

Abstract
Inorganic arsenic is a human lung carcinogen. We studied the ability of chronic inorganic arsenic (2 μM; as sodium arsenite) exposure to induce a cancer phenotype in the immortalized, non-tumorigenic human lung peripheral epithelial cell line, HPL-1D. After 38 weeks of continuous arsenic exposure, secreted matrix metalloproteinase-2 (MMP2) activity increased to over 200% of control, levels linked to arsenic-induced cancer phenotypes in other cell lines. The invasive capacity of these chronic arsenic-treated lung epithelial (CATLE) cells increased to 320% of control and colony formation increased to 280% of control. CATLE cells showed enhanced proliferation in serum-free media indicative of autonomous growth. Compared to control cells, CATLE cells showed reduced protein expression of the tumor suppressor gene PTEN (decreased to 26% of control) and the putative tumor suppressor gene SLC38A3 (14% of control). Morphological evidence of epithelial-to-mesenchymal transition (EMT) occurred in CATLE cells together with appropriate changes in expression of the EMT markers vimentin (VIM; increased to 300% of control) and e-cadherin (CDH1; decreased to 16% of control). EMT is common in carcinogenic transformation of epithelial cells. CATLE cells showed increased KRAS (291%), ERK1/2 (274%), phosphorylated ERK (p-ERK; 152%), and phosphorylated AKT1 (p-AKT1; 170%) protein expression. Increased transcript expression of metallothioneins, MT1A and MT2A and the stress response genes HMOX1 (690%) and HIF1A (247%) occurred in CATLE cells possibly in adaptation to chronic arsenic exposure. Thus, arsenic induced multiple cancer cell characteristics in human peripheral lung epithelial cells. This model may be useful to assess mechanisms of arsenic-induced lung cancer.
Keywords: Inorganic arsenic, human lung cells, transformation, adaptation, lung cancer, KRAS
Introduction
Inorganic arsenic is a known human carcinogen linked to various cancers such as skin, bladder and lung (IARC, 1980, 1987, 2004, 2012). The evidence for the association between inorganic arsenic exposure and lung cancer in humans is very robust and lung cancers are linked to multiple routes of arsenic exposure (IARC, 1980, 1987, 2004, 2012). Inhaled arsenic was shown relatively early on to be a human lung carcinogen (IARC, 1980, 1987, 2004). However, inorganic arsenic is also clearly a human pulmonary carcinogen after oral intake, primarily from naturally contaminated drinking water, which now is the major source of exposure in humans (IARC, 2004, 2012). This is important because, in humans, lung cancer is a common malignancy with a high mortality rate and is a leading cause of cancer death in the United States (Davies, 2014).
Many inorganics, including arsenic, stimulate the synthesis of metallothioneins (MTs) (Miura and Koizumi, 2007; He and Ma, 2009). MTs are low molecular weight proteins that show a high affinity for transition metals, like cadmium and zinc (Coyle et al., 2002), but also can bind the metalloid arsenic (Ngu and Stillman, 2006). MTs have an abundance of cysteine residues (Bell and Vallee, 2009), and a favorable structural arrangement to bind metallic atoms or reactive oxygen species (ROS) to manage metal toxicity or cellular oxidative stress (Ruttkay-Nedecky et al., 2013; Valko, 2005). It appears some toxic effects of inorganic arsenic occur through direct interactions of the arsenic ion with cellular components and some occur indirectly through ROS generated during cellular methylation of inorganic arsenic (Kojima et al., 2009; Tokar et al., 2014). Thus, the stimulation of MT synthesis could potentially mitigate multiple aspects of inorganic arsenic toxicity. Cells will also adapt to arsenic by induction of enzymes more directly involved in dealing with excess ROS, such as heme oxygenase-1 (HMOX1; Kumagai and Sumi, 2007).
Our laboratory has focused on developing target relevant cell models of inorganic arsenic carcinogenesis. In this regard, a human prostate epithelial cell line, RWPE-1 was successfully transformed into a cancer phenotype by chronic inorganic arsenic exposure (CAsE-PE cells; Achanzar et al., 2002) allowing study of potential genotoxic, epigenetic and stem cell-based mechanisms of arsenic-induced malignant transformation (Benbrahim-Tallaa et al., 2005; Kojima et al., 2009; Tokar et al., 2010a,b; Xu et al., 2012; Ngalame et al., 2014a,b). Furthermore, human skin keratinocyte (HaCaT) cells have been malignantly transformed following chronic low-level arsenic exposure (Pi et al., 2008) and have aided in the study of the mechanisms of co-carcinogenic effects of arsenic and ultraviolet irradiation (Sun et al., 2011). Along similar lines, other groups have established and used target cell line relevant models of arsenical carcinogenesis such as transformed human urinary bladder cells (Eblin et al., 2007; Wnek et al., 2010; Escudero-Lourdes et al., 2012; Garrett et al., 2014) or other cell lines including bronchial epithelial cell lines (Stuecke et al., 2012; Xu, et al., 2013) to help better define carcinogenic mechanisms. These have all been a great aid in advancing our knowledge of the remarkable diversity of the toxic and carcinogenic actions of this metalloid.
None-the-less, additional cell models of arsenic relevant cancer target tissues are still needed in order to help further elucidate carcinogenic mechanisms. In this regard, the HPL-1D cell line is an immortalized, non-tumorigenic human peripheral lung epithelial cell line originally developed to study events during malignant transformation of normal lung cells in vitro (Masuda et al., 1997). HPL-1D cells have made it possible for us to investigate the effects of chronic low-level exposure to inorganic agents to help define mechanisms of action in human lung cancer. Lung adenocarcinomas likely arise from the epithelia of the peripheral lung (Masuda et al., 1997; Sutherland and Berns, 2010), as would be consistent with a model developed with HPL-1D cells. Although data are limited, it appears that inhalation of inorganic arsenic, as from occupational settings, tends to produce lung adenocarcinoma while ingestion more often produces lung squamous cell carcinoma (IARC 1987, 2004; Guo et al., 2004; Chen et al., 2010), though both types of non-small cell lung tumors can occur from either route of inorganic arsenic exposure. Recently, we developed a model for cadmium-induced cancer phenotype in these HPL-1D lung cells (Person et al., 2013) and are now using these transformed cells to help further elucidate the molecular mechanisms of cadmium-induced lung cancer in humans. In this present work we sought to develop a similar model for inorganic arsenic, by chronically exposing these human lung epithelial cells to the metalloid and looking for the development of cancer characteristics.
