

Abstract
Infection with influenza virus has been a significant cause of morbidity and mortality for more than a hundred years. Severe disease and increased mortality often results from bacterial super-infection of patients with influenza virus infection. Preceding influenza infection alters the host’s innate and adaptive immune responses, allowing increased susceptibility to secondary bacterial pneumonia. Recent advances in the field have helped to define how influenza alters the immune response to bacteria through the dysregulation of phagocytes, antimicrobial peptides, and lymphocytes. Viral-induced interferons play a key role in altering the phenotype of the immune response. Potential genetic modifiers of disease will help to define additional immunologic mechanisms that predispose to viral, bacterial super-infection with the overarching goal of developing effective therapeutic strategies to prevent and treat disease.
Introduction
Infection with influenza virus is a significant cause of morbidity and mortality throughout the world. Severe disease and increased mortality can often result from bacterial super-infection primarily with the Gram-positive organisms, Staphylococcus aureus or Streptococcus pneumoniae. This review discusses the recent advances in our understanding of the immunological mechanisms by which influenza A virus infection increases the susceptibility to secondary bacterial pneumonia and how this might inform future strategies to prevent or treat this lethal combination.
Seasonal influenza infection occurs annually, and baseline immunity to seasonal influenza infections exists within communities due to prior exposure. Influenza pandemics occur when a new, highly pathogenic virus strain emerges for which there is no immunity within the human population. Pandemic viruses spread easily from person to person across a wide geographic area, affecting a large proportion of the population. During the 1918 pandemic of influenza A virus H1N1, more than 50 million people died from influenza and bacterial super-infection [1]. During the 2009 pandemic of influenza A virus H1N1, 25–50% of hospitalized virus-infected patients were super-infected with bacterial pneumonia [2–7], and super-infection was associated with higher morbidity and mortality [2,6,8–10]. Vulnerability to secondary bacterial infection peaks at approximately one week post-influenza infection (Figure 1). Influenza virus infection facilitates secondary bacterial infection through multiple immunological mechanisms. Preceding influenza virus infection leads to the dysregulation of both innate and adaptive immune responses, predisposing the host to secondary bacterial infection. Understanding the immune mechanisms that predispose patients with influenza virus infection to bacterial super-infection is paramount to preventing deaths in future influenza virus pandemics.
Figure 1.
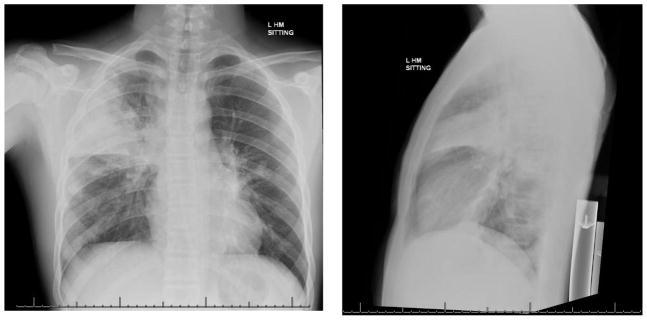
Chest radiographs of a child with influenza A H1N1 super-infected with methicillin-resistant Staphylococcus aureus. The window of vulnerability to secondary bacterial super-infection typically occurs typically one week post-influenza.
Influenza-induced defects in innate immunity against bacteria
Alveolar macrophages reside in the normal airway and are the first cells to defend against bacteria. Additionally, macrophages and neutrophils are recruited to the airways by cytokines and chemokines in order to ingest and kill bacteria. Preceding influenza infection causes dysregulation of both macrophages and neutrophils, limiting the ability to defend against subsequent bacterial infection (Figure 2).
Figure 2.

Preceding influenza attenuates innate host defense against secondary bacterial infection. In the context of bacterial infection alone, alveolar macrophages (Mac) recognize pathogens via pattern recognition receptors initiating an inflammatory cascade. Cytokines, antimicrobial peptides (AMPs), and reactive oxygen species (ROS) are generated by macrophages, recruited neutrophils, and the lung epithelium resulting in pathogen clearance. Preceding influenza (IAV) results in impaired macrophage and neutrophil killing of bacteria and decreased extracellular mediators (AMPs).
Alterations in phagocyte quantity
Recent work has investigated how preceding influenza virus infection affects the number of macrophages and neutrophils available in the airway to fight bacterial infection. It has been shown that influenza virus infection resulted in the loss of 90% of mouse resident alveolar macrophages by one week post-infection, with the remaining 10% of macrophages displaying a necrotic phenotype [11]. Cell death was thought to be related to a secondary necrotic process due to an increased number of damaged macrophages (measured by distorted nuclei and an increase in the number of cytoplasmic vacuoles) in the airspace of influenza virus-infected mice compared with mock-infected mice. Recruited inflammatory monocytes replaced the resident cells during bacterial super-infection. This alteration of innate cells in the influenza virus-infected lung resulted in an early defect in S. pneumoniae uptake at 3 hours post-bacterial challenge. The alveolar macrophage population was fully replaced by two weeks post viral infection and early innate host defense to S. pneumonia was restored.
