

Abstract
Metabolic activation and oxidant stress are key events in the pathophysiology of acetaminophen (APAP) hepatotoxicity. The initial mitochondrial oxidative stress triggered by protein adduct formation is amplified by c-jun-N-terminal kinase (JNK), resulting in mitochondrial dysfunction and ultimately cell necrosis. Apoptosis signal-regulating kinase 1 (ASK1) is considered the link between oxidant stress and JNK activation. The objective of the current study was to assess the efficacy and mechanism of action of the small-molecule ASK1 inhibitor GS-459679 in a murine model of APAP hepatotoxicity. APAP (300 mg/kg) caused extensive glutathione depletion, JNK activation and translocation to the mitochondria, oxidant stress and liver injury as indicated by plasma ALT activities and area of necrosis over a 24h observation period. Pretreatment with 30 mg/kg of GS-459679 almost completely prevented JNK activation, oxidant stress and injury without affected the metabolic activation of APAP. To evaluate the therapeutic potential of GS-459679, mice were treated with APAP and then with the inhibitor. Given 1.5h after APAP, GS-459679 was still protective, which was paralleled by reduced JNK activation and p-JNK translocation to mitochondria. However, GS-459679 treatment was not more effective than N-acetylcysteine, and the combination of GS-459679 and N-acetylcysteine exhibited similar efficacy as N-acetylcysteine monotherapy, suggesting that GS-459769 and N-acetylcysteine affect the same pathway. Importantly, inhibition of ASK1 did not impair liver regeneration as indicated by PCNA staining. In conclusion, the ASK1 inhibitor GS-459679 protected against APAP toxicity by attenuating JNK activation and oxidant stress in mice and may have therapeutic potential for APAP overdose patients.
Keywords: Acetaminophen, ASK1, hepatotoxicity, c-jun N-terminal kinase, oxidant stress
INTRODUCTION
Acetaminophen (APAP) is a widely used analgesic and antipyretic. A therapeutic dose of APAP is safe and effective while an overdose can cause severe liver injury in animals and humans (Larson, 2007; McGill et al., 2012). Hepatotoxicity is initiated by formation of a reactive metabolite, N-acetyl-p-benzoquinone imine, which is detoxified by glutathione but also binds to cellular proteins (Cohen et al., 1997). Although formation of protein adducts, especially in mitochondria, is a critical event in the pathophysiology (Tirmenstein and Nelson, 1989), it is not sufficient to cause cell death (Jaeschke and Bajt, 2006). The current hypothesis is that protein binding induces mitochondrial dysfunction including inhibition of mitochondrial respiration (Meyers et al., 1988) and formation of reactive oxygen species (ROS) (Jaeschke, 1990) and peroxynitrite (Hinson et al., 1998; Cover et al., 2005). This oxidant stress together with lysosomal iron taken up into mitochondria by the Ca2+ uniporter (Kon et al., 2010), leads to the opening of the mitochondrial membrane permeability transition (MPT) pore and collapse of the membrane potential (Kon et al., 2004; Reid et al., 2005; Ramachandran et al., 2011a; LoGuidice and Boelsterli, 2011). The massive loss of mitochondrial function triggers necrotic cell death (Gujral et al., 2002). A number of therapeutic interventions that accelerated the recovery of mitochondrial glutathione and improved the detoxification of mitochondrial ROS and peroxynitrite provided clear evidence for the critical role of the mitochondrial oxidant stress in the mechanism of cell death (Knight et al., 2002; James et al., 2003; Ramachandran et al., 2011b; Saito et al., 2010b). However, it remained unclear how this extensive mitochondrial oxidant stress ultimately can be induced.
Studies during the last decade aimed at elucidating intracellular signaling events have demonstrated a critical role of the mitogen-activated protein kinase (MAPK) c-jun-N-terminal kinase (JNK) in APAP-induced cell death in mice (Gunawan et al., 2006; Henderson et al., 2007; Latchoumycandane et al., 2007) and in human hepatocytes (Xie et al., 2014). Importantly, JNK was not only activated (phosphorylated) in the cytosol, p-JNK also translocated to the mitochondria (Hanawa et al., 2008) and amplified the mitochondrial oxidant stress (Saito et al., 2010a) through binding to an anchor protein in the outer mitochondrial membrane (Win et al., 2011). It was also shown that an early mitochondrial oxidant stress triggered JNK activation (Hanawa et al., 2008; Saito et al., 2010a). However, JNK is not a redox-sensitive kinase. In fact, the upstream kinases, apoptosis signal-regulating kinase 1 (ASK1) (Nakagawa et al., 2008) and mixed-lineage kinase 3 (MLK3) (Sharma et al., 2012) have been identified as critical upstream regulators of JNK activation. Oxidant stress can release ASK1 from its complex with reduced thioredoxin by oxidation of thioredoxin resulting in the liberation of the active kinase (Saitoh et al., 1998). Because ASK1 is thought to be mainly responsible for sustained activation of JNK, the substantial reduction in APAP-induced liver injury in ASK1-deficient mice demonstrated the critical role of ASK1 in the pathophysiology (Nakagawa et al., 2008). Recently a potent and selective small molecule inhibitor of ASK1with properties amenable for in vivo efficacy testing in rodents was developed Gerczuk et al., 2012; Toldo et al., 2012). This inhibitor enabled us to test if pharmacologic inhibition of ASK has therapeutic potential for treating APAP hepatotoxicity and to study the mechanisms of protection.
