

Abstract
Benzo[a]pyrene (BP) is mutagenic but noncarcinogenic in the murine colon. Recently, we reported rapid induction of colonic tumors by treatment of CD2F1 mice with BP (125 mg/kg for 5 days) followed by a colitis inducer, dextran sulfate sodium (DSS) (4% in drinking water for 1 or 2 weeks). However, there are no reports on detailed time course and histopathological features of colonic proliferative lesions in this model. Here, we show the detailed time course of colonic dysplasia, adenoma and adenocarcinoma induced by treatment with BP, DSS, and a combination of the two (BP/DSS). In the colon of mice exposed to BP/DSS, 14.6 dysplastic foci per mouse were present one week after DSS treatment (week 4). The number of dysplastic foci decreased with time to 3.1 at week 9 and thereafter remained almost constant. At week 4, 1.5 adenocarcinomas were also observed, with a marked increase in numbers with time, reaching 29.3 at week 14. In contrast, the number of dysplastic foci induced by DSS alone showed a time course similar to that following BP/DSS treatment; however, only a few tumors appeared. Neither dysplastic foci nor neoplastic lesions were induced by BP only. In mice exposed to BP/DSS, β-catenin was demonstrated immunohistochemically in the nucleus and/or cytoplasm of the tumor cells, and this translocation from the cell membrane was evident in subsets of dysplastic foci. In dysplastic foci induced by DSS alone, β-catenin was absent in the nucleus/cytoplasm. These finding suggest that aberrant β-catenin accumulation in dysplastic foci is associated with tumor progression in this BP/DSS model.
Keywords: benzo[a]pyrene, dextran sulfate sodium, colon, tumor, dysplastic foci, β-catenin
Introduction
Colorectal cancer is one of the most common cancers in the Western world1,2,3. Many rodent colon cancer models have been developed to study various aspects of the colonic carcinogenesis process and to investigate chemotherapeutic or chemopreventive regimens2,3,4,5. These animal models are chemically induced or genetically modified and provide many advantages, including understanding the pathogenesis of colon cancer, induction of rapid and reproducible tumors and recapitulation of the adenoma-carcinoma sequence that occurs in humans. Furthermore, they are valuable tools that can be used to better understand the underlying molecular aberrations and how these changes may relate to the pathogenesis of human colorectal cancer.
Among them, rodent colitis-associated colon cancer models using dextran sulfate sodium (DSS) in combination with or without a mutagenic carcinogen are some of the most frequently used colon cancer models. DSS, a polysulfated derivative of dextran, is non-genotoxic6 but is able to cause colonic tumors at a very low incidence/multiplicity in rodents4, 7, 8. This ability is strongly suggested to be closely associated with acute and chronic colitis induced by DSS2,3,4,5, 7, 8. Methylating agents such as azoxymethane (AOM)9,10,11 and 1,2-dimethylhydrazine (DMH)12,13,14 have often been used as the colon-specific mutagenic carcinogens in these models, and the mutagenic carcinogen and DSS are considered to be an initiator and promoter, respectively, for tumorigenesis11, 13. In such models using DSS in combination with a colon mutagenic carcinogen, generally it takes 10–20 weeks to induce colonic adenocarcinomas2,3,4,5, 9, 10, 12,13,14.
We recently established a novel accelerated mouse colon cancer model using DSS and benzo[a]pyrene (BP) and reported rapid induction of colonic adenocarcinomas at week 4 following sequential exposure to BP and DSS15. Our model comprises oral BP treatment of male CD2F1 mice at 125 mg/kg/day for 5 days, followed by a 9 day dosing-free period and subsequent administration of 4% DSS in drinking water for 1 week once (at week 2) or twice (intermittently, at weeks 2 and 5 separated by a 2-week DSS treatment free interval period). The BP used in this model is highly mutagenic16, 17 but noncarcinogenic1, 18,19,20,21,22 in the colon of mice by oral exposure. Thus, our model contrasts with other colon cancer models using DSS in combination with a colon-specific mutagenic carcinogen. Hence, a BP/DSS model is expected to provide a new tool for investigation of colon carcinogenesis, especially underling the role of genetic and epigenetic effects in colonic carcinogenesis, particularly those associated with colitis.
In our previous report15, we showed the incidence/multiplicity of colonic adenomas and adenocarcinomas only at weeks 4 and 11. In this paper, we present the detailed time course of the incidence/multiplicity of colonic dysplasia in addition to adenomas and adenocarcinomas with sampling time points at weeks 4, 7, 9, 11, 14 and 17 as well as histopathological features of these changes in this model. Furthermore, we also conducted immunohistochemical analysis of β-catenin staining in this model, since many studies have suggested that nuclear accumulation of β-catenin plays a key role in colonic carcinogenesis in animal models and humans2, 5, 7, 23, 24.
Materials and Methods
Chemicals
Benzo[a]pyrene (BP; CAS No. 50-32-8, purity of >96%) was purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA). Dextran sulfate sodium (DSS, CAS No. 9011-18-1, molecular weight; 36,000 to 50,000) was obtained from MP Biochemicals, LLC. (Aurora, OH, USA).