Materials and methods
Chemicals and reagents
Sodium arsenite (NaAsO2), p-iodonitro-tetrazolium (INT), bovine insulin, hydrocortisone and triiodothyronine were from Sigma Chemical Company (St. Louis, MO). Other chemicals and sources included: HEPES buffer (Gibco/Invitrogen, Carlsbad, CA); human transferrin (Calbiochem/EMD Chemicals, San Diego, CA); antibiotic/antimycotic solution (Gibco/Invitrogen); Ham’s F-12 media (Promocell, Heidelburg, Germany); fetal bovine serum (FBS; Gibco/Invitrogen, Carlsbad, CA); CellTiter 96 Aqueous ONE Solution Cell Proliferation Assay [3-(4,5-dimethyl-thiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS assay)] reagent (Promega, Madison, WI).
Cells, Culture Conditions and Arsenic Exposure
HPL-1D cells were established by Dr. Takashi Takahashi, Laboratory of Ultrastructure Research, Japan, and were graciously provided by Dr. Lucy Anderson of the NCI. HPL-1D cells were immortalized by transfection with large T antigen gene (Masuda et al., 1997). Cells were maintained in Ham’s F-12 medium buffered with 15 mM HEPES (pH 7.3) and 5 μg/ml bovine insulin, 5 μg/ml human transferrin, hydrocortisone 10−7 M, 2 × 10−10 M triiodothyronine, 1% antibiotic/antimycotic, and 1% FBS. Cells were passaged weekly and media refreshed every 2–3 days. To aid in the selection of a concentration for the chronic exposure, HPL-1D cells were plated in 96-well plates and exposed to 0 to 80 μM of sodium arsenite for 72 hours, at which point the MTS assay was used to assess remaining cells. The MTS assay will assess both cytotoxic and cytostatic effects. The concentration of arsenic where 50% of the cells remained after 72 hours was determined to be 30 μM. Based on these data, cells were chronically exposed to a concentration of arsenite (2 μM) where 100% of cells remained after the 72 hour exposure. This concentration of arsenic equates to approximately 150 parts per billion (ppb) arsenic, a level commonly reported in human drinking water (IARC 2004; 2012). During chronic exposure, cells were maintained in T-75 culture flasks in 10–12 mL of culture medium. At 38 weeks of arsenic exposure, cells acquired multiple physical and molecular cancer cell characteristics and are called chronic arsenic-treated lung epithelia (CATLE) cells to reflect this change in characteristics. In one experiment CATLE cells were grown for 7 days in serum-free medium to check for growth autonomous to serum-supplied growth factors, a characteristic common to many cancer cells, and compared to control HPL-1D cells grown under the same conditions. Growth in this experiment was measured by the MTS assay. HPL-1D cells were originally immortalized using SV40 (Masuda et al., 1997). Although SV40 immortalization can lead to loss of function of the p53 and retinoblastoma tumor suppressor genes, the HPL-1D cells are non-tumorigenic, retain characteristics of peripheral lung cell differentiation, and respond to growth factors that regulate proliferation and developmental processes in the peripheral lung (Masuda et al. 1997). Thus, HPL-1D is an acceptable model for studying lung cell transformation in this study. Untreated A549 cells, a human lung adenocarcinoma cell line, were used as a positive control in some experiments and are cultured as previously described (Tokar et al., 2010a) and compared to control HPL-1D cells.
Gene Expression Analyses
Transcription
Gene expression levels were determined using quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) as described by Tokar et al. (2010a). Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) and purified using RNeasy Mini Kit columns (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The purified RNA was reverse transcribed with MuLV (Moloney murine leukemia virus) (ABgene, Rockford, IL) reverse transcriptase and oligo-DT primers (ABgene, Rockford, IL). Absolute SYBR Green ROX Mix (ABgene, Rockford, IL) was used to measure mRNA levels. The cycle threshold times (Ct) were normalized to β-Actin from the same sample based on the control representing 100%. Primers were designed using Primer Express 3.0 software (Carlsbad, CA) and synthesized by Sigma Chemical Company (St. Louis, MO). To examine the cDNA we used the Bio-Rad MyiQ qRT-PCR system and quantitated the relative gene expression using the comparative CT method (2−ΔΔCt).
Western Blot
Gene expression at the protein level was assessed by western blot analysis as previously described (Person et al., 2013). Antibodies for KRAS, p-AKT1, ERK1/2, p-ERK and e-cadherin (CDH1) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for vimentin (VIM) and β-Actin were obtained from Sigma Chemical Co. (St. Louis, MO). The antibody for SLC38A3 was obtained from Abcam (Boston, MA). The primary antibodies were followed by horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Cell Signaling, Danvers, MA) as appropriate.
Immunofluorescence
To help further assess differential protein expression, cells were fixed, blocked and stained as reported (Person et al., 2013), followed by incubations in primary and secondary antibodies for VIM and CDH1. The secondary antibody was conjugated with Alexa Fluor 568, followed by DAPI nuclear staining. An automated Olympus inverted fluorescence microscope was used to capture photomicrographs. CellSens software (Olympus Corporation) was used to process the images for all groups.
Analyses of General Cancer Cell Characteristics
Zymographic Analysis for MMPs
A cancer cell phenotype is often accompanied by elevated secretions of matrix metalloproteinases (MMPs). With arsenic induced transformation this is commonly MMP2 and/or MMP9. In the present study secreted MMP2 activity was most pronounced and was measured by a standard zymographic method as previously described (Tokar et al., 2010a).
Colony Formation in Soft Agar
Colony formation was assayed by a method previously described (Tokar et al., 2010a) which assesses the anchorage-independent growth of cells, a feature typical of cancer cells.
Cellular Invasion Assay
A modified Boyden Chamber method was used to detect the invasive ability of cells as previously described (Tokar et al., 2010a).
Statistical Analysis
Data represent mean values ± SEM. All groups represent a minimum of three or more separate samples. Student’s t-tests were used to assess differences between arsenic treated cells and passage matched control cells. The tests were two-tailed with significance set at p ≤ 0.05. In one study A549 cells were assessed for MMP2 secretion and compared to control HPL-1D cells by two-tailed Student’s t-test with significance set at p ≤ 0.05.