MATERIALS AND METHODS
Animals
8 week old male C57Bl/6 mice were acquired from Jackson Laboratories for the experiments. Animals were housed in a controlled environment with a 12 hour light/dark cycle and free access to food and water. Animals were acclimatized for at least 3 days and fasted overnight before experiments. All experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center.
Inhibitors
GS-459679 and GS-444217 are potent and selective ATP competitive inhibitors of ASK1 developed by Gilead Sciences, Inc., Foster City, CA (Breckenridge et al., manuscript in preparation). In a competitive, time-resolved fluorescence resonance energy transfer immunoassay, GS-459679 and GS-444217 directly inhibit ASK1 kinase activity with IC50 (concentration that inhibits ASK1 kinase by 50%) values of 6.1 and 2.9 nM, respectively. One micromolar GS-459679 results in 99% inhibition of the ASK1 kinase but does not inhibit 20 other kinases implicated in cellular stress signaling (Gerczuk et al., 2012).
Experimental design
For pre-treatment studies, animals were administered either the ASK1 inhibitor GS-459679 (ASK1i) (10 or 30 mg/kg), or vehicle (55% PEG in H2O) 30 minutes prior to administration of APAP (300 mg/kg, i.p.). Animals were sacrificed 0.5, 6 or 24 hours after APAP. For post-treatment experiments, animals were administered APAP (300 mg/kg, i.p.) followed by ASK1i (30 mg/kg, i.p.), N-acetylcysteine (NAC) (500 mg/kg, i.p.), ASK1i + NAC or vehicle (saline) at 1.5 or 3 hours after APAP. Animals were allowed access to regular food at 6 h after APAP treatment; the animals were sacrificed at 24 h. For 48 h experiments evaluating liver regeneration, animals were given back food (control diet or diet containing 0.2% of the ASK1 inhibitor GS-444217) 6 h after APAP administration. At the end of the experiments, animals were anesthetized with isoflurane and blood was drawn from vena cava into heparinized syringes for determination of alanine aminotransferase (ALT) activity. The liver was excised and rinsed in saline before being divided for histology and subcellular fractionation, and the rest being snap frozen in liquid nitrogen and subsequently stored at −80°C.
Histology
Formalin-fixed tissue samples were embedded in paraffin and 4 μm sections were cut. Replicate sections were stained with hematoxylin and eosin (H&E) to evaluate necrosis as described (Gujral et al., 2002).
Measurement of GSH and GSSG
Total soluble GSH and GSSG were determined in the liver homogenate with a modified method of Tietze (Jaeschke and Mitchell, 1990). In brief, the frozen tissue was homogenized at 0°C in 3% sulfosalicylic acid containing 0.1mM EDTA. For measurement of GSSG, GSH was trapped with 10 mM N-ethylmaleimide. The sample was centrifuged after dilution with 0.01 N HCl and the supernatant was further diluted with 100 mM potassium phosphate buffer (KPP), pH 7.4. The samples were then assayed using dithionitrobenzoic acid. All data are expressed as GSH-equivalents.
Isolation of subcellular fractions and western blotting
Mitochondria and cytosolic fractions were isolated using differential centrifugation as described (Xie et al., 2013). Western blots were performed as described in detail (Bajt et al., 2000; Du et al., 2013), using the following antibodies: a rabbit anti-JNK antibody (Cell signaling Technology, Danvers, MA), a rabbit anti-phospho-JNK antibody #4668, which recognizes Thr183/Tyr185 phosphorylated JNK1/2 protein (Cell Signaling Technology, Danvers, MA). A horseradish peroxidase-coupled donkey anti-rabbit IgG (Santa Cruz) was used as secondary antibody. Proteins were visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech. Inc, Piscataway, NJ).
Statistics
All data are presented as mean ± SE. For normally distributed data, comparisons were made between two groups using Student’s t-test, or between three or more groups using one-way analysis of variance (ANOVA) with Tukey’s post-hoc test. For non-normally distributed data, comparisons were made between two groups using the Mann-Whitney U-test, or between three or more groups using the Kruskal-Wallis test with Dunn’s multiple comparisons. All statistical tests were performed using SigmaPlot software (Systat, San Jose, CA). P< 0.05 was considered significant.