Animals
Male Crj: CD2F1 (BALB/c x DBA/2) mice were obtained from Charles River Laboratories Japan, Inc., Tokyo, Japan, and maintained in our animal facility. All mice were housed in metal cages (one mice per cage) and were fed a basal diet (Oriental CRF-1, Oriental Yeast Co., Ltd., Tokyo, Japan) and tap water ad libitum under controlled conditions of temperature (23 ± 1°C), humidity (55 ± 10%) and light (12-h light/12-h dark cycle). They were quarantined for one week and then randomized by body weight into each group.
Experimental procedures
The designs of Experiments 1 and 2 are shown in Fig. 1. Experiment 1 consisted of DSS and BP/DSS groups, and six or eight mice in the DSS group and six or twelve mice in the BP/DSS group were necropsied at weeks 14 and 17. In this report, the first week of experiment is defined as week 0. Because mice that received both BP and DSS developed multiple adenocarcinomas as early as week 14, Experiment 2, consisting of 4 groups (vehicle, BP, DSS and BP/DSS groups), was conducted to generate data at earlier time points. Eight mice in the vehicle, BP and DSS groups and six or eight mice in the BP/DSS group were necropsied at weeks 4, 7, 9 and 11. Mice were sacrificed under anesthesia to collect the colorectum from the cecocolic junction to anal verge for histopathology or immunohistochemistry.
Fig. 1.

Outline of the protocols for the experiments for histopathology of the colon. Experiment 1 consisted of DSS and BP/DSS groups, and the mice in these groups (6 to 12 mice per group) were necropsied at weeks 14 and 17. In this report, the first week of the experiment is defined as week 0 (indicated as W0 in these figures). Experiment 2 consisted of vehicle, BP, DSS, and BP/DSS groups, and all mice were necropsied at weeks 4, 7, 9 and 11 (6 or 8 mice/group). Male CD2F1 mice were treated with BP at an oral dose of 125 mg/kg/day for 5 consecutive days, and starting 10 days after the last dose, mice were given 4% DSS in drinking water for 1 week once (at week 2) or twice (intermittently, at weeks 2 and 5 separated by a 2-week DSS treatment free interval period). Salad oil and distilled water were used as vehicle controls for BP and DSS solutions, respectively.
Seven-week-old mice were treated with BP at an oral dose of 125 mg/kg/day for 5 consecutive days (at week 0). Ten days after the last dose of BP, mice were given 4% DSS in drinking water for 1 week once (at week 2) or twice (intermittently, at weeks 2 and 5 separated by a 2-week DSS treatment free interval period). BP and DSS were dissolved in salad oil (Nisshin Oillio Group, Ltd., Tokyo, Japan) at 12.5 mg/mL and distilled water at 40 mg/mL, respectively. The salad oil and distilled water were used as vehicle controls for BP and DSS solutions, respectively. These protocols were approved by the Institutional Animal Care and Use Committee and carried out according to Eisai animal experimentation regulations.
Tissue collection and histopathology
At necropsy, the large intestine (from the ileocecal junction to the anal verge) was immediately excised, flushed with saline, infused with 10% buffered formalin, cut open longitudinally along the mesenteric attachment and grossly observed. Thereafter, tissues were stored in 10% buffered formalin, cut into three or four equal lengths from the proximal to the distal ends, processed and embedded in paraffin.
Longitudinal sections of the colon were stained with hematoxylin and eosin (H & E) for histopathological assessment of dysplastic and neoplastic lesions in the colon mucosa. These lesions were classified into dysplasia, adenoma and adenocarcinoma according to the criteria described in our previous report15 and originally reported by Riddell et al.25, Pascal26 and Ward27. The numbers of dysplastic foci, adenomas and adenocarcinomas produced in each colon were counted with the Aperio® image analysis software (Aperio Technologies, Inc., Vista, CA, USA).
Immunohistochemistry
Paraffin-embedded sections of mouse colons with dysplastic and neoplastic lesions from the BP/DSS group at weeks 4, 7, 9 or 11 were examined for immunohistochemical staining for β-catenin and Ki-67. For comparison, dysplastic foci and neoplastic lesions found in the colon of a DSS-treated mouse at weeks 4 and 11, the colon of a BP-treated mouse and the normal colon from a mouse treated with vehicle at week 11 were also examined by these methods.
Monoclonal mouse anti-mouse β-catenin (clone 14, BD Transduction Laboratories, Lexington, KY, USA) was used at 1/1000, and monoclonal rat anti-mouse Ki-67 (clone TEC-3, DAKO, Glostrup, Denmark) for detection of cell proliferation was used at 1/40. After autoclave or microwave antigen retrieval of 4 μm sections, the EnvisionTM+Dual Link System or a streptavidin biotin-peroxidase complex method (biotinylated rabbit anti-rat immunoglobulin and peroxidase-labeled streptavidin, DAKO) was used to examine their expression and localization. These sections were further counterstained with Mayer’s hematoxylin solution for microscopic examination. Sections from the same blocks without the addition of primary antibodies served as negative controls.
Results
Mortality, clinical findings and body weight
Most of the mice in the BP/DSS and DSS groups showed soft or bloody stools during the latter part of each week-long DSS treatment with resolution of these clinical signs after a few days following treatment cessation. Bloody stool was also observed in mice with tumors in the BP/DSS group at and after week 7. There was also a significant decrease in the mean body weights of mice in the BP/DSS and DSS groups (as compared with mice in the vehicle and BP groups) during the latter part of each week-long DSS treatment, with recovery after about 2 weeks. At and after week 8, there were no significant differences in the mean body weights between any treatment groups (Fig. 2).