Earlier work has demonstrated that influenza virus super-infection with S. aureus or S. pneumoniae resulted in enhanced neutrophilic inflammation in the lungs at time points mimicking human susceptibility to co-infection. More recent studies have confirmed these data. Mice infected with S. aureus six days after administration of influenza virus had higher numbers of neutrophils in bronchoalveolar lavage fluid 24 hours later compared with mice that received bacteria alone [12]. Mice infected with S. pneumoniae seven days after receiving influenza virus also had higher numbers of neutrophils in the bronchoalveolar lavage and lung tissue and higher bacterial load in the lung compared to mice that received bacteria alone [13]. In these studies, higher numbers of neutrophils recruited to the airways correlated with increased mortality. However, neutrophil depletion (using an anti-Ly6G-specific antibody) showed that neutrophils did not have a significant effect on S. pneumoniae burden or morbidity, assessed by body weight loss, during super-infection, suggesting a neutrophil-independent mechanism for pathogenesis [13].
A potential mechanism by which Panton-Valentine leukocidin (PVL)-producing USA 300 clonotype S. aureus may be taking advantage of neutrophil recruitment during co-infection is through the lysis of neutrophils resulting in the release of proteases. PVL is a pore-forming exotoxin produced by certain strains of S. aureus. The PVL-producing USA 300 clonotype of S. aureus (also known as methicillin-resistant S. aureus; MRSA) has emerged globally in recent years as an important human pathogen [14]. Human neutrophils exposed to influenza virus for three hours and subsequently incubated with media from cultured MRSA showed an increased rate of neutrophil cell death compared to neutrophils exposed to MRSA alone. In addition, there was an upregulation of the cell surface marker CD11b, indicating neutrophil activation, in the neutrophils exposed to both influenza virus and MRSA compared to either pathogen alone [15]. Influenza and bacterial super-infection with MRSA could potentially increase neutrophil recruitment to the lung and augment PVL-related damage to neutrophils.
Other studies using a pre-clinical murine model of bacterial superinfection have shown suppression of neutrophil recruitment in response to bacterial infection following the resolution of influenza infection. Mice challenged with S. pneumoniae at both two and six weeks post-infection with murine adapted influenza virus (PR8) had substantially reduced numbers of neutrophils in bronchoalveolar lavage fluid 24 hours after bacterial infection compared with mice that received bacteria alone. At these time points, influenza virus was undetectable in the lungs, and pre-bacterial challenge lung cellularity and cytokine levels had returned to the pre-influenza infection levels. The decreased recruitment of neutrophils was linked to a sustained desensitization of macrophages to Toll-like receptor (TLR) ligands [16], suggesting that influenza virus induced a prolonged refractory state of the innate immune response. Consistent with this, nuclear translocation of the p65 subunit of nuclear factor-κB (NF-κB) was inhibited in macrophages, but not in airway epithelial cells, in response to a TLR5 agonist (flagellin). There was further evidence that TLR2 and TLR4, in addition to TLR5, were similarly affected by influenza virus infection. In summary, the level of neutrophil recruitment after bacterial super-infection was dependent on the timing of bacterial challenge relative to influenza infection. At days 3 – 7 days post-influenza infection, enhanced neutrophil recruitment was seen with secondary bacterial challenge. At weeks 2 – 6 post-influenza infection, bacterial super-infection resulted in lower numbers of neutrophils recruited to the co-infected airways, which was associated with sustained desensitization of alveolar macrophages to bacterial toll-like receptor ligands. Despite increased numbers of neutrophils during influenza complicated by bacterial super-infection, mouse models in which neutrophils have been depleted show no difference in bacterial burden compared to those with no neutrophil depletion, suggesting that dysregulation of phagocyte function or phagocyte-independent mechanisms play a more important role during influenza and bacterial super-infection.
Alterations in phagocyte function
Ongoing investigation has explored the function of both macrophages and neutrophils in the airway during influenza and bacterial super-infection. Recent work has demonstrated that influenza virus does not induce a defect in S. aureus uptake by alveolar macrophages ex vivo or in vivo 24 hours after challenge, suggesting a phagocyte-independent mechanism for pathogenesis [17]. Somewhat contrary to these findings, other investigators have demonstrated functional impairment of both macrophages and neutrophils associated with a decreased generation of intracellular reactive oxygen species (ROS) [18]. Oxidative burst is a process by which nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) produces ROS, toxic compounds used to kills bacteria within phagocytes. NADPH oxidase-deficient mice, gp91(phox −/−), failed to clear S. aureus sufficiently from the lungs, both with and without preceding influenza virus infection. In addition, mosaic gp91(phox +/−) mice, that have both gp91phox WT and deficient neutrophils, also failed to clear S. aureus from the lungs, both with and without preceding influenza virus infection. These data suggest a functional defect in phagocytes during super-infection. In further support of a role for reactive oxygen species (ROS), overexpression of granulocyte/macrophage colony-stimulating factor resulted in an elevated production of ROS by macrophages and decreased morbidity, mortality and bacterial burden in influenza virus-S. aureus super-infection [19]. Although different laboratories have demonstrated disparate findings regarding phagocyte function during co-infection, these differences can be explained by timing of infection, timing of phagocyte analysis, virus and bacteria dosing, and different strains of virus and bacteria.