RESULTS
Pharmacological inhibition of ASK1 protects against APAP-induced liver injury
As an initial attempt to investigate the effect of ASK1 inhibition on APAP hepatotoxicity, mice were treated with 10 or 30 mg/kg of GS-459679 (ASK1i), 30 min prior to administration of 300 mg/kg APAP. Liver injury was then examined 6 and 24 h later. APAP induced significant liver injury as indicated by the increase in plasma ALT activities, which were elevated at 6 h and further increased by 24 h (Figure 1A). This was accompanied by development of centrilobular necrosis (Figure 1B, C). Pretreatment with 10 mg/kg ASK1i did not achieve significant protection against APAP hepatotoxicity up to 24 h. However, pre-treatment with 30 mg/kg ASK1i resulted in a pronounced protection as indicated by the 93% (6 h) and 95.2% (24 h) reduction in plasma ALT activities (Figure 1A) and 100% (6 h) and 84% (24 h) less centrilobular necrosis (Figure 1B, C), indicating that pharmacological blockage of ASK1 activity could prevent APAP-induced liver injury. Evaluation of plasma concentrations of ASK1i at 6 h post-APAP administration confirmed a higher exposure of ASK1i in animals administered 30 mg/kg compared to 10 mg/kg (Supplemental Figure 1). Since the 30 mg/kg dose seemed to be most effective, this dose was used for further experiments. In order to investigate whether the ASK1i treatment protects against the injury by inhibiting drug metabolism, short term experiments were carried out by measuring the depletion of liver GSH at 30 min after APAP as a surrogate marker for NAPQI formation. APAP caused 86% depletion of hepatic GSH levels within 30 min. The initial loss of liver GSH was comparable in animals treated with APAP alone or in combination with either the vehicle or ASK1i, suggesting that the inhibitor did not affect metabolic activation of APAP (Figure 2). Assessment of GSH content and oxidant stress (GSSG-to-GSH ratio) after APAP treatment indicated a partial recovery of hepatic GSH levels at 6 and 24 h (Figure 3A), a significant increase in GSSG levels at 24 h (Figure 3B) and a significantly higher GSSG-to-GSH ratio at 6 and 24 h (Figure 3C). Treatment with 30 mg/kg ASK1i remarkably enhanced the recovery of GSH levels (Figure 3A) and prevented (6 h) or attenuated the oxidant stress (24 h) (Figure 3C).
Figure 1.

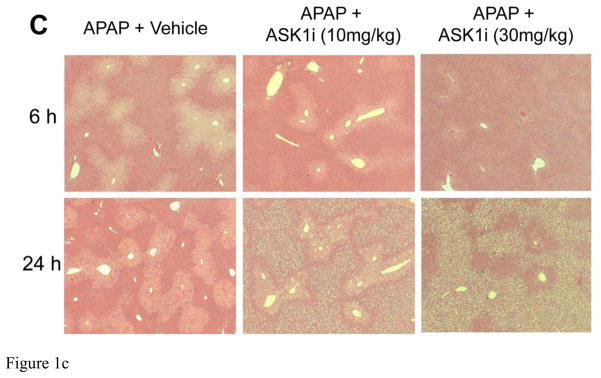
Protective effect of the ASK1 inhibitor GS-459679 against APAP -induced liver injury. C57BL/6 mice were pretreated with either 10 or 30 mg/kg GS-459679 or an equivalent dose of the vehicle (55% PEG in water) 30 min before 300 mg/kg APAP administration. (A) Plasma alanine aminotransferase (ALT) activities measured at 6 and 24 h after APAP treatment. (B) Quantitation of the areas of necrosis in H&E-stained liver sections. (C) Representative sections from each group with H&E staining. Data represent means ± SE of n=4–11 animals per group. *P<0.05 compared with control (Ctrl) groups, #P<0.05 compared with corresponding APAP+Vehicle groups.
Figure 2.

Effect of ASK1 inhibitor GS-459679 on APAP-induced GSH depletion. Mice were treated with 30 mg/kg GS-459679 or equivalent amount of vehicle (55% PEG in water) 30 min before 300 mg/kg APAP administration. GSH levels (μmol/g liver) were measured 30 min after APAP treatment. Data represent means ± SE on n=3–4 animals per group. *P<0.05 compared with the control group.
Figure 3.

Effect of the ASK1 inhibitor GS-459679 on hepatic GSH and GSSG levels after APAP treatment. Mice were treated with 30 mg/kg GS-459679 or equivalent amount of vehicle (55% PEG in water) 30 min before 300 mg/kg APAP administration. GSH levels were measured either 6 or 24 h after APAP treatment. (A) Total liver glutathione (GSH+GSSG) in μmol/g liver. (B) Glutathione disulfide (GSSG) in nmol/g liver. (C) GSSG-to-GSH ratio. Data represent means ± SE of n=4–7 animals per group.*P<0.05 compared with control (Ctrl) groups, #P<0.05 compared with APAP+Vehicle groups.
ASK1 inhibition prevents activation of JNK subsequent to APAP overdose
JNK activation has been implicated in APAP hepatotoxicity and ASK1 is suggested to function upstream of JNK in this pathological context (Nakagawa et al., 2008). To confirm the mechanism of action of the pharmacological ASK inhibitor, the next series of experiments evaluated JNK activation after APAP overdose. APAP treatment caused significant JNK activation as demonstrated by elevated levels of the Thr183/Tyr185 phosphorylated protein, which peaked by 6 h and decreased by 24 h (Figure 4A). Pretreatment with the ASK1i almost completely prevented JNK activation at 6 h (Figure 4A). The changes in JNK phosphorylation were not due to alterations in total JNK protein, since these were unaltered at both 6 and 24 hours after APAP (Figure 4A). Quantitative densitometric analysis confirmed the changes in the p-JNK and total JNK levels (Figure 4B, C).
Figure 4.