Fig. 2.

Body weights of mice. The symbols (▼) indicate the time of tissue collection (necropsy) for histopathology.
In the Experiment 1, three mice in the BP/DSS group were found dead or were sacrificed due to poor physical condition/health at weeks 12 or 13. Clinically, bloody stool and a marked reduction in body weights were noted, and multiple neoplastic masses obstructing the large intestine were noted at necropsy. These findings suggest that impaired bowel movement contributed to poor physical condition/mortality. There were no deaths in Experiment 2.
Gross findings
All mice from the BP/DSS group showed shortening of the large intestine accompanied by irregular thickening of the intestinal wall. At week 4, the mucosal surface of the distal colon was rough (Fig. 3A), and at week 7, several masses were observed in the distal colon (Fig. 3B). At week 9, many nodular or polypoid masses were evident predominantly in the distal and middle parts of the colon in all mice from the BP/DSS group (Fig. 3C). At week 11, the masses became larger in size, and some of the masses appeared to have fused (Fig. 3D).
Fig. 3.
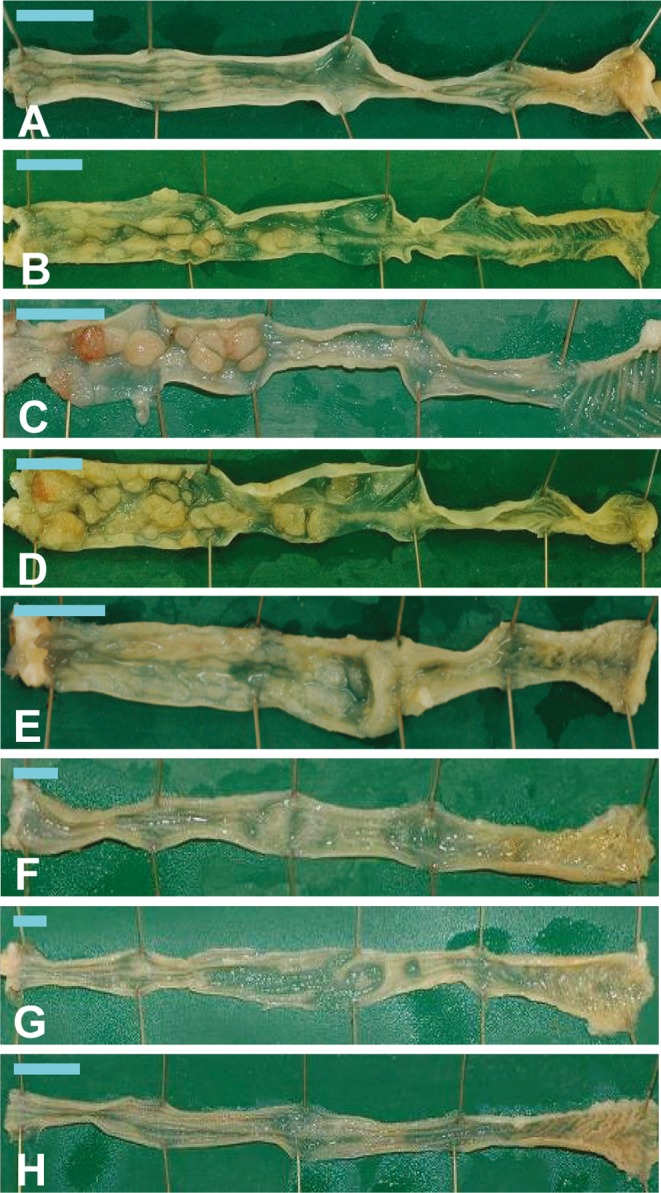
Macroscopic view of the colons of mice from the BP/DSS group at weeks 4 (A), 7 (B), 9 (C) and 11 (D), DSS group at weeks 7 (E) and 9 (F), vehicle group at week 7 (G) and BP group at week 7 (H). In the BP/DSS group, a rough surface was seen in the distal colon at week 4 (A), and nodular or polypoid masses were found in the distal and/or middle colons at weeks 7 (B), 9 (C) and 11 (D). No obvious changes were found in the vehicle (G) and BP groups (H), while masses were sporadically observed in the DSS group (E and F). Bar: 1 cm.
In the mice of the DSS group, findings similar to the BP/DSS group, i.e., shortening of the large bowel, irregular thickening of the intestinal wall and a rough surface of the colonic mucosa, were observed at week 4. In contrast with the BP/DSS group, masses were sporadically observed only in a few mice at or after week 7 (Figs. 3E and 3F), and these masses were smaller than those found in the BP/DSS group. There were no noteworthy gross changes in any of the mice from the vehicle (Fig. 3G) or BP (Fig. 3H) group at any time points.
Histopathology
Erosive or ulcerative colitis with severe inflammatory cell infiltration and edema in the lamina propria and/or submucosa was observed in the distal and middle parts of the colon of mice exposed to DSS (BP/DSS and DSS groups) at weeks 4 and 7. At and after week 9, erosions with focal infiltration of inflammatory cells and foamy accumulations of macrophages were sporadically observed in the lamina propria.