Results
HPL-1D cells exhibit cancer cell characteristics after chronic arsenic treatment
In order to develop a model of arsenic-induced lung carcinogenesis, HPL-1D cells were continuously exposed to a low level of inorganic arsenic (2 μM). MMP2, an enzyme commonly secreted by tumor cells to degrade the extracellular matrix and assist in local invasion or metastasis, was assessed as a marker of acquired cancer cell phenotype. Elevations of secreted MMP2 activity occurred as a consequence of chronic arsenic exposure starting at 36 weeks (Figure 1A). After 38 weeks of arsenic exposure, secreted MMP2 activity was increased to 230% of control. Increases in the levels of MMP activity of this magnitude are commonly associated with arsenic-induced acquired cancer phenotype and were similar to A549 cells, an aggressive human lung adenocarcinoma cell line (Figure 1A).
Fig. 1.

Chronic exposure to inorganic arsenic as sodium arsenite (2 μM; see Methods) causes HPL-1D to acquire cancer cell characteristics consistent with a malignant phenotype. (A) Zymographic analysis of secreted MMP2 activity over time in HPL-1D cells. The aggressive lung adenocarcinoma cell line A549 as compared to control HPL-1D cells is shown as a positive control. (B) Invasive capacity of arsenic treated HPL-1D cells after 38 weeks of exposure. (C) Soft agar assay of colony formation in arsenic treated HPL-1D cells after 38 weeks of exposure. (D) Impact of serum deprivation on cell growth in control cells and cells chronically treated with arsenic. Cell growth was assessed after 7 days in appropriate medium. Data represent the mean ± SEM (n = 3 or more). Treated data were compared in all cases with passage-matched controls. Statistical analysis performed by Student’s t-test with p ≤ 0.05 considered significant. An asterisk (*) indicates significance from passage matched control or from control HPL-1D cells for the A549 lung carcinoma cells for MMP2 secretion.
Increased invasive capacity of cells is also commonly associated with an acquired cancer cell phenotype. The invasive capacity of the chronic arsenic treated lung epithelial cells was markedly increased to 320% of control after 38 weeks of arsenic exposure (Figure 1B). The ability of cells to form anchorage-independent colonies in soft agar was examined (Figure 1C). Chronic exposure to arsenic caused an increase in colony formation to 280% of control. Enhanced colony formation is frequently associated with acquired cancer phenotype. The chronic arsenic exposed cells were also grown for 7 days in serum-free medium (Figure 1D) in order to observe potential growth autonomous to serum-supplied growth factors. The ability to grow unabated in serum-free media is a characteristic common to many cancer cells. Compared to control cells grown in serum-free medium the arsenic-treated cells showed increased growth.
Compared to control, arsenic exposure at 38 weeks also markedly reduced expression of the putative tumor suppressor gene SLC38A3 (14% of control) at the protein level (Figure 2; blot top inset). Expression of PTEN, a widely recognized tumor suppressor gene that is commonly lost in arsenic-induced malignant transformation, was markedly reduced to 27% of control with chronic arsenic exposure (Figure 2; blot top inset).
Fig. 2.

Effect of chronic exposure to arsenic on tumor suppressor gene expression in HPL-1D cells. Quantitative protein expression of tumor suppressor gene PTEN and the putative tumor suppressor gene SLC38A3 was assessed after 38 weeks of continuous arsenic exposure. Quantitative protein data were normalized to β-Actin with control set to 100% and represent the mean ± SEM (n = 3 or more). Statistical analysis performed by Student’s t-test with p ≤ 0.05 considered significant. An asterisk (*) indicates significance from passage matched control.
Based on the elevated secreted MMP2 activity, increased invasive capacity, increased colony formation, autonomous growth, and generalized loss of tumor suppressor gene expression, it appeared that by 38 weeks of continuous arsenic exposure these human lung epithelial cells had acquired multiple cancer cell characteristics. Thus, after 38 weeks of arsenic exposure the cells are called chronic arsenic-treated lung epithelia (CATLE) cells to signify these changes towards a cancer cell phenotype.
EMT in CATLE cells
Alterations in cell morphology are often observed following the transition to cancer including epithelial-to-mesenchymal transition (EMT), which occurs with distinct changes in gene expression. CATLE cells showed clear evidence of EMT at both morphological and molecular levels (Figure 3). With EMT the normal rounded features typical of epithelial cells shift to an elongated spindle-like, more motile structure (Figure 3A inset), and thereby enhancing metastatic capacity, consistent with increased invasiveness (see Figure 1B). There are also alterations in EMT marker proteins, including CDH1, which decreases with EMT, and VIM which increases. CDH1 protein decreased to 16% of control while VIM increased to 353% in CATLE cells (Figure 3A). Immunoflourescence staining confirmed that the reduction of CDH1 (Figure 3B) and the increases in VIM (Figure 3C) proteins were both wide-spread and generalized in all cells. Thus, arsenic exposure caused acquisition of additional morphological and molecular characteristics typical for cancer cells.
Fig. 3.
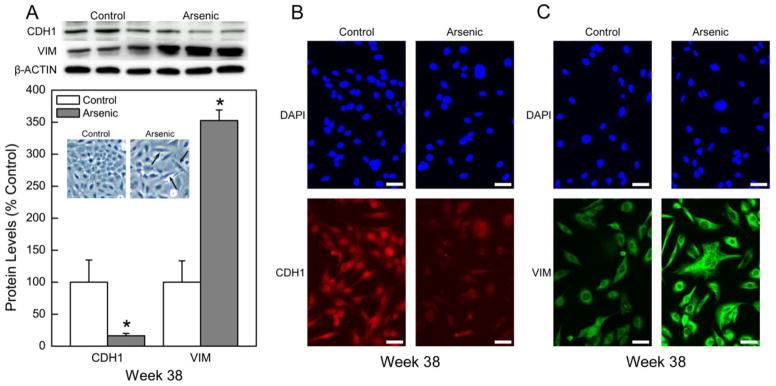
Evidence of epithelial to mesenchymal transition (EMT) in chronic arsenic treated CATLE cells. (A) Quantitative expression of EMT markers including loss of CDH1 protein and increased VIM protein in CATLE cells (lower figure). Quantitative protein data were normalized to β-Actin with control set to 100% and represent the mean ± SEM (n = 3 or more). Statistical analysis performed by Student’s t-test with p ≤ 0.05 considered significant. An asterisk (*) indicates significance from control. Note also the inset showing frequent morphologic change to spindle-shape in CATLE cells typical of EMT. Top panel shows Western blot results with CDH1 loss and VIM increase with EMT. (B) Immunostaining of CDH1 in CATLE cells. Staining with DAPI shows a similar number of viable cells in the fields in both control and treated groups (upper panels). There is a clear loss of CDH1 protein throughout the entire cell population of arsenic-treated CATLE cells (lower panels). (C) Immunostaining of VIM in CATLE cells. Staining with DAPI shows a similar number of viable cells in the fields in both control and treated groups (upper panels). There is a clear increase in VIM protein throughout the entire cell population of arsenic-treated CATLE cells (lower panels). The white bars in panel B and C equal 50 microns.