Effect of the ASK1 inhibitor on JNK activation. Western blot analysis (A) of p-JNK and total JNK in the cytosolic fraction of livers from untreated mice (Ctrl), mice treated with 30 mg/kg ASK1 inhibitor or mice treated with the same volume of vehicle (55% PEG in water) 30 min prior to 300 mg/kg of APAP administration. Liver samples were obtained 6 or 24 h after APAP treatment. Densitometric quantification was performed on bands of p-JNK (B) and total JNK (C). Data represent means ± SE of n=4 animals. *P<0.05 compared with control groups. #P<0.05 compared with APAP+Vehicle group.
Inhibition of ASK1 prevents APAP-induced translocation of JNK to mitochondria
JNK translocation to mitochondria is a key mechanistic feature of APAP-induced hepatotoxicity and mitochondrial permeability transition (Hanawa et al., 2008). Since administration of ASK1i prevented JNK activation, the effect of this intervention on mitochondrial JNK translocation was examined. Treatment with APAP resulted in translocation of activated JNK (p-JNK) to the mitochondria by 6 h and this was significantly inhibited by ASK1i pretreatment (Figure 5). The APAP-induced elevation in p-JNK was accompanied by increases in total JNK in mitochondria and this was prevented by administration of ASK1i (Figure 5). This suggests that JNK translocation to mitochondria occurs subsequent to activation, implying that ASK1 inhibition in the context of APAP overdose prevents JNK activation and downstream translocation of p-JNK to mitochondria.
Figure 5.

Western blots and densitometric analysis of p-JNK translocation to the mitochondria at 6 h. Mice were treated with 30 mg/kg ASK1 inhibitor GS-459679 or the vehicle (55% PEG in water) 30 min before 300 mg/kg APAP injection. Western blotting was performed for p-JNK and total JNK in mitochondria (A). Densitometric quantification was carried out on band intensity of both total JNK (B) and p-JNK (C). Data represent means ± SE of n=4 animals per group. *P<0.05 compared with control (Ctrl) group, #P<0.05 compared with APAP+Vehicle group.
ASK1i administration can protect against liver injury even when given after APAP overdose
To examine if ASK1i administration was also effective in a therapeutic setting, the next series of experiments compared ASK1i with N-acetylcysteine, which is the current standard of care in the clinic for APAP overdose. Animals treated with a single dose of 500 mg/kg NAC 1.5 h after APAP were significantly protected against liver injury at 24 h as indicated by both ALT activities and area of necrosis (Figure 6A). However, this protection was significantly eroded when NAC was administered 3 h after APAP, although ALT levels were still significantly less than controls (Figure 6A). Animals treated with ASK1i 1.5 h after APAP also showed significant protection against liver injury when compared to vehicle-treated animals but when ASK1i administration was delayed to 3 h post APAP, no protection against injury was evident (Figure 6B). When animals were treated with both NAC and ASK1i, protection was almost complete when the combination was given at 1.5 h after APAP (Figure 6C). However, delaying the drug administration to either 3 h (Figure 6C) or 2 h and 15 min (data not shown) after APAP negated this protection. This suggests that ASK1 inhibition is only effective early after APAP administration (within 1.5 hours in the mouse) and loses its efficacy relatively rapidly after that time.
Figure 6.

Effect of ASK1 inhibitor GS-459679 post-treatment against APAP hepatotoxicity at 24 h. Mice were treated with 500 mg/kg N-acetylcysteine (NAC) (A), 30 mg/kg ASK1i (B) and the combination of both (C) either 1.5 or 3 h after 300 mg/kg of APAP administration. The vehicle for NAC was PBS, for ASK1i was 55% PEG, and for ASK1i+NAC it was PBS+55%PEG. Plasma ALT activities and the areas of necrosis were quantified. Data represent means ± SE of 3–5 animals per group and time point. *P<0.05 compared with the respective APAP+Vehicle treated group.
ASK1i administration after APAP overdose can also prevent JNK activation in the cytosol and its translocation to the mitochondria. To examine if the mechanism of protection by ASK1i as a 1.5 h post treatment also involved prevention of JNK activation, cytosolic JNK activation and its translocation to mitochondria was examined by western blotting at 6 h after APAP. The APAP-induced JNK activation in the cytosol was significantly reduced by NAC, ASK1i or NAC+ASK1i post treatment, all of which also effectively prevented p-JNK translocation to mitochondria (Figure 7). To evaluate the reason for the limited therapeutic window for the ASK1i inhibitor, early JNK activation and mitochondrial p-JNK translocation was determined (Figure 8). Limited APAP-induced JNK activation in the cytosol was observed as early as 1 h with almost no p-JNK translocation to the mitochondria. However, by 2 h, massive JNK activation and extensive mitochondrial p-JNK translocation had occurred (Figure 8). This suggests that ASK1 inhibition is only effective if it occurs before mitochondrial p-JNK translocation.
Figure 7.

Effect of ASK1 inhibitor GS-459679 post-treatment on APAP-induced JNK activation. Western blots (A) and densitometry analysis of p-JNK in the cytosolic (B) and mitochondrial (C) fractions. Mice were treated with 500 mg/kg N-acetylcysteine (NAC) (A), 30 mg/kg ASK1i (B) and the combination of both (C) 1.5 h after administration of 300 mg/kg APAP. The vehicle for NAC was PBS, for ASK1i was 55% PEG in water, and for ASK1i+NAC it was PBS+55%PEG. Data represent means ± SE of 3–5 animals per group. *P<0.05 compared with APAP+Vehicle treated group.