In the BP/DSS group, an irregularly thickened mucosa accompanied by colitis and a regenerative crypt epithelium were observed in the distal part of the colon at week 4 (Fig. 4A), and there were many dysplastic foci characterized by irregular branching, distorted architecture with cellular and nuclear pleomorphism, nuclear enlargement and hyperchromatism and a paucity of goblet cells (Fig. 5A and 5D). Morphologically, these dysplastic foci clearly differed from regenerating crypts. While the regenerative crypts also showed an abnormal crypt architecture with distortion, dilatation, branching and/or shortening of crypts, they were present in close proximity to erosion and/or ulceration, lacked distinct cellular atypia and showed goblet cells with maturation towards the surface even though mucin production was decreased. Neoplastic lesions, adenomas as well as adenocarcinomas, were first observed in the distal part of the colonic mucosa as early as week 4 (Fig. 4A) and week 7 (Fig. 4B) and were subsequently noted in the distal and middle parts of the colonic mucosa. A gradual increase in the size of neoplastic lesions with time was noted at weeks 9 (Fig. 4C) and 11 (Fig. 4D). The neoplasms were polypoid masses composed of tubular and papillary proliferations that protruded into the intestinal lumen, extended into the lamina propria and compressed the adjacent mucosa and were diagnosed as tubular adenomas or well/moderately differentiated adenocarcinomas27. Adenomas (Fig. 6A) consisted of variably sized glands lined by single/multiple layers of epithelial cells, while adenocarcinomas (Fig. 6D) consisted of variably sized distorted glands, which branched irregularly and were lined by marked stratified epithelial cells characterized by cellular and nuclear pleomorphism and loss of nuclear polarity, and were occasionally accompanied by squamous metaplasia (Fig. 8A). Paneth cell differentiation (with eosinophilic cytoplasmic granules) was observed occasionally in the adenomas and adenocarcinomas (Fig. 8B). Cellular invasion into the lamina propria was observed in some adenocarcinomas; however, true invasion into the submucosa or metastasis was not observed.
Fig. 4.

Histopathology of the colons of mice from the BP/DSS group at weeks 4, 7, 9 and 11. Neoplastic lesions including tubular adenoma and polypoid tubular adenocarcinoma can be seen in the distal part of the colonic mucosa at week 4 (A). Thereafter, the size of the neoplastic lesions gradually increases with time at weeks 7 (B), 9 (C) and 11 (D). H& E stain.
Fig. 5.
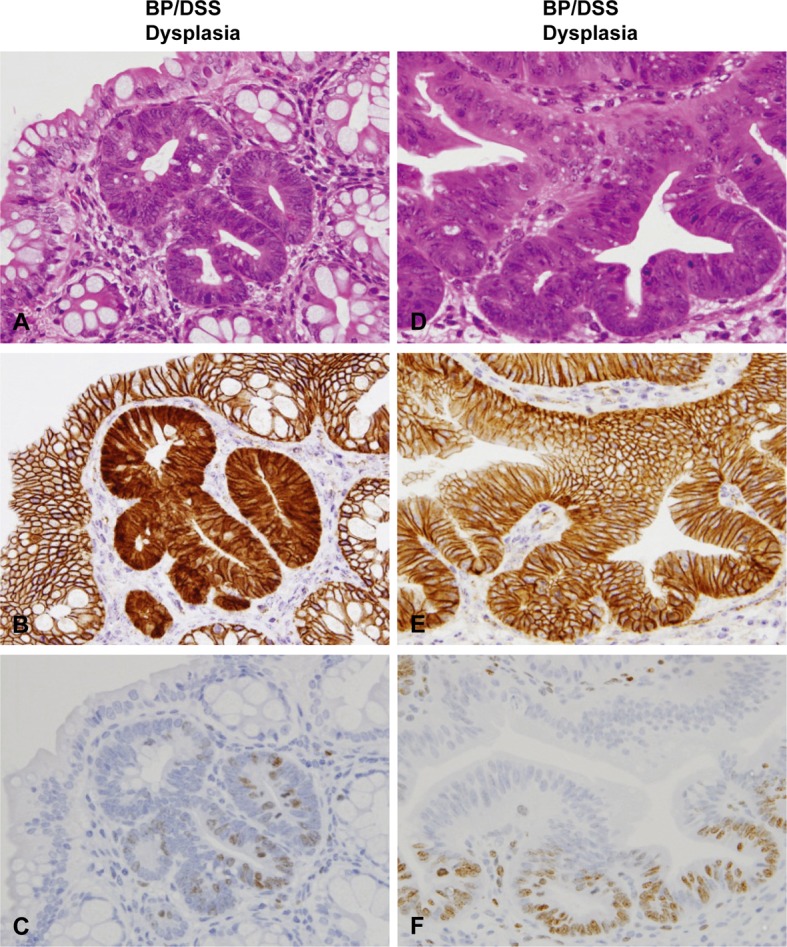
Histopathology with H & E stain (A and D) and immunohistochemistry of β-catenin (B and E) and Ki-67 (C and F) in dysplasia of mice from the BP/DSS group. Figs. (A) to (C) and Figs. (D) to (F) are serial sections of different dysplasias, respectively. Dysplasia was characterized by irregular crypt branching and distortion of the crypt architecture. β-catenin was localized on the cell membrane of most dysplastic foci (E) and distributed in the nucleus and/or cytoplasm of a small number of dysplastic foci (B). Ki-67-positive cells were diffusely distributed in dysplastic foci (C and F).