KRAS, ERK1/2, and p-ERK protein expression in CATLE cells
KRAS, an oncogene that regulates cell division and is commonly activated in arsenic-induced cell transformation and in human lung cancers, also showed a marked, nearly 300% increase in CATLE cells compared to control cells (Figure 4A). The extracellular signal-regulated kinases (ERKs) are mitogen-activated protein kinases critical to cell proliferation and apoptosis. These serine/threonine kinases are ubiquitously expressed in mammalian cells and aberrantly active in numerous cancers (Samatar and Poulikakos, 2014). ERK1/2 and p-ERK increased in CATLE cells to 274% and 152% of control, respectively (Figure 4B). Another gene of oncogenic relevance that was assessed in CATLE cells was AKT1 (Figure 5). The expression of AKT1 at the transcript level was elevated to 242% of control (Figure 5A). This equated to a significant elevation in p-AKT1 protein levels of 170% of control (Figure 5B). Together, these gene expression changes all support acquisition of cancer cell characteristics in CATLE cells.
Fig. 4.

Effect of chronic exposure to arsenic on KRAS, ERK1/2, and p-ERK expression. (A) Quantitative protein expression of KRAS oncogene after 38 weeks of arsenic exposure. (B) Quantitative protein expression of ERK1/2 and p-ERK after 38 weeks of arsenic exposure. Data were first normalized to β-Actin with control set to 100% and represent the mean ± SEM (n = 3 or more). Statistical analysis performed by Student’s t-test with p ≤ 0.05 considered significant. An asterisk (*) indicates significance from passage matched control.
Fig. 5.

Effect of chronic exposure to arsenic on AKT expression. (A) Quantitative transcript expression of AKT1 after 38 weeks of arsenic exposure. (B) Quantitative protein expression of p-AKT after 38 weeks of arsenic exposure. Data were first normalized to β-Actin with control set to 100% and represent the mean ± SEM (n = 3 or more). Statistical analysis performed by Student’s t-test with p ≤ 0.05 considered significant. An asterisk (*) indicates significance from passage matched control.
MT and oxidant stress related genes in CATLE cells
Following chronic exposure (38 weeks) to arsenic, the major MT isoforms, MT1A and MT2A were increased in CATLE cells to more than 350% and 640% of control, respectively (Figure 6A). The increase in MT1A and MT2A suggests that these MTs are produced in response to arsenic and may be involved in adaptation to chronic arsenic exposure. MT can also be expressed in malignant lung cells.
Fig. 6.

Effect of chronic exposure to arsenic on expression of MT and antioxidant response genes. Quantitative transcript expression of the MT isoforms, MT1A and MT2A, and HMOX1 and HIF1A were assessed in CATLE cells as compared to control. Values represent the mean ± SEM (n = 3 or more) as determined by quantitative real time RT-PCR normalized to β-Actin. Treated cells were compared to passage-matched control cells with p ≤ 0.05 considered significant. An asterisk (*) indicates significance from control.
The exposure of cells to carcinogenic inorganics, like inorganic arsenic, will often induce oxidative stress response genes like heme oxygenase-1 (HMOX1) and hypoxia inducible factor-1α (HIF1A) as part of an adaptive mechanism. Indeed, HMOX1 was increased 690% in CATLE cells compared to control while HIF1A increased 247% (Figure 6B).
Discussion
Strong evidence supports a link between human inorganic arsenic exposure and cancer of the lung (IARC 1980, 1987, 2004, 2012). Various populations have been exposed throughout their life time or in relevant occupations to arsenic and developed lung cancers (IARC 1980, 1987, 2004, 2012). In addition, relatively recent findings indicate high cancer mortality, including a high rate of death from lung cancer, occurs in young adults after in utero and childhood exposure to arsenic in the drinking water in a population in Chile where high arsenic exposure was abruptly stopped when an arsenic removal plant commenced operations (e.g. Smith et al., 2012). This indicates that inorganic arsenic is also a highly potent early life human carcinogen that targets the lung. Mouse inorganic arsenic transplacental or whole life exposure studies also often show the lung as a target site (IARC 2012; Tokar et al., 2011; Waalkes et al., 2014). Thus, there is a clear predilection for the lung cells to be negatively altered towards a malignant phenotype by inorganic arsenic exposure. In the present work, the human lung peripheral epithelial cell line, HPL-1D, acquired multiple physical and molecular characteristics typical of cancer cells. After 38 weeks of inorganic arsenic exposure the chronic arsenic treated lung epithelial (CATLE) cells showed increased MMP2 secretion, invasion, colony formation, and evidence of growth factor-independent (autonomous) growth, and clear morphological and molecular evidence of EMT, all characteristics of cancer cells. CATLE cells also showed loss of expression of the tumor suppressor gene PTEN (Carracedo et al., 2011; Cooper et al., 2013), and the putative tumor suppressor gene SLC38A3 (Kholodnyuk et al., 2006), together with increased expression of the oncogene KRAS and increased expression of AKT1, a central member of a frequently activated signaling pathway in cancer (Carpten et al., 2007). Loss of PTEN expression and over-expression of KRAS are common in human lung cancer (Zhu et al., 2014; Kadara et al., 2012; Cooper et al. 2013) and common in arsenic-induced malignant transformation of other cell types (Benbrahim-Tallaa et al., 2005; Tokar et al., 2010a; Ngalame et al., 2014a,b; Xu et al., 2014). ERK1/2 and p-ERK, which are critical to cell proliferation, are commonly activated in cancers and linked to RAS activation (e.g. Neuzillet et al., 2013; Samatar and Poulikakos, 2014), and were also activated in CATLE cells. In addition, activation of the AKT pathway is thought to have a direct impact on human carcinogenesis (Carpten et al., 2007), and CATLE cells show both increased AKT1 and p-AKT1, the activated form of the protein. Together, these various changes indicate arsenic induced acquisition of multiple cancer cell qualities in these human peripheral lung cells. These data augment recent reports of arsenic-induced transformation of two human bronchial epithelial cell lines (HBE and BEAS-2B; Xu et al., 2013; Stueckle et al., 2012) and should provide an additional model to help define the complex factors involved in the mechanisms of arsenic-induced lung carcinogenesis.