In addition to phagocytosis and ROS-dependent killing, neutrophils can kill bacteria by extruding neutrophil extracellular traps (NETs) [20,21]. The cytotoxic effects of NETs are related to their antimicrobial protein components such as histones, defensins and myeloperoxidase. It has been reported that NETs entangle with alveolar-capillary surfaces of the lungs during influenza virus infection, potentially causing lung damage [22]. Recent work has shown that influenza virus infection that is complicated by secondary S. pneumoniae was associated with increased NET generation [23]. However, despite the high levels of NETs released in super-infection, they did not participate in bacterial killing ex vivo. In fact, they may promote increased pulmonary pathology owing to the release of neutrophil-associated cytotoxic proteins. Extracellular histones released by NETs are known to contribute to inflammation during sepsis [24]. In summary, current evidence supports the concept that defects in macrophage and neutrophil function are induced by primary influenza virus infection in certain contexts.
Antimicrobial peptides
In addition to phagocyte-dependent mechanisms enhancing susceptibility to secondary bacterial infections, there are phagocyte-independent mechanisms of pathogenesis. Recent work has shown that preceding influenza virus infection suppressed the production of interleukin-17 (IL-17)- and IL-22-associated antimicrobial peptides (such as lipocalin 2, CAMP, REG3B, S100A8 and S100A9) in mouse lungs in response to secondary S. aureus challenge [17]. Furthermore, exogenous supplementation of the antimicrobial peptide lipocalin 2 improved S. aureus clearance and reduced inflammation in super-infected mice. These data suggest an additional innate defect in the setting of influenza virus infection that increases susceptibility and severity of bacterial super-infection. Regulation of antimicrobial peptide production during super-infection in humans has not been evaluated.
Antiviral interferons
Seminal work has identified the antiviral IFN pathway as an important mechanism by which influenza virus suppresses extracellular pathogen host defense. Studies have investigated both type I IFNs (IFNα and IFNβ) and type II IFN (IFNγ) and their respective roles in influenza virus and bacterial super-infection. Mice deficient in type I IFN receptor 1 (Ifnar1−/− mice) that were infected with influenza virus and then, 5 days later, challenged with S. pneumoniae were protected from secondary bacterial infection. Of note, Ifnar1−/− and wild-type mice infected only with influenza virus showed similar lung viral burden or weight loss at days 5 or 7 post-infection. However, wild-type mice had attenuated production of CXCL1 and CXCL2 during super-infection compared with mice infected with S. pneumoniae alone, which was thought to be a potential mechanism for increased sensitivity to bacterial pneumonia [25]. More recently, the protein lysine methyltransferase Setdb2 was characterized as an interferon-stimulating gene and found to regulate CXCL1 expression and recruitment of neutrophils. Setdb2GT/GT mice with influenza super-infected with S. penumoniae 5 days later demonstrated increased CXCL1, increased neutrophil recruitment, and lower bacterial burden compared to wild-type mice [26].
Influenza virus-induced type I IFNs have also been shown to inhibit IL-23 production, which is required for the generation of type 17 immunity and for effective clearance of secondary S. aureus pneumonia [12]. Indeed, influenza virus-infected mice that were challenged (6 days later) with S. aureus had reduced numbers of IL-17-producing γδT cells and IL-17-producing CD4+ T cells compared with mice challenged with bacteria alone [12]. Similar results were found for S. pneumoniae super-infection [27]. Moreover, adoptive transfer of Ifnar1−/− γδT cells into wild-type mice reduced their susceptibility to secondary S. pneumoniae infection [27]. Interestingly, mice that were colonized with S. pneumoniae and subsequently infected with influenza virus showed synergistic stimulation of type I IFNs and this diminished the recruitment of macrophages through decreased levels of CC-chemokine ligand 2 (CCL2; also known as MCP1) production, resulting in increased bacterial colonization [28]. Therefore, type I IFN signalling potentially interferes with both neutrophil recruitment and IL-17 responses, suggesting a mechanism for decreased bacterial clearance. However, the question arises - is type I IFN production sufficient for exacerbation of secondary bacterial infection? Poly I:C and TLR7 ligands used to induce type I IFNs in the absence of virus exacerbated both S. aureus and S. pneumoniae burden in the lungs [29]. However, studies in our laboratory using exogenous IFNβ did not show increased susceptibility to S. aureus infection (Robinson & Alcorn, unpublished observations). So, although type I IFNs are probably necessary for exacerbation of secondary bacterial infection, it remains unclear whether they are sufficient.
Additional work has been done to identify the role of IFNγ in the immunopathogenesis of super-infection. It has been shown that mice deficient in IFNγ-mediated signalling were protected from influenza virus-S. pneumoniae super-infection. Influenza virus-induced IFNγ production was shown to inhibit phagocytosis of bacteria by macrophages and direct application of IFNγ downregulated the expression of the class A scavenger receptor MARCO (macrophage receptor with collagenous structure) on macrophages [30]. More recent work has shown that IL-13 downregulates the production of IFNγ at early time points (3 days post-influenza), when susceptibility to super-infection with S. aureus was reduced. At later time points during influenza infection (7 days post-influenza), mice had low IL-13 levels and elevated IFNγ levels, but were more susceptible to S. aureus super-infection. Blocking of the decoy receptor IL-13Rα2 resulted in reduced bacterial burden when given to mice during the period of increased susceptibility (6 days post-influenza) to bacterial super-infection, consistent with increased levels of IL-13[31]. Conversely, another investigation has shown that Ifng−/− mice were susceptible to influenza virus-S. aureus super-infection, similar to wild-type mice [12]. This difference may be explained by the use of different strains of bacteria, different doses of influenza and/or bacteria or different timing of infection. In summary, IFNs are highly expressed during viral infections and likely have an important role in the inhibition of bacterial host defense by influenza virus, but the molecular mechanisms involved warrant further exploration.