Fig. 6.
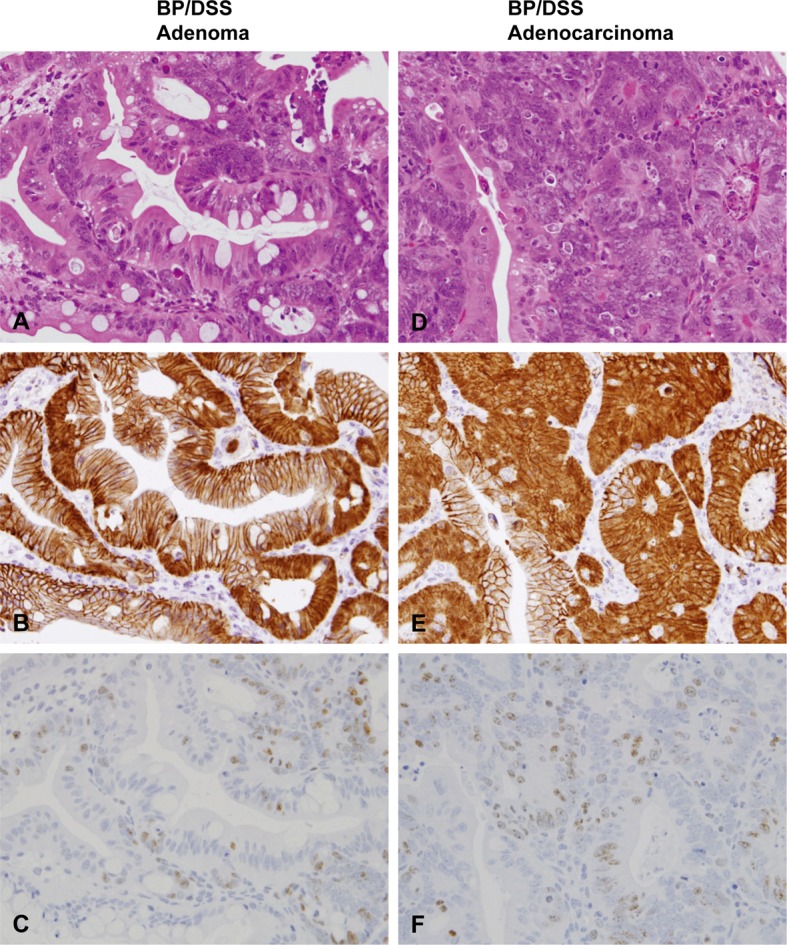
Histopathology with H & E stain (A and D) and immunohistochemistry of β-catenin (B and E) and Ki-67 (C and F) in adenomas and adenocarcinomas of mice from the BP/DSS group. Adenomas consisted of variably sized glands lined by single/multiple layers of epithelial cells (A). Adenocarcinomas were characterized by variable sized, irregularly branched and distorted glands lined by marked stratified epithelial cells (D). β-catenin was distributed in the nucleus and cytoplasm of adenoma (B) and adenocarcinoma cells (E). Ki-67-positive cells were most numerous in colonic adenoma (C) and adenocarcinoma cells (F).
Fig. 8.
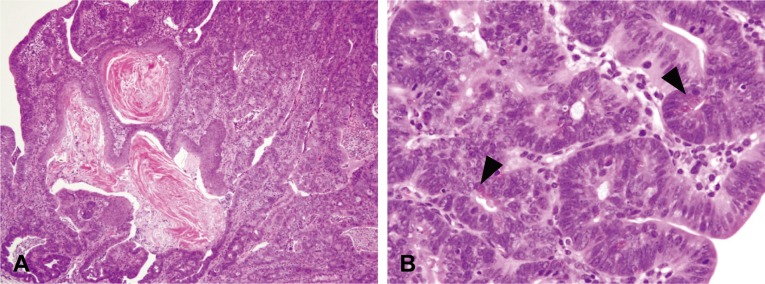
Histopathology of the colons of mice from the BP/DSS group. Adenocarcinomas were occasionally accompanied by squamous metaplasia (A) or differentiation into Paneth cells (B, arrowhead). H& E stain.
In the DSS group, dysplastic foci were predominantly observed in the distal parts of the colonic mucosa at week 4 and in the distal and middle parts at and after week 7. Dysplastic foci observed in mice of this group were morphologically similar to those observed in mice of the BP/DSS group (Fig. 7G). Solitary adenomas (Fig. 7J) or adenocarcinomas similar to those observed in the BP/DSS group were noted in a few mice at and after week 7. There were no noteworthy changes in mice from the vehicle (Fig. 7A) and BP (Fig. 7D) groups, as reported in our previous paper16.
Fig. 7.