In humans, it is noteworthy that both inhalation and ingestion of inorganic arsenic are definitively linked to cancer of the lung (IARC 1980, 1987, 2004, 2012). However, the resultant histological type of lung cancer appears to vary with route of arsenic exposure, as inorganic arsenic inhalation via occupational settings produces a higher portion of lung adenocarcinoma while ingestion of arsenic is mostly associated with lung squamous cell carcinoma (IARC 1980; 1987, 2004, 2012; Guo et al., 2004; Chen et al., 2010). This is not an absolute as both these histological types of lung cancer may be induced by either ingestion or inhalation of arsenic in humans (Guo et al., 2004; Chen et al., 2010). It is thought that lung adenocarcinoma and squamous cell carcinoma originate from different cell types within the lung (Sutherland and Berns, 2010; Kadara et al., 2012). Lung adenocarcinomas likely arise from the cells of the epithelia of the peripheral lung (Masuda et al., 1997; Sutherland and Berns, 2010; Kadara et al., 2012), consistent with the CATLE cell model which was developed from cultured peripheral lung cells in the present study. A lung squamous cell carcinoma is typically preceded by squamous metaplasia with dysplasia in respiratory epithelium of the central airways (bronchi, etc. Sutherland and Berns, 2010; Kadara et al., 2012; Klebe and Henderson, 2013) and would be modeled by cultured bronchial epithelial cells (Xu et al., 2013; Stueckle et al., 2012). In fact, it has been proposed that different types of cancer stem cells may reside in different compartments of the lung (central or peripheral) and produce different types of cancers (i.e. squamous cell carcinoma or adenocarcinoma; Klebe and Henderson, 2013). The observation that arsenic exposures tend to induce different histological forms of lung cancer, that likely arise from different cell types (Sutherland and Berns, 2010; Kadara et al., 2012; Klebe and Henderson, 2013), based on portal of entry creates the underlying possibility of route-specific target cells. This possibility has lead to the proposal that different mechanisms may be involved in generating the different histological types of lung cancer after oral or inhalation exposure to arsenic (Guo et al., 2004), an intriguing concept. One could envision route-specific differences in cell disposition as an issue, at least in part. In any event, this potential disunity of mechanism in the same tissue is an important possibility and the existence of multiple cell models will greatly aid in defining any potentially different mechanisms (Xu et al., 2013; Stueckle et al., 2012; present study). In fact, gene expression analyses in arsenic transformants do indicate some major differences, such as in expression of the lung tumor suppressor gene PTEN (Cooper et al., 2013), which is over-expressed in arsenic transformed BEAS-2B bronchial epithelial cells (B-As cells; Stueckle et al., 2012) but markedly suppressed in the arsenic-converted CATLE cells (present study). In addition, RAS signaling appears blocked and KRAS is minimally increased in bronchial epithelial cells transformed by arsenic (Stueckle et al., 2012), which is distinctly different from the robust increases that are seen in KRAS expression in arsenic-converted peripheral epithelial CATLE cells of the present study. In this regard, it is noteworthy in humans that a large portion of lung adenocarcinoma, unlike lung squamous cell carcinoma, have either an activating mutation or activation of wild type KRAS (Zhu et al., 2014; Kadara et al., 2012; Cooper et al., 2013). These data are consistent with the increased expression of KRAS in arsenic-transformed CATLE cells and support the notion that these cells may be a model for arsenic-induced adenocarcinomatous-type lung cancers. Since we did not attempt to form xenograft tumors with CATLE cells it is impossible to say with certainty what type of tumor they might form. However, further comparative work should be carried out with these arsenic cell models of lung cancer.
AKT1 is a key member of the PI3K/AKT survival and proliferation signaling pathway and is frequently activated in diverse human cancers (Carpten et al., 2007), including lung cancers (Vivanco and Sawyers, 2002). Activation of AKT in cancer cells can alter proliferation, apoptosis, invasion, and survival (Vivanco and Sawyers, 2002). Elevated activity of the AKT1 protein kinase, a key member of this signaling pathway, occurs in various human cancers (Vivanco and Sawyers, 2002), and in arsenic-transformed cells (Wang et al., 2012), and is consistent with its activation in the arsenic treated CATLE cells of the present study. In particular, pAKT1 was elevated in CATLE cells and it is the phosphorylated form that has been shown to be the activated form of AKT (Chen et al., 2001). AKT activation in cancer is often associated with PTEN inactivation including inactivation in human lung cancer (Vivanco and Sawyers, 2002), a relationship similarly observed in CATLE cells. AKT can have effects on tumor angiogenesis mediated, in part, through HIF1A (Carpten et al., 2007) and CATLE cells showed marked increases in expression of HIF1A. Thus, in all, AKT activation may be a critical event in arsenic transformation of the peripheral lung cells observed in the present study.
MMPs are secreted enzymes that act to degrade components of the extracellular matrix and the basement membranes (Hua et al., 2011; Qian et al., 2010). MMPs normally function in tissue development and remodeling (Hua et al., 2011), but there are clear correlations between tumorigenesis or cellular malignant transformation and increased secretion of various MMPs (Hua et al., 2011; Qian et al., 2010; Kojima et al., 2009; Tokar et al., 2013; Xu et al., 2014). MMPs have roles in early cancer development, such as in tumor initiation, angiogenesis and growth, and in later cancer progression, such as invasion and metastases (Hua et al., 2011). MMPs are also involved in inflammation and cell signaling and will activate specific receptors or growth factors causing aberrant cellular proliferation, apoptosis, differentiation or migration (Hua et al., 2011). In this regard, arsenic toxicity is often through aberrant signal transduction or transcription factor activation (Kumagai and Sumi, 2007). The CATLE cells that had been chronically exposed to inorganic arsenic in the present study secreted MMP2 at levels similar to a highly aggressive human lung adenocarcinoma cell line (A549). These data are consistent with the hypersecretion of MMP2 coinciding with oncogenic transformation induced by chronic arsenic exposure in other epithelial cell lines, like human prostate (Kojima et al., 2009), human breast (Xu et al., 2014), rat liver (Kojima et al., 2009), and rat kidney (Tokar et al., 2013) cell lines. Interestingly, two recent meta-analyses of published studies on MMP2 in lung cancer patients concluded that high MMP2 expression levels are correlated with a poorer survival than low expression (Guo et al., 2007; Qian et al., 2010; Wang and Cai et al., 2012) and that this correlation may be specific for lung adenocarcinomas (Qian et al., 2010). Indeed, MMP2 expression levels are higher in human lung adenocarcinoma than in lung squamous cell carcinoma (Hida and Hamada, 2012). Therefore, the hypersecretion of MMP2 by CATLE cells in the present study is consistent with an acquired human lung cancer phenotype.
