stiff but not soft substrate respectively. These findings are in agreement with previous observations that report the capacity of MSC differentiation in vascular lineages by individual stimuli such as elasticity [8] or biological ligand [4, 39–41]. For instance, the PECAM level affected by collagen I-related environments independently of the matrix elasticity may be explained by the lack of collagen I in normal endothelium (PECAM+) and increased collagen I content [48] in damaged endothelium to promote trans-differentiation of endothelial cells to mesenchymal cells [49]. We note that unlike previous studies, our data revealed the potential to augment or suppress cellular functions by coupling discrete protein combinations with appropriate matrix elasticity. Recent evidence suggests a significant crosstalk between ECM protein and underlying matrix elasticity. Fibronectin exhibits greater unfolding on stiffer substrates [20], improving expression of cell binding domains and cellular attachment of fibroblasts [18]. Further, it has recently been recognized that focal-adhesion kinase (FAK), a mechano-sensing integrin signal important for cellular differentiation [50], can be differentially activated based on ECM protein combined with substrate stiffness [51]. In line with that, ECM stiffness alone was found to induce malignant phenotypes in normal mammary epithelial cells, but this effect could be abrogated when accompanied by an increase in basement-membrane ligands [47]. These led to a mechanism whereby substrate stiffness coupled with ECM composition can modulate cellular phenotype, suggesting that substrate mechanical cues may instruct diverse effects on cell behavior depending on the presence and type of integrins presented. Our study reinforce the fact that distinct protein environments signal cellular phenomenon diversely when presented on matrices of different stiffness. This group represents MSC expression of PECAM that responds differently to the protein environments examined, depending on the matrix elasticity, and therefore MSC differentiation can be synergistically or antagonistically regulated by matrix elasticity and protein environments.
While different protein combinations induced differential cellular processes, we also demonstrated a significant dependence of matrix elasticity on the up-regulation of PECAM independent of the protein combination. It is widely accepted that matrix elasticity directs MSC differentiation and commitment into different lineages [5, 8]. These findings have been further validated with recent observations that stem cell differentiation is predominantly driven by the underlying substrate stiffness even in the presence of protein tethering [52]. Our data revealed a significant (p<0.05) up-regulation of vascular marker PECAM on stiff substrate (20 kPa) in comparison to soft substrate (4.6 kPa). The stiffness we report here for optimal vascular commitment of MSCs is in agreement with recent studies. For instance, Kshitiz et al linked optimum levels of PECAM expression of cardiac progenitor cells cultured in 3-dimensional-matrices approaching an elasticity of 16 kPa [53]. The authors suggested a mechanism by which the expression of VEGF receptor (VEGFR2) was enhanced under the effective elasticity observed, a mechanosensing pathway previously described for human microvascular endothelial (HMVE) cells [54]. Another recent study reported a pro-angiogenic secretome for MSCs cultured on substrates of 20 kPa stiffness when compared to softer substrates (2 kPa) [55]. These studies are complimentary to the findings we report here, whereby appropriate stiffness is likely crucial in the fate decisions afforded by stem cells in response to their ECM environment and specifically in the context of vascular regeneration.
5. Conclusion
We developed a high throughput method that allows for the rapid screening of a diversity of engineered microenvironments with tunable matrix elasticity and geometry, combined with specific ECM protein combination. This work highlights the importance and necessity of employing a systemic approach, whereby incorporating several environmental signals becomes necessary to establish optimal MSC differentiation protocols. Collectively, our data suggests that a complex milieu exists coupling protein functional behavior with substrate elasticity and that this phenomenon may potentially be exploited through proper application of high-throughput screening methodologies. Future studies will be focused on adapting this technology to instruct specific MSC differentiation processes by expanding the library of ECM proteins and experimental parameters employed.
Supplementary Material
Rat primary aorta smooth muscle cells (PASMCs) cultured on neotissue subarray a9 with elasticity 4.6 kPa; scale bar 500 μm (A). Magnification of PASMC cellular islands pertaining to specific protein conditions; scale bar 50 μm (A, right). Rat mesenchymal stem cells (MSCs) cultured on neotissue subarray a9 with elasticity 4.6 kPa for 72 h; scale bar 500 μm (B). Magnification of MSC cellular islands pertaining to specific protein conditions after 72 h culture (B, right).
(A, B) Results of 27 full factorial ANOVA of significant (p < 0.05) main and interaction effects for MSC adhesion (A) and spreading (B) and compared against either soft (4.6 kPa) or stiff (20 kPa) matrix conditions (n = 3 neotissue arrays). Colored dashed line represents significance (p < 0.05).