Histopathology with H & E stain and immunohistochemistry of β-catenin and Ki-67 in the colonic mucosa of mice from the vehicle group (A–C) and BP group (D–F) and in dysplasias (G–I) and adenomas (J–L) of mice from the DSS group. No obvious histological changes were found in the vehicle (A) and BP group (D). Histological characteristics of dysplastic foci (G) and adenomas (J) found in the DSS group were similar to those observed in the BP/DSS group. β-catenin was localized on the cell membrane of the normal mucosal cells in the vehicle (B) and BP groups (E) and dysplastic foci (H) in the DSS group. The β-catenin was distributed in the nucleus and cytoplasm of adenomas (K) in the DSS group. Ki-67-positive cells were mainly localized in the lower zone of normal colonic crypts (C and F) and in diffusely distributed dysplastic foci (I) and adenomas (L) in the DSS group.
Incidence and multiplicity of dysplastic foci, adenomas and adenocarcinomas
Table 1 lists the incidence of dysplastic foci, adenomas and adenocarcinomas noted in all treatment groups at each observation time point, while Table 2 shows the multiplicity of these lesions.
Table 1. Incidence* of Dysplastic Foci, Adenomas, and Adenocarcinomas.
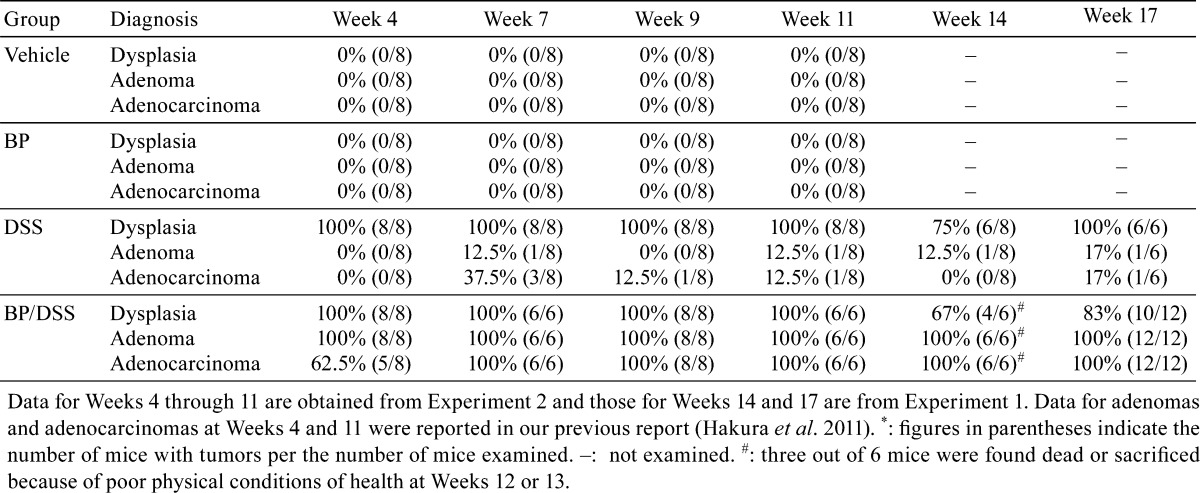
Table 2. Multiplicity* of Dysplastic Foci, Adenomas, and Adenocarcinomas.
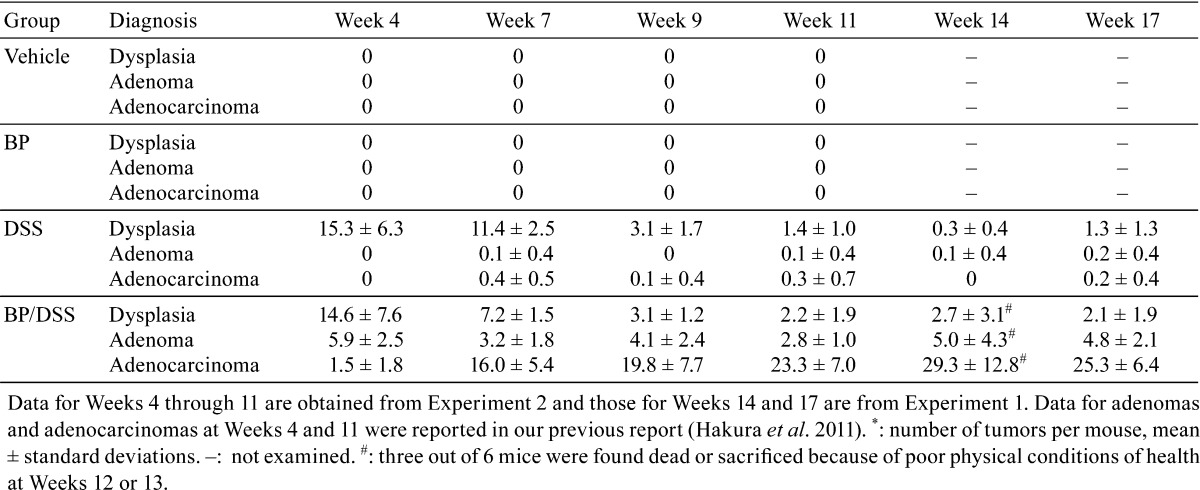
In the BP/DSS group at week 4, the incidence of dysplastic foci, adenomas and adenocarcinomas was 100%, 100% and 62.5%, respectively. From weeks 7 through 17, the incidence of adenomas and adenocarcinomas was 100%. The dysplastic foci were observed at an incidence of 100% up to week 11 and thereafter were still observed in 67% or 83% animals. The number (multiplicity) of dysplastic foci was 14.6 ± 7.6 at week 4 and decreased with time to 3.1 ± 1.2 at week 9, thereafter remaining constant. The multiplicity of adenomas was 5.9 ± 2.5 at week 4 and remained almost constant throughout the observation period. In contrast, the multiplicity of adenocarcinomas was 1.5 ± 1.8 at week 4 and increased time dependently to a peak of 29.3 ± 12.8 at week 14 followed by a slight reduction to 25.3 ± 6.4 at week 17, possibly due to fusion with adjacent adenocarcinomas.