Two of the major forms of the metal-binding protein MT, MT1A and MT2A, were over-expressed in CATLE cells in the present study. This over-expression could be part of an adaptive response to arsenic (e.g. Tokar et al., 2010b; Qu and Waalkes, 2015). Inorganic arsenic binds to MT (Irvine et al., 2013) and MT will reduce arsenic toxicity in vivo and in cell culture (Liu et al., 2000; Miao et al., 2013; Qu and Waalkes, 2015). MT may also directly interact with ROS generated by inorganics, like arsenic, and thereby act as antioxidants to prevent oxidative stress (Bell and Vallee, 2009). Indeed, we recently found MT blocked arsenic-induced oxidative stress and DNA damage in human embryonic liver cells (Qu and Waalkes, 2015). An adaptive antioxidant response would also be consistent with the increases seen in HMOX1 and HIF1A expression seen in CATLE cells. However, in many cases MT is also frequently over-expressed in human non-small cell lung cancers (i.e. primarily adenocarcinomas and squamous cell carcinomas; Theocharis et al., 2002; Eckschlager et al., 2009; Werynska et al., 2013). In this regard MT expression is considered a prognostic factor for enhanced human lung tumor progression and poor outcome (Eckschlager et al., 2009; Werynska et al., 2013). Multiple forms of MT have been shown to be positive in both human lung adenocarcinoma and squamous cell carcinoma but not small-cell lung carcinoma (Eckschlager et al., 2009; Theocharis et al., 2002). In rodent models, mice exposed in utero to inorganic arsenic via the mother’s drinking water will develop lung adenocarcinoma as adults and also show aberrant gene expression relevant to pulmonary cancer during development, which includes markedly increased levels of MT expression in fetal lung (Shen et al., 2007). Thus, the elevated MT levels that are observed in the present study in arsenic treated lung epithelial cells when they acquired cancer cell characteristics support an association between MT and a malignant lung phenotype or as an adaptive change.
In summary, the present work demonstrates that chronic exposure to low-level arsenic was associated with acquisition of multiple tumor cell characteristics in human peripheral lung epithelial cells. These data suggest that arsenic may directly impact lung epithelial cells inducing a cancer phenotype that could lead to adenocarcinomas. These arsenic treated cells could provide another valuable model for the study of mechanisms of arsenic-induced lung cancers.
Supplementary Material
Highlights.
Chronic arsenic exposure transforms a human peripheral lung epithelia cell line
Cells acquire characteristics in common with human lung adenocarcinoma cells
These transformed cells provide a valuable model for arsenic-induced lung cancer
Acknowledgments
The authors wish to thank Drs. Alex Merrick, Humphrey Yao, Michael DeVito, Darlene Dixon and John Bucher for their critical comments. This work was totally supported by Intramural Program funds from National Institute of Environmental Health Sciences, Division of the National Toxicology Program.
Abbreviations
- CDH1
e-cadherin
- EMT
epithelial-to-mesenchymal transition
- ERKs
extracellular signal-regulated kinases
- FBS
fetal bovine serum
- HIF1A
hypoxia inducible factor-1α
- HMOX1
heme oxygenase-1
- MMPs
Matrix metalloproteinases
- MT
metallothionein
- MTS
3-(4,5-dimethyl-thiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- ppb
parts per billion
- ROS
reactive oxygen species
- VIM
vimentin
Footnotes
Conflict of interest statement
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achanzar WE, Brambila EM, Diwan BA, Webber MM, Waalkes MP. Inorganic arsenite-induced malignant transformation of human prostate epithelial cells. J Natl Cancer Inst. 2002;94:1888–1891. doi: 10.1093/jnci/94.24.1888. [DOI] [PubMed] [Google Scholar]
- Bell SG, Vallee BL. The metallothionein/thionein system: An oxidoreductive metabolic zinc link. Chembiochem. 2009;10:55–62. doi: 10.1002/cbic.200800511. [DOI] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Waterland RA, Styblo M, Achanzar WE, Webber MM, Waalkes MP. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol Appl Pharmacol. 2005;206:288–298. doi: 10.1016/j.taap.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: how much is too little? Cancer Res. 2011;71:629–633. doi: 10.1158/0008-5472.CAN-10-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Chiou HY, Hsu LI, Hsueh YM, Wu MM, Chen CJ. Ingested arsenic, characteristics of well water consumption and risk of different histological types of lung cancer in northeastern Taiwan. Environ Res. 2010;110:455–462. doi: 10.1016/j.envres.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276:31858–31862. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- Cooper WA, Lam DC, O’Toole SA, Minna JD. Molecular biology of lung cancer. J Thorac Dis Suppl. 2013;5:S479–490. doi: 10.3978/j.issn.2072-1439.2013.08.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: The multipurpose protein. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M. New modalities of cancer treatment for NSCLC: focus on immunotherapy. Cancer Manag Res. 2014;6:63–75. doi: 10.2147/CMAR.S57550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckschlager T, Adam V, Hrabeta J, Figova K, Kizek R. Metallothioneins and cancer. Curr Protein Pept Sci. 2009;10:360–375. doi: 10.2174/138920309788922243. [DOI] [PubMed] [Google Scholar]
- Eblin KE, Bredfeldt TG, Buffington S, Gandolfi AJ. Mitogenic signal transduction caused by monomethylarsonous acid in human bladder cells: role in arsenic-induced carcinogenesis. Toxicol Sci. 2007;95:321–330. doi: 10.1093/toxsci/kfl160. [DOI] [PubMed] [Google Scholar]
- Escudero-Lourdes C, Wu T, Camarillo JM, Gandolfi AJ. Interleukin-8 (IL-8) over-production and autocrine cell activation are key factors in monomethylarsonous acid [MMA(III)]-induced malignant transformation of urothelial cells. Toxicol Appl Pharmacol. 2012;258:10–18. doi: 10.1016/j.taap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett SH, Somji S, Sens DA, Zhang KK. Prediction of the number of activated genes in multiple independent Cd(+2)- and As(+3)-induced malignant transformations of human urothelial cells (UROtsa) PLoS One. 2014;9:e85614. doi: 10.1371/journal.pone.0085614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo HR, Wang NS, Hu H, Monson RR. Cell type specificity of lung cancer associated with arsenic ingestion. Cancer Epidemiol Biomarkers Prev. 2004;13:638–643. [PubMed] [Google Scholar]