Acknowledgments
The authors would like to thank Mr. Markham and Linda Crnic Institute for Down Syndrome at University of Colorado at Denver Anschutz for use of the microarraying equipment and thank Dr. Stephanie Bryant and her laboratory for help with the PEGDM imaging. The authors would also like to thank the BioFrontiers Advanced Light Microscopy Core for their excellent microscopy and imaging support. This work is finically supported through NIH funding sources K25HL097246 (W.T.) and R01 HL119371 (W.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reilly GC, Engler AJ. Intrinsic extracellular matrix properties regulate stem cell differentiation. Biomechanics. 2010;43:55–62. doi: 10.1016/j.jbiomech.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 2.da Silva Meirelles L, Caplan AI, Nardi NB. In search of the in vivo identity of mesenchymal stem cells. Stem Cells. 2008;26:2287. doi: 10.1634/stemcells.2007-1122. [DOI] [PubMed] [Google Scholar]
- 3.Bajpai VK, Mistriotis P, Loh YH, Daley GQ, Andreadis ST. Functional vascular smooth muscle cells derived from human induced pluripotent stem cells via mesenchymal stem cell intermediates. Cardiovasc Res. 2012;96:391–400. doi: 10.1093/cvr/cvs253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki S, Narita Y, Yamawaki A, Murase Y, Satake M, Mutsuga M, et al. Effects of extracellular matrix on differentiation of human bone marrow-derived mesenchymal stem cells into smooth muscle cell lineage: utility for cardiovascular tissue engineering. Cells Tissues Organs. 2010;191(4):269–280. doi: 10.1159/000260061. [DOI] [PubMed] [Google Scholar]
- 5.Engler JA, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 6.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310(5751):1139, e43. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 7.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, et al. Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate. Nat Mater. 2010;9(6):518. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wingate K, Bonani W, Tan Y, Bryant S, Tan W. Compressive elasticity of threedimensional nanofiber matrix directs mesenchymal stem cell differentiation to vascular cells with endothelial or smooth muscle cell markers. Acta Biomater. 2012;8:1440. doi: 10.1016/j.actbio.2011.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Titmarsh DM, Chen H, Wolvetang EJ, Cooper-White J. Arrayed cellular environments for stem cells and regenerative medicine. Biotechnol J. 2013;8:167–179. doi: 10.1002/biot.201200149. [DOI] [PubMed] [Google Scholar]
- 10.Flaim CJ, Chien S, Bhatia SN. An extracellular matrix microarray for probing cellular differentiation. Nat Methods. 2005;2:119–125. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 11.Ghaedi M, Tuleuova N, Zern MA, Wu J, Revzin A. Bottom-up signaling from HGF-containing surfaces promotes hepatic differentiation of mesenchymal stem cells. Biochem Biophys Res Commun. 2011;407:295–300. doi: 10.1016/j.bbrc.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson DG, Levenberg S, Langer R. Nanoliter-scale synthesis of arrayed biomaterials and application to human embryonic stem cells. Nat Biotechnol. 2004;22:863–866. doi: 10.1038/nbt981. [DOI] [PubMed] [Google Scholar]
- 13.Mei Y, Saha K, Bogatyrev SR, Yang J, et al. Combinatorial development of biomaterials for clonal growth of human pluripotent stem cells. Nat Mater. 2010;9:768–778. doi: 10.1038/nmat2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gobaa S, Hoehnel S, Roccio M, Negro A, Kobel S, Lutolf MP. Artificial niche microarrays for probing single stem cell fate in high throughput. Nat Meth. 2011;8:949. doi: 10.1038/nmeth.1732. [DOI] [PubMed] [Google Scholar]
- 15.Wingate K, Floren M, Tan Y, Tseng Pi Ou N, Tan W. Tissue Engineering Part A. 2014;20:2503–2512. doi: 10.1089/ten.tea.2013.0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fozdar DY, Soman P, Lee JW, Han LH, Chen S. Three-dimensional polymer constructs exhibiting a tunable negative Poisson’s ratio. Adv Funct Mater. 2011;21:2712–2720. doi: 10.1002/adfm.201002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu J, Tang C, Kottke-Marchant K, Marchant RE. Design and synthesis of biomimetic hydrogel scaffolds with controlled organization of cyclic RGD peptides. Bioconjug Chem. 2009;20:333–339. doi: 10.1021/bc800441v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seo JH, Sakai K, Yui N. Adsorption state of fibronectin on poly(dimethylsiloxane) surfaces with varied stiffness can dominate adhesion density of fibroblasts. Acta Biomater. 