In the DSS group, dysplastic foci were observed at an incidence of 100% up to week 11 and thereafter were observed still in 75% or 100% mice. The mean number of dysplastic foci was 15.3 ± 6.3 at week 4, decreasing with time to 3.1 ± 1.7 at week 9 and thereafter remaining almost constant. Adenomas or adenocarcinomas were found in only a few mice at and after week 7. Neither dysplastic foci nor neoplastic lesions were found in any mice of the vehicle and BP groups, consistent with our previous report16.
Immunohistochemistry
In the normal colonic mucosal epithelial cells of vehicle-treated mice (control) (Fig. 7B) and most of the dysplastic foci from the BP/DSS group (Fig. 5E), β-catenin was exclusively localized on the cell membrane. In contrast, β-catenin was present in the nucleus and/or cytoplasm of the rest of a small number of dysplastic foci (Fig. 5B), colonic adenoma (Fig. 6B) and adenocarcinoma (Fig. 6E) cells in the BP/DSS group. There were no apparent differences in the intensity of cytoplasmic β-catenin expression between adenoma and adenocarcinoma cells; however, the intensity of nuclear β-catenin expression in adenocarcinoma cells and the number of adenocarcinoma cells demonstrating nuclear positivity was greater than in dysplastic foci or adenoma cells. Nuclear staining of β-catenin in tumors (Fig. 6B and 6E) and dysplastic foci (Fig. 5B) showed a scattered heterogeneous pattern. In the DSS group, β-catenin was exclusively localized on the cell membrane in all dysplastic foci (Fig. 7H) and was present in the nucleus and/or cytoplasm of colonic adenoma (Fig. 7K) and adenocarcinoma cells in the same pattern. Immunopositivity for β-catenin was also observed on cell membranes of vascular endothelial cells and ganglion cells in the submucosal and myenteric (Meissner’s and Auerbach’s) plexus (Fig. 7B).
Ki-67-positive cells were mainly localized in the lower zone of normal colonic crypts of a vehicle-treated mouse (Fig. 7C). In mice of the DSS or BP/DSS group, the number of Ki-67-positive cells greatly increased in colonic dysplastic foci (Fig. 5C, 5F and 7I), adenomas (Fig. 6C and 7L) and adenocarcinomas (Fig. 6F) compared with normal crypts (Fig. 7C).
Discussion
BP is mutagenic16, 17 but not carcinogenic in the murine colon1, 18,19,20,21,22. Treatment of CD2F1 mice with BP (125 mg/kg/day) for 5 days (at week 0) and subsequent exposure to DSS (4% in drinking water) for 1 week once (at week 2) or twice (at weeks 2 and 5) rapidly induced dysplasia, adenoma and adenocarcinoma in the colonic mucosa as early as week 4 (one week after treatment with DSS); dysplastic foci, adenomas and adenocarcinomas were present at incidences of 100%, 100% and 62.5% and at multiplicities of 14.6 ± 7.6, 5.9 ± 2.5 and 1.5 ± 1.8, respectively. The BP/DSS treatment also induced erosive or ulcerative lesions of the colonic epithelium during the period of DSS treatment for one week, followed by subsequent regeneration of the colonic mucosa for the next week (at week 4). Because induction of mutations has previously been reported in the colon of MutaTM Mouse transgenic CD2F1 mice (carrying the E. coli lacZ gene for detection of mutations) 2 and 26 weeks following oral treatment with 125 mg/kg/day BP for 5 days16, 17, the colonic neoplasms were suggested to be derived from BP-mutated colonic epithelial cells through progression of dysplastic foci to adenomas within a very short period of one week following exposure to DSS. This rapid induction of adenocarcinomas may support a single-hit model or “de novo” type of tumorigenesis after exposure to DSS rather than a multi-hit model (sequential genetic alterations), which has been proposed in many studies on colorectal cancer in human or animal models2, 5, 28, 29. Namely, the induction of adenocarcinomas might be a consequence of direct transformation of BP-initiated colonic epithelial stem cells or progenitor cells into adenocarcinoma cells through progression of transiently appearing dysplastic foci and subsequent adenomas after DSS exposure30, 31.