- Guo CB, Wang S, Deng C, Zhang DL, Wang FL, Jin XQ. Relationship between matrix metalloproteinase 2 and lung cancer progression. Mol Diagn Ther. 2007;11:183–192. doi: 10.1007/BF03256240. [DOI] [PubMed] [Google Scholar]
- He X, Ma Q. Induction of metallothionein I by arsenic via metal-activated transcription factor 1: critical role of c-terminal cysteine residues in arsenic sensing. J Biol Chem. 2009;284:12609–12621. doi: 10.1074/jbc.M901204200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida Y, Hamada J. Differential expressions of matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs and their endogenous inhibitors among histologic subtypes of lung cancers. Anticancer Agents Med Chem. 2012;12:744–752. doi: 10.2174/187152012802650156. [DOI] [PubMed] [Google Scholar]
- Hua H, Li M, Luo T, Yin Y, Jiang Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68:3853–3868. doi: 10.1007/s00018-011-0763-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. Some Metals and Metallic Compounds. Vol. 23. IARC Press; Lyon: 1980. International Agency for Research on Cancer (IARC) Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans; pp. 39–143. [Google Scholar]
- IARC. Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs. Suppl 7. 1 to 42. IARC Press; Lyon: 1987. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; pp. 100–106. [PubMed] [Google Scholar]
- IARC. Some Drinking-water Disinfectants and Contaminants, Including Arsenic. Vol. 84. IARC Press; Lyon: 2004. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; pp. 39–267. [PMC free article] [PubMed] [Google Scholar]
- IARC. Arsenic, Metals, Fibres, and Dusts. 100C. IARC Press; Lyon: 2012. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; pp. 121–141. [PMC free article] [PubMed] [Google Scholar]
- Irvine GW, Summers KL, Stillman MJ. Cysteine accessibility during As3+ metalation of the α- and β-domains of recombinant human MT1a. Biochem Biophys Res Commun. 2013;433:477–483. doi: 10.1016/j.bbrc.2013.03.026. [DOI] [PubMed] [Google Scholar]
- Kadara H, Kabbout M, Wistuba II. Pulmonary adenocarcinoma: a renewed entity in 2011. Respirology. 2012;17:50–65. doi: 10.1111/j.1440-1843.2011.02095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodnyuk ID, Kozireva S, Kost-Alimova M, Kashuba V, Klein G, Imreh S. Down regulation of 3p genes, LTF, SLC38A3 and DRR1, upon growth of human chromosome 3-mouse fibrosarcoma hybrids in severe combined immunodeficiency mice. Int J Cancer. 2006;119:99–107. doi: 10.1002/ijc.21794. [DOI] [PubMed] [Google Scholar]
- Klebe S, Henderson DW. Facts and fiction: premalignant lesions of lung tissues. Pathology. 2013;45:305–315. doi: 10.1097/PAT.0b013e32835f45fd. [DOI] [PubMed] [Google Scholar]
- Kojima C, Ramirez DC, Tokar EJ, Himeno S, Drobná Z, Stýblo M, Mason RP, Waalkes MP. Requirement of arsenic biomethylation for oxidative DNA damage. J Natl Cancer Inst. 2009;101:1670–1681. doi: 10.1093/jnci/djp414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y, Sumi D. Arsenic: signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annual Rev Pharmacol Toxicol. 2007;47:243–262. doi: 10.1146/annurev.pharmtox.47.120505.105144. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Goyer RA, Achanzar W, Waalkes MP. Metallothionein-I/II null mice are more sensitive than wild-type mice to the hepatotoxic and nephrotoxic effects of chronic oral or injected inorganic arsenicals. Toxicol Sci. 2000;55:460–467. doi: 10.1093/toxsci/55.2.460. [DOI] [PubMed] [Google Scholar]
- Masuda A, Kondo M, Saito T, Yatabe Y, Kobayashi T, Okamoto M, Suyama M, Takahashi T, Takahashi T. Establishment of human peripheral lung epithelial cell lines (HPL1) retaining differentiated characteristics and responsiveness to epidermal growth factor, hepatocyte growth factor, and transforming growth factor beta1. Cancer Res. 1997;57:4898–4904. [PubMed] [Google Scholar]
- Miao X, Tang Z, Wang Y, Su G, Sun W, Wei W, Li W, Miao L, Cai L, Tan Y, Liu Q. Metallothionein prevention of arsenic trioxide-induced cardiac cell death is associated with its inhibition of mitogen-activated protein kinases activation in vitro and in vivo. Toxicol Lett. 2013;220:277–285. doi: 10.1016/j.toxlet.2013.04.025. [DOI] [PubMed] [Google Scholar]
- Miura N, Koizumi S. Heavy metal responses of the human metallothionein isoform genes. Yakugaku Zasshi. 2007;127:665–673. doi: 10.1248/yakushi.127.665. [DOI] [PubMed] [Google Scholar]
- Neuzillet C, Hammel P, Tijeras-Raballand A, Couvelard A, Raymond E. Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013;32:147–162. doi: 10.1007/s10555-012-9396-2. [DOI] [PubMed] [Google Scholar]
- Ngalame NN, Tokar EJ, Person RJ, Xu Y, Waalkes MP. Aberrant microRNA expression likely controls RAS oncogene activation during malignant transformation of human prostate epithelial and stem cells by arsenic. Toxicol Sci. 2014a;138:268–277. doi: 10.1093/toxsci/kfu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngalame NN, Tokar EJ, Person RJ, Waalkes MP. Silencing KRAS overexpression in arsenic-transformed prostate epithelial and stem cells partially mitigates malignant phenotype. Toxicol Sci. 2014b;142:489–496. doi: 10.1093/toxsci/kfu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngu T, Stillman M. Arsenic binding to human metallothionein. J Am Chem Soc. 2006;128:12473–12483. doi: 10.1021/ja062914c. [DOI] [PubMed] [Google Scholar]
- Person RJ, Tokar EJ, Xu Y, Orihuela R, Ngalame NN, Waalkes MP. Chronic cadmium exposure in vitro induces cancer cell characteristics in human lung cells. Toxicol Appl Pharmacol. 2013;273:281–288. doi: 10.1016/j.taap.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, Styblo M, Waalkes MP. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic Biol Med. 2008;45:651–658. doi: 10.1016/j.freeradbiomed.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Q, Wang Q, Zhan P, Peng L, Wei SZ, Shi Y, Song Y. The role of matrix metalloproteinase 2 on the survival of patients with non-small cell lung cancer: a systematic review with meta-analysis. Cancer Invest. 2010;28:661–669. doi: 10.3109/07357901003735634. [DOI] [PubMed] [Google Scholar]