Figure 8.

Time course of JNK activation after APAP treatment. P-JNK was determined in cytosol (A, B) and in mitochondrial fraction (C, D) after treatment with 300 mg/kg APAP. Data represent means ± SE of 3 animals per group. *P<0.05 compared with control (Ctrl).
ASK1 inhibitor administration does not impair liver regeneration after APAP
A major concern about inhibition of the JNK pathway is the fact that it may lead to impairment of liver regeneration (Schwabe et al., 2003). To test whether pharmacological inhibition of ASK1 reduces the regenerative capacity of the liver, expression of proliferating cell nuclear antigen (PCNA), which is a marker of regeneration, was assessed. APAP hepatotoxicity caused significant up-regulation of PCNA expression at 24 h, an effect which was amplified by administration of ASK1i either as a 1.5 h post-treatment (Figure 9A, B) or a 30 min pretreatment (data not shown). Histological assessment of PCNA staining confirmed the western blot data (Figure 9C). We also evaluated if continuous inhibition of ASK1 following APAP-induced liver injury impaired liver regeneration. To that end, animals were administered APAP and then 6 h later provided food with 0.2% by weight of the ASK1 inhibitor GS-444217 and PCNA staining was evaluated 48 h post-APAP administration. GS-444217 was utilized for this study because it has excellent absorption properties such that high steady state plasma concentrations are achieved when it is administered in rodent chow (Supplemental Figure 2 and Gilead unpublished data). Consistent with the data from pretreatment or post-treatment of ASK1i (GS-459679), the increased PCNA expression was not impaired by GS-444217 in the diet following APAP administration (Figure 10A, B). These results were confirmed by PCNA staining in histological sections (Figure 10C). We confirmed that GS-444217 had a similar protective effect on APAP-induced liver injury as GS-459679 when administered 30 min prior to APAP (Supplemental Figure 3). Together, these data suggest that ASK1 inhibition does not interfere with the regeneration process after APAP hepatotoxicity.
Figure 9.

Effect of the ASK1 inhibitor on the regeneration response after APAP-induced liver injury. Proliferating Cell Nuclear Antigen (PCNA) levels was determined by western blotting (A) and immunohistochemistry (C) 24 h after APAP in animals treated either with vehicle (55% PEG in water) or 30 mg/kg ASK1 inhibitor 1.5h after APAP. Densitometry data (B) represent means ± SE of 4 animals per group. *P<0.05 compared with control (Ctrl) group, #P<0.05 compared with APAP+Vehicle group.
Figure 10.

Effect of chronic ASK1 inhibitor (GS-444217) treatment on the regeneration response after APAP-induced liver injury. Proliferating Cell Nuclear Antigen (PCNA) levels were determined by western blotting (A) and immunohistochemistry (C) 48 h after APAP in animals fed an ASK1 inhibitor-containing diet or control diet beginning at 6 h after 300 mg/kg APAP. Densitometry data (B) represent means ± SE of 4 animals per group. *P<0.05 compared with control (Ctrl) group, #P<0.05 compared with APAP+control diet.
DISCUSSION
The objective of the investigation was to evaluate the protective effect of the ASK1 inhibitor GS-459679 in a murine model of APAP hepatotoxicity. Our data showed that pretreatment with the ASK1 inhibitor and treatment during a limited therapeutic window after APAP overdose attenuated liver injury.
The ASK1 inhibitor protects against APAP hepatotoxicity by inhibition of JNK
ASK1 is a serine/threonine protein kinase, which can activate the MAPK cascade resulting in the activation of p38 and JNK. ASK1 is expressed in both cytoplasm and mitochondria and can be activated by ROS or cytokines such as tumor necrosis factor-α (Ichijo et al., 1997; Nishitoh et al., 1998; Matsuzawa et al., 2005). Under basal conditions, ASK1 is bound and repressed by the reduced form of thioredoxin (Saitoh et al., 1998). However, during oxidative stress, ASK1 is released from the oxidized form of thioredoxin and activated by an autocatalytic mechanism (Liu et al., 2000), allowing it to induce the phosphorylation of MAP2 kinases that phosphorylate and activate downstream targets such as JNK. It has been demonstrated that mice deficient in ASK1 were significantly protected against APAP hepatotoxicity (Nakagawa et al., 1998). Our data show that pharmacological inhibition of ASK1 with a potent and highly selective small molecule ASK1 inhibitor can attenuate APAP-induced liver injury as indicated by reduced JNK activation, lower oxidant stress and ALT release and limited centrilobular necrosis.