Influenza-induced defects in lymphocytes and antibacterial host defense
Peak susceptibility to secondary bacterial pneumonia occurs approximately one week into influenza infection. At this time, innate γδ T cells, NK cells, innate lymphoid cells, and adaptive αβ T cells have migrated to the airways as part of the immune response to viral infection. Preceding influenza infection alters the immune response of these T cells when challenged with secondary bacterial infection (Figure 3).
Figure 3.

Influenza infection results in inhibition of Type 17 immunity in the lung. Bacterial infection induces robust IL-17, IL-22, and TNF-α production in the lung. This process mediates inflammation and antimicrobial host defense. In the context of preceding influenza (IAV), type I IFNs inhibit Type 17 immunity by attenuating IL-1β and IL-23 production by macrophages and dendritic cells. IL-17A and IL-22 production by γδ and CD4+ T cells is markedly reduced resulting in impaired host defense against bacterial challenge.
Type 17 immunity
Both γδ T cells and T helper 17 cells are part of the type 17 immune response, releasing IL-17 and IL-22 in response to pathogens. Prior infection with influenza virus attenuates S. aureus-induced (or S. pneumoniae-induced) IL-17 production by TH17 cells and γδ T cells in mouse lungs [12] and transfer of exogenous γδ T cells improves the clearance of bacteria following super-infection [27]. Compared with wild-type mice, Il22−/− mice display increased bacterial burden and increased mortality upon S. pneumoniae super-infection [32]. Manipulation of this pathway during influenza virus infection by addition of the type 17-inducing cytokines IL-1β or IL-23, rescued IL-17 and IL-22 production following bacterial challenge and improved S. aureus clearance [12,33]. In outbred mice, the ability to generate IL-1β in response to secondary S. aureus infection was correlated with decreased morbidity [34]. IL-27, known to inhibit the development of Type 17 cells through the induction of STAT1, has recently been shown to play a role in influenza and bacterial super-infection. Mice deficient in IL-27 signaling (IL-27R) were less susceptible to secondary S. pneumoniae and S. aureus during influenza infection [35] (Robinson & Alcorn, unpublished data). In summary these data indicate an important role for the suppression of type 17 immunity as a mechanism of secondary bacterial co-infection susceptibility and severity.
Natural Killer, T regulatory, and Innate Lymphoid Cells
Additional work has indicated that influenza virus infection may attenuate natural killer (NK) cell responses to S. aureus in the lungs [36]. Compared with control mice, suppression of NK cells in influenza virus-infected mice resulted in reduced tumor necrosis factor (TNF) production and increased S. aureus burden during bacterial super-infection. NK cell-derived TNF led to the induction of alveolar macrophage activity against S. aureus. Macrophages were otherwise attenuated by NK cells through cell to cell interactions but could be activated by exposure to exogenous TNF or IL-15 (which recruits NK cells). Furthermore, recent work has shown that the loss of CD200R (cd200r −/− mice) from antigen presenting cells rescued secondary S. pneumonia exacerbation during influenza infection. NK cells were increased in these mice compared to wild-type [37].
A possible role for regulatory T (TReg) cells in susceptibility to secondary bacterial pneumonia has also emerged, as production of the anti-inflammatory cytokine IL-10 has been shown to be increased during influenza virus-S. pneumoniae super-infection [38]. In addition, neutralization of IL-10 with specific antibody decreased S. pneumoniae growth and mortality in mice [39]. Although TReg cells are known to produce IL-10 during influenza virus infection [40], it is not clear whether TReg cells are responsible for the IL-10 production during super-infection. Interestingly, treatment of super-infected mice with an indoleamine 2,3-dioxygenase (IDO) inhibitor resulted in decreased IL-10 production and improved S. pneumoniae clearance [41].
Finally, a potential role for innate lymphoid cells (ILCs) in super-infection has been examined. ILCs expressing ST2 (also known as IL-1RL1) termed ILC2s have been found to accumulate in lung tissue following influenza virus infection, and depletion of ILC2s or blockade of ST2 resulted in loss of integrity of the airway epithelium [42]. Il1rl1−/− mice have deficient ILC2 function, and influenza virus-S. pneumoniae super-infection in these mice resulted in increased bacterial burden compared with wild-type mice. Increased inflammation is suggested by elevated levels of IL-6, IL-1β, CXCL1, IL-10 and IL-33 in Il1rl1−/− mice compared with wild-type mice [43]. It is possible that ILC2s protect against denudation of the airway epithelium during influenza virus infection, allowing for a more severe secondary bacterial infection. Although ST2 only played a limited role in the development of bacterial super-infection in this investigation, the secondary bacterial challenge was performed at 14 days post-influenza infection, well outside the typical window of susceptibility to secondary bacterial pneumonia.
Together, these data suggest NK cells, TReg cells and ILCs may potentially play a role in host defense against secondary bacterial infection during influenza infection. Overall, it remains likely that dysregulated inflammation is the key to increased susceptibility to secondary bacterial pneumonia.