- Qu W, Waalkes MP. Metallothionein blocks oxidative DNA damage induced by acute inorganic arsenic exposure. Toxicol Appl Pharmacol. 2015;282:267–274. doi: 10.1016/j.taap.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruttkay-Nedecky B, Nejdl L, Gumulec J, Zitka O, Masarik M, Eckschlager T, Stiborova M, Adam V, Kizek R. The role of metallothionein in oxidative stress. Int J Mol Sci. 2013;14:6044–6066. doi: 10.3390/ijms14036044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Shen J, Liu J, Xie Y, Diwan BA, Waalkes MP. Fetal onset of aberrant gene expression relevant to pulmonary carcinogenesis in lung adenocarcinoma development induced by in utero arsenic exposure. Toxicol Sci. 2007;95:313–320. doi: 10.1093/toxsci/kfl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Liaw J, Yuan Y, Ferreccio C, Steinmaus C. Mortality in young adults following in utero and childhood exposure to arsenic in drinking water. Environ Health Perspect. 2012;120:1527–1531. doi: 10.1289/ehp.1104867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stueckle TA, Lu Y, Davis ME, Wang L, Jiang BH, Holaskova I, Schafer R, Barnett JB, Rojanasakul Y. Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxicol Appl Pharmacol. 2012;261:204–216. doi: 10.1016/j.taap.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Kojima C, Chignell C, Mason R, Waalkes MP. Arsenic transformation predisposes human skin keratinocytes to UV-induced DNA damage yet enhances their survival apparently by diminishing oxidant response. Toxicol Appl Pharmacol. 2011;255:242–250. doi: 10.1016/j.taap.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Berns A. Cell of origin of lung cancer. Mol Oncol. 2010;4:397–403. doi: 10.1016/j.molonc.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharis S, Karkantaris C, Philipides T, Agapitos E, Gika A, Margeli A, Kittas C, Koutselinis A. Expression of metallothionein in lung carcinoma: correlation with histological type and grade. Histopathology. 2002;40:143–151. doi: 10.1046/j.1365-2559.2002.01325.x. [DOI] [PubMed] [Google Scholar]
- Tokar EJ, Diwan BA, Waalkes MP. Arsenic exposure transforms human epithelial stem/progenitor cells into a cancer stem-like phenotype. Environ Health Perspect. 2010a;118:108–115. doi: 10.1289/ehp.0901059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokar EJ, Qu W, Liu J, Liu W, Webber MM, Phang JM, Waalkes MP. Arsenic-specific stem cell selection during malignant transformation. J Natl Cancer Inst. 2010b;102:638–649. doi: 10.1093/jnci/djq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokar EJ, Diwan BA, Ward JM, Delker DA, Waalkes MP. Carcinogenic effects of “whole-life” exposure to inorganic arsenic in CD1 mice. Toxicol Sci. 2011;119:73–83. doi: 10.1093/toxsci/kfq315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokar EJ, Person RJ, Sun Y, Perantoni AO, Waalkes MP. Chronic exposure of renal stem cells to inorganic arsenic induces a cancer phenotype. Chem Res Toxicol. 2013;26:96–105. doi: 10.1021/tx3004054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokar EJ, Kojima C, Waalkes MP. Methylarsonous acid causes oxidative DNA damage in cells independent of the ability to biomethylate inorganic arsenic. Arch Toxicol. 2014;88:249–261. doi: 10.1007/s00204-013-1141-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M. Metals, Toxicity and Oxidative Stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Qu W, Tokar EJ, Kissling GE, Dixon D. Lung tumors in mice induced by “whole-life” inorganic arsenic exposure at human-relevant doses. Arch Toxicol. 2014;88:1619–1629. doi: 10.1007/s00204-014-1305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cai Y. Matrix metalloproteinase 2 polymorphisms and expression in lung cancer: a meta-analysis. Tumour Biol. 2012;33:1819–1828. doi: 10.1007/s13277-012-0441-0. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang J, Fisher T, Xiao H, Jiang Y, Yang C. Akt activation is responsible for enhanced migratory and invasive behavior of arsenic-transformed human bronchial epithelial cells. Environ Health Perspect. 2012;120:92–97. doi: 10.1289/ehp.1104061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werynska B, Pula B, Muszczynska-Bernhard B, Gomulkiewicz A, Piotrowska A, Prus R, Podhorska-Okolow M, Jankowska R, Dziegiel P. Metallothionein 1F and 2A overexpression predicts poor outcome of non-small cell lung cancer patients. Exp Mol Pathol. 2013;94:301–308. doi: 10.1016/j.yexmp.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Wnek SM, Jensen TJ, Severson PL, Futscher BW, Gandolfi AJ. Monomethylarsonous acid produces irreversible events resulting in malignant transformation of a human bladder cell line following 12 weeks of low-level exposure. Toxicol Sci. 2010;116:44–57. doi: 10.1093/toxsci/kfq106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Tokar EJ, Sun Y, Waalkes MP. Arsenic-transformed malignant prostate epithelia can convert noncontiguous normal stem cells into an oncogenic phenotype. Environ Health Perspect. 2012;120:865–871. doi: 10.1289/ehp.1204987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhao Y, Xu W, Luo F, Wang B, Li Y, Pang Y, Liu Q. Involvement of HIF-2α-mediated inflammation in arsenite-induced transformation of human bronchial epithelial cells. Toxicol Appl Pharmacol. 2013;272:542–550. doi: 10.1016/j.taap.2013.06.017. [DOI] [PubMed] [Google Scholar]
- Xu Y, Tokar EJ, Waalkes MP. Arsenic-induced cancer cell phenotype in human breast epithelia is estrogen receptor-independent but involves aromatase activation. Arch Toxicol. 2014;88:263–274. doi: 10.1007/s00204-013-1131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Golay HG, Barbie DA. Targeting pathways downstream of KRAS in lung adenocarcinoma. Pharmacogenomics. 2014;15:1507–1518. doi: 10.2217/pgs.14.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.