The importance of JNK activation and p-JNK translocation to the mitochondria as a mechanism to amplify the mitochondrial oxidant stress has been well established in mice (Hanawa et al., 2008; Saito et al., 2010a) and also recently in primary human hepatocytes (Xie et al., 2014). The fact that the ASK1 inhibitor attenuated JNK activation in the cytosol, largely prevented the translocation of p-JNK to the mitochondria and reduced the mitochondrial oxidant stress was consistent with an effect upstream of JNK activation. Several other kinases including mixed-lineage kinase 3 (MLK3) (Sharma et al., 2012) and glycogen synthase kinase 3β (GSK3β) (Shinohara et al., 2010) were shown to be involved in JNK activation during APAP-induced liver injury. Although MLK3 can also be activated by oxidant stress, MLK3 is responsible for GSK3β and JNK activation (Sharma et al., 2012). In contrast, ASK1 is considered to be mainly involved in the prolonged late JNK activation (>4h post-APAP administration) (Nakagawa et al., 2008). The importance of the prolonged JNK activation for APAP-induced liver injury is supported by studies in JNK-deficient mice (Gunawan et al., 2006, Henderson et al., 2007), other studies demonstrating that liver injury is decreased when oxidative stress and sustained JNK activation are decreased; for example in female mice (Du et al., 2014), in Fas receptor-deficient lpr mice (Williams et al., 2013), or in mice treated with allopurinol (Williams et al., 2014). In addition, the increased liver injury after APAP overdose in animals deficient in phosphatases, which are known to counteract JNK phosphorylation, further supports the critical role of the ASK1-induced JNK activation cascade at least in the mouse (Mobasher et al., 2013; Wancket et al., 2012).
The role of ASK1 and JNK activation during APAP overdose in humans remains unclear. More recent translational studies support the hypothesis that protein adducts formation and mitochondrial dysfunction are important for the human pathophysiology (Davern et al., 2006; McGill et al., 2012, 2014). Although APAP causes extensive JNK activation and mitochondrial p-JNK translocation in primary human hepatocytes, the protection by inhibition of JNK was modest (Xie et al., 2014). In contrast, APAP did not induce JNK activation in the metabolically competent human hepatoma cell line HepaRG (Xie et al., 2014), which is, however, still susceptible to APAP-induced cell injury involving oxidant stress and mitochondrial dysfunction (McGill et al., 2011). Thus, whereas the relevance of ASK1 and JNK activation is well documented in the murine system, the importance of ASK1 in the human pathophysiology of APAP-induced liver injury remains to be further investigated.
The therapeutic window of pharmacological ASK1 inhibition in APAP hepatotoxicity
In addition to the efficacy of pretreatment with the ASK1 inhibitor, our study also demonstrated that administration of the ASK1 inhibitor during a narrow therapeutic window after APAP overdose can still be effective. However, the ASK1 inhibitor was not more effective than NAC treatment, the standard of care in patients. In addition, the combined treatment of NAC and ASK1 inhibitor did not show a relevant additive effect. These results indicate that both interventions target the same mechanism in the pathophysiology. The faster recovery of hepatic GSH levels mediated by treatment of NAC after the metabolism phase leads to enhanced scavenging of reactive oxygen and peroxynitrite (Knight et al., 2002; James et al., 2003; Saito et al., 2010b). Inhibition of ASK1 reduces JNK activation, which is critical for the amplification of the mitochondrial oxidant stress (Hanawa et al., 2008; Saito et al, 2010a). Thus, both interventions attenuate the mitochondrial oxidant stress, which is responsible for the MPT pore opening and necrosis (Kon et al., 2004; Reid et al., 2005).
The limited therapeutic window of ASK1 inhibition in mice may be related to the rapid JNK activation and subsequent translocation to the mitochondria. The ASK1 inhibitor was effective only when it was administered before significant p-JNK had translocated to the mitochondria, suggesting the primary mechanism of action of the ASK1 inhibitor was to prevent JNK activation and translocation. After p-JNK has translocated to the mitochondria, the amplification of the oxidant stress may involve other kinases in a feed-forward mechanism, which no longer depends on ASK1 alone or may involve JNK-independent mechanisms (Saberi et al., 2014). In either case, these data are not consistent with the original hypothesis that ASK1 is only responsible for the late JNK activation. Our data suggest that ASK1 inhibition is required relatively early (<1.5 h after APAP) in order to be effective.
The limited therapeutic window for ASK1 inhibition in mice may not translate to the human pathophysiology, which is generally delayed compared to mice with peak injury at 36–48 h in humans and around 12 h in mice. The reason for the delay in human hepatocytes appears to be related to delay in mitochondrial adduct formation, JNK activation and p-JNK translocation to the mitochondria after APAP overdose (Xie et al., 2014). As a consequence, NAC treatment is still 100% protective even if initiated 6 h after APAP and still partially effective when given 15 h after APAP exposure (Xie et al., 2014). In addition, a JNK inhibitor was still effective when administered 3 h after APAP in human hepatocytes (Xie et al., 2014). Thus, the therapeutic window for the ASK1 inhibitor needs to be assessed in humans but it can be expected to be substantially larger than in mice.