Genetic susceptibility to infection
Over the past decade, human and mouse genetic studies have identified pathways that are required for host immunity to S. pneumoniae and S. aureus, which may provide clues to the pathways that are perturbed by influenza virus infection. Patients with mutations that affect TLR/IL-1 receptor signaling, such as in the adaptor proteins myeloid differentiation primary-response gene 88 (MYD88) and IL-1R-associated kinase 4 (IRAK4), have increased susceptibility to pyogenic infections, including meningitis, sepsis and abscesses due to S. pneumoniae and S. aureus [44]. Interestingly, these patients can mount pathogen-specific T and B cell responses but are unable to make glycan-specific antibody responses to pneumococcus. In mouse models, MyD88 is essential for innate immunity to S. pneumoniae [45]. Consistent with a defect in innate immunity, patients that have mutations in NF-κB essential modulator (NEMO) and IKBA, which result in impaired NF-κB signalling downstream of TLRs and cytokine receptors, also suffer from pyogenic infections with S. pneumoniae and S. aureus. These patients also have impaired immunity against mycobacterial disease.
S. aureus pneumonia is also a complication of hyper IgE syndrome (HIES) which is due to mutations in signal transducer and activator of transcription 3 (STAT3) [46]. Patients with HIES lack bacteria-specific TH17 cells and have reduced STAT3 signalling in non-myeloid cells. Supernatants from cultures of activated T cells from these patients failed to mediate S. aureus-induced antimicrobial activity, including chemokines and antimicrobial peptides, in skin keratinocytes and lung epithelial cells [47]. These data suggest a link between defective TH17 cell responses and abnormal mucosal immunity against S. aureus in humans. It has recently been shown that the acute phase response to S. pneumoniae is regulated by hepatic expression of STAT3 and RELA and this could explain in part the increased susceptibility of HIES patients to infection. In addition, MRSA can be cleared from the lungs in the absence of T cells, B cells or NK cells but clearance was attenuated in the absence of STAT3 expression in the lung epithelium [48]. One downstream regulator of this response was REG3γ, a STAT3-dependent soluble C-type lectin that can bind S. aureus [48]. Thus the susceptibility of HIES patients to S. aureus pulmonary infection could represent defects in STAT3 signalling in both the myeloid and non-myeloid compartment. Although there is currently no evidence that the aforementioned genetic mutations have a role in influenza virus-bacteria super-infection, we speculate that they may influence super-infection in addition to bacterial infection alone. Consistent with this hypothesis, MyD88 is known to be indispensable for host defense against primary influenza virus infection [49] and NEMO-deficient patients are susceptible to other severe viruses such as adenovirus, herpes simplex virus and cytomegalovirus [44]. STAT3 is a negative regulator of type I IFNs [50], and as mentioned, type I IFNs are known to inhibit IL-23 and subsequent S. aureus-induction of type 17 immunity, allowing for enhanced susceptibility to secondary bacterial pneumonia [12].
Therapeutic opportunities
Various approaches for preventing and treating influenza and secondary bacterial pneumonia have been tested in mice. A wide range of antibiotics and anti-inflammatories have recently been investigated. The steroid dexamethasone was shown to limit inflammation during influenza virus-S. pneumoniae super-infection, but did not improve morbidity or the outcome of disease [13]. Furthermore, early dexamethasone treatment during influenza virus infection worsened viral burden [51]. However, the combination of dexamethasone with ampicillin or azithromycin improved S. pneumoniae clearance, decreased lung pathology and improved survival in these two studies. Ampicillin treatment alone resulted in increased neutrophil recruitment and mortality in a TLR2-dependent manner, likely due to excessive S. pneumoniae lysis [52]. Azithromycin alone reduced S. pneumonia burden, but did not improve lung injury [13,52]. Linezolid, an antibiotic that is active against MRSA by binding to the 50S ribosomal subunit and inhibiting protein synthesis, has also been shown to reduce secondary bacterial pneumonia in experimental models by attenuating IFNγ expression [53]. Mice treated with recombinant IFNγ prior to bacterial challenge with S. pneumoniae seven days post-influenza virus infection showed partially reversed protective effects of linezolid. Thus, some antibiotic approaches may work through immune modulation in addition to their direct antimicrobial properties. Triple therapy with linezolid, clindamycin and vancomycin decreased inflammatory cytokine production, S. aureus burden, and morbidity during influenza virus super-infection [54]. These data suggest that antibiotic or steroid therapies that limit inflammation may be of some benefit during influenza and secondary bacterial co-infection.
However, as we understand more about the immunological susceptibility to bacterial super-infection, new potential immunotherapeutic strategies may become available. Therapies aimed at inhibiting chemokines, chemokine receptors, or TLRs warrant investigation. Recently, small molecule inhibitor of CCR2 (the receptor for CCL2) was evaluated in influenza virus-S. pneumoniae infection [55]. Inhibition of CCR2 decreased lung injury, morbidity and mortality in mice without impacting bacterial burden or later antibody responses to influenza virus. However, attempts to limit neutrophil recruitment with neutralizing antibody specific for CXCL2 during super-infection provided no improvement [13]. A TLR4 agonistic monoclonal antibody, UT12, has also been investigated during influenza virus-S. pneumoniae super-infection. Mice received UT12 prior to both influenza virus administration and S. pneumoniae bacterial challenge two days post-influenza infection. In this case, UT12 hastened macrophage recruitment induced by C-JUN N-terminal kinase (JNK) and NF-κB pathway-dependent CCL2 production [56]. While we continue to investigate the potential of developing targeted prophylaxis, rapid clinical recognition of bacterial pneumonia complicating influenza infection and rapid implementation of therapeutic antibiotics by clinicians remains the most prudent strategies to reduce post-influenza bacterial pneumonia.