2013;9:5493–5501. doi: 10.1016/j.actbio.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Trappmann B, et al. Extracellular-matrix tethering regulates stem-cell fate. Nature Mater. 2012;11:742–749. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 20.Antia M, Baneyx G, Kubow KE, Vogel V. Fibronectin in aging extracellular matrix fibrils is progressively unfolded by cells and elicits an enhanced rigidity response. Faraday Discuss. 2008;139:229–249. doi: 10.1039/b718714a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klotzsch E, Schoen I, Ries J, Renn A, Sandoghdare V, Vogel V. Biomater. Sci. 2014;2:883. doi: 10.1039/c3bm60262a. [DOI] [PubMed] [Google Scholar]
- 22.Hynes RO. The extracellular matrix: Not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole MA, Voelcker NH, Thissen H, Griesser HJ. Stimuli-responsive interfaces and systems for the control of protein-surface and cell-surface interactions. Biomaterials. 2009;30:1827–1850. doi: 10.1016/j.biomaterials.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 24.Carlson AL, Florek CA, Kim JJ, Neubauer T, Moore JC, Cohen CI, et al. Microfibrous substrate geometry as a critical trigger for organization, self-renewal, and differentiation of human embryonic stem cells within synthetic 3-dimensional microenvironments. FASEB. 2012;26:3240–3251. doi: 10.1096/fj.11-192732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim SH, Mao HQ. Electrospun scaffolds for stem cell engineering. Adv Drug Deliv Rev. 2009;61:1084–1096. doi: 10.1016/j.addr.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 26.MacBeath G, Schreiber SL. Printing Proteins as Microarrays for High-Throughput Function Determination. Science. 2000;289:1760. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 27.Soen Y, Mori A, Palmer TD, Brown PO. Exploring the regulation of human neural precursor cell differentiation using arrays of signaling microenvironments. Mol Syst Biol. 2006;2:37. doi: 10.1038/msb4100076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis GE, Senger DR. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 29.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89:957–989. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Battista S, Guarnieri D, Borselli C, Zeppetelli S, Borzacchiello A, Mayol L, Gerbasio D, Keene DR, Ambrosio L, Netti PA. The effect of matrix composition of 3D constructs on embryonic stem cell differentiation. Biomaterials. 2005;26:6194–6207. doi: 10.1016/j.biomaterials.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Fernandes TG, Kwon SJ, Bale SS, Lee MY, Diogo MM, Clark DS, et al. Three-Dimensional Cell Culture Microarray for High Throughput Studies of Stem Cell Fate. Biotechnol Bioeng. 2010;106:106–118. doi: 10.1002/bit.22661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolatshahi-Pirouz A, Nikkhah M, Gaharwar AK, Hashmi B, Guermani E, Aliabadi H, et al. A combinatorial cell-laden gel microarray for inducing osteogenic differentiation of human mesenchymal stem cells. Sci Rep. 2014;4:3896. doi: 10.1038/srep03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranga A, Lutolf MP. High-throughput approaches for the analysis of extrinsic regulators of stem cell fate. Curr Opin Cell Biol. 2012;24:236–244. doi: 10.1016/j.ceb.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Hui EE, Bhatia SN. Micromechanical control of cell–cell interactions. Proc Natl Acad Sci. 2007;104:5722. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khetani SR, Bhatia SN. Microscale Human Liver Tissue for Drug Development. Nature Biotechnology. 2007;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- 36.Oswald J, Boxberger S, Jørgensen B, Feldmann S, Ehninger G, et al. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells. 2004;22:377–384. doi: 10.1634/stemcells.22-3-377. [DOI] [PubMed] [Google Scholar]
- 37.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 38.Ahsan T, Nerem RM. Fluid shear stress promotes an endothelial-like phenotype during the early differentiation of embryonic stem cells. Tissue Eng Part A. 2010;16:3547–53. doi: 10.1089/ten.tea.2010.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kniazeva E, Kachgal S, Putnam AJ. Effects of extracellular matrix density and mesenchymal stem cells on neovascularization in vivo. Tissue Eng Part A. 2011;17:905–914. doi: 10.1089/ten.tea.2010.0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lozito TP, Taboas JM, Kuo CK, Tuan RS. Mesenchymal stem cell modification of endothelial matrix regulates their vascular differentiation. J Cell Biochem. 2009;107:706–713. doi: 10.1002/jcb.22166. [DOI] [PubMed] [Google Scholar]