Effect of the ASK1 inhibitor on regeneration
Although JNK activation has been associated with mechanisms of liver injury in many disease models including APAP hepatotoxicity (Czaja, 2003; Gunawan et al., 2006), JNK can phosphorylate c-jun and activate the transcription factor AP-1, which can promote regeneration by Cyclin D1 gene expression (Schwabe et al., 2003). Because regeneration is critical for recovery and survival after APAP-induced liver injury (Apte et al., 2009; Bajt et al., 2003; Bhushan et al., 2013), and because other kinases can activate JNK independently of ASK1, it was important to evaluate if the ASK1 inhibitor affects regeneration as assessed by the expression of PCNA in the recovering liver. Similar to previous interventions such as GSH or NAC (Bajt et al., 2003), which reduced APAP-induced liver injury, the ASK1 inhibitor actually enhanced PCNA expression. However, using a second ASK1 inhibitor with high oral availability in the diet starting 6 h after APAP had no effect of PCNA expression during the recovery phase at 48 h. Thus, our data demonstrated that 2 different ASK1 inhibitors protected against APAP hepatotoxicity and did not inhibit regeneration. Second, the inhibitor GS-444217, which has better oral availability than GS-459679, allowed continuous inhibition of ASK1 for several days. The fact that no effect on regeneration was observed supports the hypothesis that this inhibitor could be used therapeutically. Together, these findings indicate that inhibition of ASK1-mediated JNK activation can attenuate liver injury but does not affect cell cycle activation and regeneration after APAP-induced liver injury.
In summary, our data demonstrate that the specific and selective ASK1 inhibitor GS-459679 protected against APAP-induced liver injury by inhibiting JNK activation, p-JNK translocation to the mitochondria and oxidant stress. The inhibitor was effective in this murine model as pretreatment and during a limited therapeutic window also when administered after APAP. In addition, the ASK1 inhibitor did not inhibit regeneration after APAP-induced liver injury. Because of the delayed JNK activation and delayed cell injury process in humans, small molecule ASK1 inhibitors may be suitable as therapeutic interventions against APAP-induced liver injury.
Supplementary Material
HIGHLIGHTS.
Two ASK1 inhibitors protected against acetaminophen-induced liver injury
The ASK1 inhibitors protect when used as pre- or post-treatment
Protection by ASK1 inhibitor is not due to inhibition of APAP metabolism
The ASK1 inhibitor prevents JNK activation and translocation to mitochondria
Treatment with ASK1 inhibitors does not impair liver regeneration after APAP
Acknowledgments
This work was supported in part by a grant from Gilead Science Inc., by National Institutes of Health Grants AA12916 and DK070195 and by grants from the National Center for Research Resources (5P20RR021940-07) and the National Institute of General Medical Sciences (8 P20 GM103549-07) from the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
David Breckenridge and John Liles are employees of Gilead Sciences, Inc. Hartmut Jaeschke received a grant from Gilead Sciences, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, Monga SP. Beta-catenin activation promotes liver regeneration after acetaminophen-induced injury. Am J Pathol. 2009;175:1056–1065. doi: 10.2353/ajpath.2009.080976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajt ML, Lawson JA, Vonderfecht SL, Gujral JS, Jaeschke H. Protection against Fas receptor-mediated apoptosis in hepatocytes and nonparenchymal cells by a caspase-8 inhibitor in vivo: evidence for a postmitochondrial processing of caspase-8. Toxicol Sci. 2000;58:109–117. doi: 10.1093/toxsci/58.1.109. [DOI] [PubMed] [Google Scholar]
- Bajt ML, Knight TR, Farhood A, Jaeschke H. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. 2003;307:67–73. doi: 10.1124/jpet.103.052506. [DOI] [PubMed] [Google Scholar]
- Bhushan B, Borude P, Edwards G, Walesky C, Cleveland J, Li F, Ma X, Apte U. Role of bile acids in liver injury and regeneration following acetaminophen overdose. Am J Pathol. 2013;183:1518–1526. doi: 10.1016/j.ajpath.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1–12. doi: 10.1006/taap.1996.8074. [DOI] [PubMed] [Google Scholar]
- Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315:879–87. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- Czaja MJ. The future of GI and liver research: editorial perspectives. III JNK/AP-1 regulation of hepatocyte death. Am J Physiol Gastrointest Liver Physiol. 2003;284:G875–9. doi: 10.1152/ajpgi.00549.2002. [DOI] [PubMed] [Google Scholar]
- Davern TJ, 2nd, James LP, Hinson JA, Polson J, Larson AM, Fontana RJ, Lalani E, Munoz S, Shakil AO, Lee WM Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687–94. doi: 10.1053/j.gastro.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Du K, Williams CD, McGill MR, Xie Y, Farhood A, Vinken M, Jaeschke H. The gap junction inhibitor 2-aminoethoxy-diphenyl-borate protects against acetaminophen hepatotoxicity by inhibiting cytochrome P450 enzymes and c-jun N-terminal kinase activation. Toxicol Appl Pharmacol. 2013;273:484–491. doi: 10.1016/j.taap.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Williams CD, McGill MR, Jaeschke H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol Appl Pharmacol. 2014;281:58–66. doi: 10.1016/j.taap.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerczuk PZ, Breckenridge DG, Liles JT, Budas GR, Shryock JC, Belardinelli L, Kloner RA, Dai W. An apoptosis signal-regulating kinase 1 inhibitor reduces cardiomyocyte apoptosis and infarct size in a rat ischemia-reperfusion model. J Cardiovasc Pharmacol. 2012;60:276–82. doi: 10.1097/FJC.0b013e31825ea0fa. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Knight TR, Farhood A, Bajt ML, Jaeschke H. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–78. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–77. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56:982–90. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–41. [PubMed] [Google Scholar]
- Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Mitchell JR. Use of isolated perfused organs in hypoxia and ischemia/reperfusion oxidant stress. Methods Enzymol. 1990;186:752–9. doi: 10.1016/0076-6879(90)86175-u. [DOI] [PubMed] [Google Scholar]
- James LP, McCullough SS, Lamps LW, Hinson JA. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 2003;75:458–67. doi: 10.1093/toxsci/kfg181. [DOI] [PubMed] [Google Scholar]
- Knight TR, Ho YS, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303:468–75. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–9. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- Kon K, Kim JS, Uchiyama A, Jaeschke H, Lemasters JJ. Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen-induced toxicity to mouse hepatocytes. Toxicol Sci. 2010;117:101–8. doi: 10.1093/toxsci/kfq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–48. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–21. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Molecular and cellular biology. 2000;20:2198–208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoGuidice A, Boelsterli UA. Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin D-regulated permeability transition. Hepatology. 2011;54:969–78. doi: 10.1002/hep.24464. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nature Immunol. 2005;6:587–92. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–83. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Staggs VS, Sharpe MR, Lee WM, Jaeschke H Acute Liver Failure Study Group. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology. 2014;60:1336–45. doi: 10.1002/hep.27265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill MR, Yan HM, Ramachandran A, Murray GJ, Rollins DE, Jaeschke H. HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 2011;53:974–82. doi: 10.1002/hep.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers LL, Beierschmitt WP, Khairallah EA, Cohen SD. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol Appl Pharmacol. 1988;36:1193–6. doi: 10.1016/0041-008x(88)90040-3. [DOI] [PubMed] [Google Scholar]
- Mobasher MA, González-Rodriguez A, Santamaría B, Ramos S, Martín MÁ, Goya L, Rada P, Letzig L, James LP, Cuadrado A, Martín-Pérez J, Simpson KJ, Muntané J, Valverde AM. Protein tyrosine phosphatase 1B modulates GSK3β/Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity. Cell Death Dis. 2013;4:e626. doi: 10.1038/cddis.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, Ichijo H, Omata M. Deletion of apoptosis signal-regulating kinase 1 attenuates acetaminophen-induced liver injury by inhibiting c-Jun N-terminal kinase activation. Gastroenterology. 2008;135:1311–21. doi: 10.1053/j.gastro.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Molecular Cell. 1998;2:389–95. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 2011a;45:156–64. doi: 10.3109/10715762.2010.520319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2011b;251:226–33. doi: 10.1016/j.taap.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated hepatocytes. J Pharmacol Exp Ther. 2005;312:509–516. doi: 10.1124/jpet.104.075945. [DOI] [PubMed] [Google Scholar]
- Saberi B, Ybanez MD, Johnson HS, Gaarde WA, Han D, Kaplowitz N. Protein kinase C (PKC) participates in acetaminophen hepatotoxicity through c-jun-N-terminal kinase (JNK)-dependent and -independent signaling pathways. Hepatology. 2014;59:1543–1554. doi: 10.1002/hep.26625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito C, Lemasters JJ, Jaeschke H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2010a;246:8–17. doi: 10.1016/j.taap.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010b;51:246–254. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- Sharma M, Gadang V, Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol. 2012;82:1001–7. doi: 10.1124/mol.112.079863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Ybanez MD, Win S, Than TA, Jain S, Gaarde WA, Han D, Kaplowitz N. Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1. J Biol Chem. 285:8244–8255. doi: 10.1074/jbc.M109.054999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9. [PubMed] [Google Scholar]
- Toldo S, Breckenridge DG, Mezzaroma E, Van Tassell BW, Shryock J, Kannan H, Phan D, Budas G, Farkas D, Lesnefsky E, Voelkel N, Abbate A. Inhibition of apoptosis signal-regulating kinase 1 reduces myocardial ischemia-reperfusion injury in the mouse. J Am Heart Assoc. 2012;1:e002360. doi: 10.1161/JAHA.112.002360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wancket LM, Meng X, Rogers LK, Liu Y. Mitogen-activated protein kinase phosphatase (Mkp)-1 protects mice against acetaminophen-induced hepatic injury. Toxicol Pathol. 2012;40:1095–105. doi: 10.1177/0192623312447551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, McGill MR, Farhood A, Jaeschke H. Fas receptor-deficient lpr mice are protected against acetaminophen hepatotoxicity due to higher glutathione synthesis and enhanced detoxification of oxidant stress. Food Chem Toxicol. 2013;58:228–35. doi: 10.1016/j.fct.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CD, McGill MR, Lebofsky M, Bajt ML, Jaeschke H. Protection against acetaminophen-induced liver injury by allopurinol is dependent on aldehyde oxidase-mediated liver preconditioning. Toxicol Appl Pharmacol. 2014;274:417–424. doi: 10.1016/j.taap.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem. 2011;286:5071–5078. doi: 10.1074/jbc.M111.276089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Williams CD, McGill MR, Lebofsky M, Ramachandran A, Jaeschke H. Purinergic receptor antagonist a438079 protects against acetaminophen-induced liver injury by inhibiting p450 isoenzymes, not by inflammasome activation. Toxicol Sci. 2013;131:325–335. doi: 10.1093/toxsci/kfs283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279:266–274. doi: 10.1016/j.taap.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.