Conclusions
In recent years, there has been renewed interest in understanding the immunologic mechanisms of influenza virus complicated by bacterial super-infection. The role of phagocytes remains a critical area of investigation. Resident alveolar macrophages may be depleted by influenza infection, allowing for enhanced susceptibility to bacterial super-infection. Enhanced neutrophilia has been demonstrated during the window of susceptibility to bacterial super-infection, although neutrophil depletion does not alter the outcome of mortality or bacterial burden. Preceding influenza may alter the function of phagocytes related to a decreased generation of intracellular reactive oxygen species, although different laboratories continue to find inconsistencies in the data supporting the dysregulation of phagocyte-mediated clearance of bacteria. This may be a result of the fine details of experimental models. Antimicrobial peptides have recently been shown to play a key role in the pathogenesis of viral, bacterial super-infection. The mechanisms by which virus-induced interferons interfere with normal antibacterial defense remains an active area of research. Both Type I (αβ) and Type II (γ) interferons have been show to play a key role, with Type I IFN interfering with both neutrophil recruitment and Type 17 immunity, and Type II IFN interfering with the expression of the scavenger receptor MARCO. Although interferons are necessary for the development of influenza, bacterial super-infection, more investigation needs to be done to determine the sufficiency of interferons in the observed disease pathogenesis. The role of T cells, both innate and adaptive, has also emerged as a mechanism for susceptibility. The altered T cell phenotypes that develop during resolving influenza infection likely impact innate antibacterial immunity against secondary bacterial infection. As additional genetic mutations and genetic modifiers of human disease are discovered, we will likely uncover additional immunologic mechanisms that predispose to both viral and bacterial infections. Finally, the challenge remains to develop effective therapeutic approaches that will limit acute lung injury during viral infection, while preserving bacterial clearance pathways. Basic science approaches to dissect the molecular and cellular pathways of influenza and complicating bacterial super-infection will inform novel clinical strategies to strategically target disease pathogenesis.
Highlights.
Influenza complicated by bacterial infection increases morbidity and mortality
Preceding influenza causes defects in antibacterial phagocyte function
Preceding influenza attenuates antibacterial antimicrobial peptide production
Preceding influenza causes defects in lymphocyte-mediated immunity
Genetic mutations can predispose to both viral and bacterial infections
Acknowledgments
Funding sources include Parker B. Francis Fellowship (KMR), NIH NHLBI R01 HL107380 (JFA) and R37 HL079142 (JKK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. The Journal of infectious diseases. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rice TW, Rubinson L, Uyeki TM, Vaughn FL, John BB, Miller RR, 3rd, Higgs E, Randolph AG, Smoot BE, Thompson BT. Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Critical care medicine. 2012;40:1487–1498. doi: 10.1097/CCM.0b013e3182416f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shannon S. Surveillance for Pediatric Deaths Associated with 2009 Pandemic Influenza A (H1N1) Virus Infection --- United States, April--August 2009. CDC MMWR Weekly. 2009;58:941–947. [PubMed] [Google Scholar]
- 4.Shieh WJ, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, Sumner J, Liu L, Patel M, Batten B, et al. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. The American journal of pathology. 2010;177:166–175. doi: 10.2353/ajpath.2010.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blyth CC, Webb SA, Kok J, Dwyer DE, van Hal SJ, Foo H, Ginn AN, Kesson AM, Seppelt I, Iredell JR. The impact of bacterial and viral co-infection in severe influenza. Influenza and other respiratory viruses. 2013;7:168–176. doi: 10.1111/j.1750-2659.2012.00360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randolph AG, Vaughn F, Sullivan R, Rubinson L, Thompson BT, Yoon G, Smoot E, Rice TW, Loftis LL, Helfaer M, et al. Critically ill children during the 2009–2010 influenza pandemic in the United States. Pediatrics. 2011;128:e1450–1458. doi: 10.1542/peds.2011-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gill JR, Sheng ZM, Ely SF, Guinee DG, Beasley MB, Suh J, Deshpande C, Mollura DJ, Morens DM, Bray M, et al. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Archives of pathology & laboratory medicine. 2010;134:235–243. doi: 10.5858/134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palacios G, Hornig M, Cisterna D, Savji N, Bussetti AV, Kapoor V, Hui J, Tokarz R, Briese T, Baumeister E, et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PloS one. 2009;4:e8540. doi: 10.1371/journal.pone.0008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen T, Kyle UG, Jaimon N, Tcharmtchi MH, Coss-Bu JA, Lam F, Teruya J, Loftis L. Coinfection with Staphylococcus aureus increases risk of severe coagulopathy in critically ill children with influenza A (H1N1) virus infection. Critical care medicine. 2012;40:3246–3250. doi: 10.1097/CCM.0b013e318260c7f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ampofo K, Herbener A, Blaschke AJ, Heyrend C, Poritz M, Korgenski K, Rolfs R, Jain S, da Carvalho MG, Pimenta FC, et al. Association of 2009 pandemic influenza A (H1N1) infection and increased hospitalization with parapneumonic empyema in children in Utah. The Pediatric infectious disease journal. 2010;29:905–909. doi: 10.1097/INF.0b013e3181df2c70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11**.Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. Journal of immunology. 2013;191:1250–1259. doi: 10.4049/jimmunol.1300014. The authors demonstrate that influenza virus infection results in the loss of 90% of murine resident alveolar macrophages by one week post-influenza infection. They use new methodology to differentiate between resident alveolar macrophages, resident interstitial macrophages, and recruited macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. Journal of immunology. 2011;186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Damjanovic D, Lai R, Jeyanathan M, Hogaboam CM, Xing Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. The American journal of pathology. 2013;183:868–880. doi: 10.1016/j.ajpath.2013.05.016. The authors show that neutrophil depletion does not have a significant effect on morbidity (weight loss) or bacterial burden during influenza, S. pneumoniae super-infection. [DOI] [PubMed] [Google Scholar]