- 41.Wang CH, Wang TM, Young TH, Lai YK, Yen ML. The critical role of ECM proteins within the human MSC niche in endothelial differentiation. Biomaterials. 2013;34:4223–4234. doi: 10.1016/j.biomaterials.2013.02.062. [DOI] [PubMed] [Google Scholar]
- 42.Portalska K, Leferink A, Groen N, Fernandes H, Moroni L, van Blitterswijk C, et al. Endothelial differentiation of mesenchymal stromal cells. PLoS One. 2012;7:e46842. doi: 10.1371/journal.pone.0046842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu CC, Chao YC, Chen CN, Chien S, Chen YC, Chien CC, et al. Synergism of biochemical and mechanical stimuli in the differentiation of human placenta-derived multipotent cells into endothelial cells. J Biomech. 2008;41:813–2. doi: 10.1016/j.jbiomech.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Yang F, Cho SW, Son SM, Hudson SP, Bogatyrev S, Keung L, et al. Combinatorial extracellular matrices for human embryonic stem cell differentiation in 3D. Biomacromolecules. 2010;11:1909–1914. doi: 10.1021/bm100357t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdeen AA, Weiss JB, Lee J, Kilian KA. Matrix composition and mechanics direct proangiogenic signaling from mesenchymal stem cells. Tissue Eng Part A. 2014;20:2737–45. doi: 10.1089/ten.tea.2013.0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cukierman E, Pankov R, Yamada KM. Cell interactions with three-dimensional matrices. Curr Opin Cell Biol. 2002;14:633–639. doi: 10.1016/s0955-0674(02)00364-2. [DOI] [PubMed] [Google Scholar]
- 47.Chaudhuri O, Koshy ST, Branco da Cunha C, Shin J, Verbeke CS, Allison KH, et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nature Mater. 2014;13:970–978. doi: 10.1038/nmat4009. [DOI] [PubMed] [Google Scholar]
- 48.Dean RG, Balding LC, Candido R, Burns WC, Cao Z, Twigg SM, et al. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53:1245–1256. doi: 10.1369/jhc.4A6560.2005. [DOI] [PubMed] [Google Scholar]
- 49.Breitbach M, Bostani T, Roell W, Xia Y, Dewald O, Nygren JM, et al. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood. 2007;110:1362–1369. doi: 10.1182/blood-2006-12-063412. [DOI] [PubMed] [Google Scholar]
- 50.Liao X, Lu S, Wu Y, Xu W, Zhuo Y, Peng Q, et al. The effect of differentiation induction on FAK and Src activity in live HMSCs visualized by FRET. PLoS One. 2013;27:8. doi: 10.1371/journal.pone.0072233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seong J, Tajik A, Sun J, Guan JL, Humphries MJ, Craig SE, et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc Natl Acad Sci. 2013;110:19372–19377. doi: 10.1073/pnas.1307405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, et al. Interplay of matrix stiffness and protein tethering in stem cell differentiation. Nature Mater. 2014;13:979–987. doi: 10.1038/nmat4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kshitiz ME, Hubbi EH, Ahn J, Downey J, Kim A, et al. Matrix rigidity controls endothelial differentiation and morphogenesis of cardiac precursors. Sci Signal. 2015;5:ra41. doi: 10.1126/scisignal.2003002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, et al. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature. 2009;457:1103–1108. doi: 10.1038/nature07765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seib FP, Prewitz M, Werner C, Bornhäuser M. Matrix elasticity regulates the secretory profile of human bone marrow-derived multipotent mesenchymal stromal cells (MSCs) Biochem Biophys Res Commun. 2009;389:663. doi: 10.1016/j.bbrc.2009.09.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rat primary aorta smooth muscle cells (PASMCs) cultured on neotissue subarray a9 with elasticity 4.6 kPa; scale bar 500 μm (A). Magnification of PASMC cellular islands pertaining to specific protein conditions; scale bar 50 μm (A, right). Rat mesenchymal stem cells (MSCs) cultured on neotissue subarray a9 with elasticity 4.6 kPa for 72 h; scale bar 500 μm (B). Magnification of MSC cellular islands pertaining to specific protein conditions after 72 h culture (B, right).
(A, B) Results of 27 full factorial ANOVA of significant (p < 0.05) main and interaction effects for MSC adhesion (A) and spreading (B) and compared against either soft (4.6 kPa) or stiff (20 kPa) matrix conditions (n = 3 neotissue arrays). Colored dashed line represents significance (p < 0.05).