- 14.Mediavilla JR, Chen L, Mathema B, Kreiswirth BN. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA) Current opinion in microbiology. 2012;15:588–595. doi: 10.1016/j.mib.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Niemann S, Ehrhardt C, Medina E, Warnking K, Tuchscherr L, Heitmann V, Ludwig S, Peters G, Loffler B. Combined action of influenza virus and Staphylococcus aureus panton-valentine leukocidin provokes severe lung epithelium damage. The Journal of infectious diseases. 2012;206:1138–1148. doi: 10.1093/infdis/jis468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt LS, Lambrecht BN, Sirard JC, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. The Journal of experimental medicine. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17**.Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, Enelow RI, Chan YR, Kolls JK, Alcorn JF. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. The Journal of infectious diseases. 2014;209:865–875. doi: 10.1093/infdis/jit527. Authors demonstrate that preceding influenza infection augments the production of IL-17- and IL-22- associated antimicrobial peptides in response to secondary S. aureus super-infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Sun K, Metzger DW. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. Journal of immunology. 2014;192:3301–3307. doi: 10.4049/jimmunol.1303049. In this study, authors show that preceding influenza likely causes a functional impairment of both macrohpages and neutrophils against bacteria associated with a decreased generation of intracellular reactive oxygen species. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramaniam R, Barnes PF, Fletcher K, Boggaram V, Hillberry Z, Neuenschwander P, Shams H. Protecting Against Post-influenza Bacterial Pneumonia by Increasing Phagocyte Recruitment and ROS Production. The Journal of infectious diseases. 2014;209:1827–1836. doi: 10.1093/infdis/jit830. [DOI] [PubMed] [Google Scholar]
- 20.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 21.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. The Journal of cell biology. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. The American journal of pathology. 2011;179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayana Moorthy A, Narasaraju T, Rai P, Perumalsamy R, Tan KB, Wang S, Engelward B, Chow VT. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Frontiers in immunology. 2013;4:56. doi: 10.3389/fimmu.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nature medicine. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. The Journal of clinical investigation. 2009;119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26**.Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminathan S, Bosnjak B, Bauer L, Kandasamy RK, Griesshammer IM, Kosack L, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nature immunology. 2015;16:67–74. doi: 10.1038/ni.3046. The authors demonstrate that the protein lysine methyltransferase Setdb2 regulates neutrophil recruitment during influenza and bacterial super-infection through the expression of the chemokine CXCL1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. Journal of virology. 2012;86:12304–12312. doi: 10.1128/JVI.01269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. The Journal of clinical investigation. 2011;121:3657–3665. doi: 10.1172/JCI57762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian X, Xu F, Lung WY, Meyerson C, Ghaffari AA, Cheng G, Deng JC. Poly I:C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PloS one. 2012;7:e41879. doi: 10.1371/journal.pone.0041879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nature medicine. 2008;14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 31*.Rynda-Apple A, Harmsen A, Erickson AS, Larson K, Morton RV, Richert LE, Harmsen AG. Regulation of IFN-gamma by IL-13 dictates susceptibility to secondary postinfluenza MRSA pneumonia. European journal of immunology. 2014;44:3263–3272. doi: 10.1002/eji.201444582. In this study, authors show that IL-13 alters susceptibility to post-influenza bacterial pneumonia through regulation of IFNγ production. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, Blanc F, De Trez C, Van Maele L, Dumoutier L, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. Journal of virology. 2013;87:6911–6924. doi: 10.1128/JVI.02943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. Journal of immunology. 2013;191:5153–5159. doi: 10.4049/jimmunol.1301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.McHugh KJ, Mandalapu S, Kolls JK, Ross TM, Alcorn JF. A novel outbred mouse model of 2009 pandemic influenza and bacterial co-infection severity. PloS one. 2013;8:e82865. doi: 10.1371/journal.pone.0082865. In this study, authors used an outbred mouse model to study influenza and bacterial super-infection. The ability to generate IL-1β in response to secondary bacterial challenge was associated with morbidity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao J, Wang D, Xu F, Gong Y, Wang H, Song Z, Li D, Zhang H, Zhang L, Xia Y, et al. Activation of IL-27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO molecular medicine. 2014;6:120–140. doi: 10.1002/emmm.201302890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Small CL, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG, Arck P, Jordana M, Kaushic C, Ashkar AA, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. Journal of immunology. 2010;184:2048–2056. doi: 10.4049/jimmunol.0902772. [DOI] [PubMed] [Google Scholar]
- 37.Goulding J, Godlee A, Vekaria S, Hilty M, Snelgrove R, Hussell T. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. The Journal of infectious diseases. 2011;204:1086–1094. doi: 10.1093/infdis/jir467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infection and immunity. 2006;74:6707–6721. doi: 10.1128/IAI.00789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Sluijs KF, van Elden LJ, Nijhuis M, Schuurman R, Pater JM, Florquin S, Goldman M, Jansen HM, Lutter R, van der Poll T. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. Journal of immunology. 2004;172:7603–7609. doi: 10.4049/jimmunol.172.12.7603. [DOI] [PubMed] [Google Scholar]
- 40.Bedoya F, Cheng GS, Leibow A, Zakhary N, Weissler K, Garcia V, Aitken M, Kropf E, Garlick DS, Wherry EJ, et al. Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus-infected mice. Journal of immunology. 2013;190:6115–6125. doi: 10.4049/jimmunol.1203302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Sluijs KF, Nijhuis M, Levels JH, Florquin S, Mellor AL, Jansen HM, van der Poll T, Lutter R. Influenza-induced expression of indoleamine 2,3-dioxygenase enhances interleukin-10 production and bacterial outgrowth during secondary pneumococcal pneumonia. The Journal of infectious diseases. 2006;193:214–222. doi: 10.1086/498911. [DOI] [PubMed] [Google Scholar]
- 42*.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nature immunology. 2011;12:1045–1054. doi: 10.1031/ni.2131. Through the use of Il1rl1−/− mice, authors show that innate lymphoid cells may play a role in influenza virus-S. pneumoniae super-infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blok DC, van der Sluijs KF, Florquin S, de Boer OJ, van ’t Veer C, de Vos AF, van der Poll T. Limited anti-inflammatory role for interleukin-1 receptor like 1 (ST2) in the host response to murine postinfluenza pneumococcal pneumonia. PloS one. 2013;8:e58191. doi: 10.1371/journal.pone.0058191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clinical microbiology reviews. 2011;24:490–497. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albiger B, Sandgren A, Katsuragi H, Meyer-Hoffert U, Beiter K, Wartha F, Hornef M, Normark S, Normark BH. Myeloid differentiation factor 88-dependent signalling controls bacterial growth during colonization and systemic pneumococcal disease in mice. Cellular microbiology. 2005;7:1603–1615. doi: 10.1111/j.1462-5822.2005.00578.x. [DOI] [PubMed] [Google Scholar]
- 46.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, et al. STAT3 mutations in the hyper-IgE syndrome. The New England journal of medicine. 2007;357:1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 47.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. The Journal of experimental medicine. 2009;206:1291–1301. doi: 10.1084/jem.20082767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi SM, McAleer JP, Zheng M, Pociask DA, Kaplan MH, Qin S, Reinhart TA, Kolls JK. Innate Stat3-mediated induction of the antimicrobial protein Reg3gamma is required for host defense against MRSA pneumonia. The Journal of experimental medicine. 2013;210:551–561. doi: 10.1084/jem.20120260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo SU, Kwon HJ, Song JH, Byun YH, Seong BL, Kawai T, Akira S, Kweon MN. MyD88 signaling is indispensable for primary influenza A virus infection but dispensable for secondary infection. Journal of virology. 2010;84:12713–12722. doi: 10.1128/JVI.01675-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang WB, Levy DE, Lee CK. STAT3 negatively regulates type I IFN-mediated antiviral response. Journal of immunology. 2011;187:2578–2585. doi: 10.4049/jimmunol.1004128. [DOI] [PubMed] [Google Scholar]
- 51.Ghoneim HE, McCullers JA. Adjunctive corticosteroid therapy improves lung immunopathology and survival during severe secondary pneumococcal pneumonia in mice. The Journal of infectious diseases. 2014;209:1459–1468. doi: 10.1093/infdis/jit653. [DOI] [PubMed] [Google Scholar]
- 52.Karlstrom A, Heston SM, Boyd KL, Tuomanen EI, McCullers JA. Toll-like receptor 2 mediates fatal immunopathology in mice during treatment of secondary pneumococcal pneumonia following influenza. The Journal of infectious diseases. 2011;204:1358–1366. doi: 10.1093/infdis/jir522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Breslow-Deckman JM, Mattingly CM, Birket SE, Hoskins SN, Ho TN, Garvy BA, Feola DJ. Linezolid decreases susceptibility to secondary bacterial pneumonia postinfluenza infection in mice through its effects on IFN-gamma. Journal of immunology. 2013;191:1792–1799. doi: 10.4049/jimmunol.1300180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X, He Y, Xiao K, White JR, Fusco DN, Papanicolaou GA. Effect of linezolid on clinical severity and pulmonary cytokines in a murine model of influenza A and Staphylococcus aureus coinfection. PloS one. 2013;8:e57483. doi: 10.1371/journal.pone.0057483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. Journal of immunology. 2011;186:508–515. doi: 10.4049/jimmunol.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka A, Nakamura S, Seki M, Fukudome K, Iwanaga N, Imamura Y, Miyazaki T, Izumikawa K, Kakeya H, Yanagihara K, et al. Toll-like receptor 4 agonistic antibody promotes innate immunity against severe pneumonia induced by coinfection with influenza virus and Streptococcus pneumoniae. Clinical and vaccine immunology : CVI. 2013;20:977–985. doi: 10.1128/CVI.00010-13. [DOI] [PMC free article] [PubMed] [Google Scholar]