Colonic adenocarcinomas found in mice from the BP/DSS group increased time dependently in both incidence and multiplicity. Adenocarcinomas were found at an incidence of 62.5% at week 4 and at an incidence of 100% at and after week 7. Their multiplicity was 1.5 ± 1.8 at week 4 and increased time dependently, reaching a peak of 29.3 ± 12.8 at week 14. To our knowledge, the induction of colonic adenocarcinomas observed in this model is faster and/or yields a significantly higher number of malignant tumors as compared with other reported colon cancer models including chemically induced and genetically engineered models2, 4, 9,10,11,12,13,14, 32. A possible reason for the high incidence/multiplicity of tumor formation in such a short period of time is use of the strain of CD2F1 mice, which allowed us to treat the mice with DSS at a relatively high concentration of 4% in drinking water as compared with other strains. There are no other reports on use of this strain of mice in a DSS-associated colon cancer model. Male CD2F1 mice tolerated this dose, with induction of extensive necrosis of the colorectal mucosa.
The histopathological findings noted in this model show similarities to those reported in other DSS-induced colitis-associated rodent colon cancer models with and without a colon mutagenic carcinogen such as AOM, DMH, or 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)2, 4, 5, 7, 8, 32. Neoplasms found in this model were histologically diagnosed as tubular adenomas and well- or moderately differentiated adenocarcinomas. Furthermore, colonic neoplasms were occasionally accompanied by the occurrence of squamous metaplasia or Paneth cells, as reported in other models12, 13. The Paneth cells might be driven in intestinal crypts by a maturation program through induction of Wnt signaling33 and might be related to promotion/progression of initiated/mutated cells to tumor cells by constituting the niche for stem cells in intestinal crypts34. In this model, adenocarcinomas invaded into the lamina propria; however, invasion into the submucosa and metastasis were not observed. While submucosal invasion and metastasis is frequent in human colorectal cancer, rodent colon tumor models tend to nonmetastatic and rarely show evidence of tumor cell invasion35, 36. Adenomas and adenocarcinomas predominantly appeared in the region of middle to distal colon, where erosive or ulcerative colitis were distributed, as reported in other DSS models2, 4, 5. Such tumors were not induced only by mechanical disruption of colonic epithelial cells, i.e., scraping with a cotton-tipped swab until the cotton was slightly stained with blood once or twice (2 weeks apart) in place of DSS treatment following BP treatment (our unpublished data). Therefore, enhanced cell proliferation alone due to regeneration of the mucosa was not a critical factor for tumor induction. Thus, inflammation is probably one of the important factors for the induction of colon cancer in this model.
β-Catenin accumulated in the nucleus/cytoplasm of the colonic adenomas and adenocarcinomas in the BP/DSS-treated mice. In vehicle-treated mice, β-catenin was expressed exclusively on the cell membrane of normal colon mucosal epithelial cells. This translocation of β-catenin into the nucleus/cytoplasm was consistent with that found in other DSS-induced colon cancer models in combination with a carcinogen such as AOM, DMH or PhIP2, 4, 11,12,13,14, 32. Tumor development in mice exposed to BP and DSS was considered in these studies to be, at least in part, associated with activation of the β-catenin/Tcf signaling pathway, because β-catenin is known to accumulate when the β-catenin or APC genes are mutated or the Wnt signaling pathway is activated2, 5, 23, 24, 29. This may be supported by a recent study showing that BP increases the number of colon tumors in ApcMin/+ mice37, which have a point mutation at the recessive Apc tumor suppressor gene29.
Similar to adenomas and adenocarcinomas, the morphological characteristics of dysplastic foci observed in mice exposed to DSS alone were comparable to those found in the BP/DSS-treated mice, and the multiplicities of dysplastic foci that appeared in mice exposed to DSS alone had a time course similar to that observed in the BP/DSS-treated mice. However, the neoplastic potential of these dysplastic foci varied between BP/DSS- and DSS-induced foci, since the number of adenocarcinomas increased time dependently in the BP/DSS-treated mice, reaching a mean of 29.3 at week 14, whereas in the mice exposed to DSS alone, only a few tumors (with a mean of 0.1–0.4) were noted during the observation period. In addition to tumors, abnormal β-catenin expression was observed in subsets of dysplastic foci in the BP/DSS-treated mice. Certain types of β-catenin accumulated dysplastic crypts, such as BCAC (β-catenin-accumulated crypts), β-catenin-accumulated dysplastic ACF (aberrant crypt foci) and dysplasia-associated lesion or mass (DALM), were shown to be present in the colon of rats or mice with a predisposition to develop colon cancer in response to some colonic carcinogens (AOM, DMH, PhIP and DSS)7, 12, 38,39,40,41. Although such β-catenin-accumulated dysplastic foci/crypts have been identified as preneoplastic or neoplastic lesions, all of these foci/crypts do not always develop into tumors. Because aberrant β-catenin accumulation was found in dysplastic foci/crypts without β-catenin mutations12, 39, 42, 43 and was caused by β-catenin mutations selected during malignant transformation in colon carcinogenesis12, 43, it is considered that all or subsets of β-catenin-accumulated dysplastic foci induced by treatment with both BP and DSS develop into tumors. The multiplicity (29.3) of adenocarcinomas at week 14 was higher than that (14.6) of dysplastic foci. The reasons for this gap between the multiplicity of preneoplastic and neoplastic lesions are not fully understood; however, it should be noted that every dysplastic foci induced in this study could not be counted because the multiplicity of the neoplastic lesions was evaluated using one longitudinal H&E section of the colon. On the other hand, there is a possibility that dysplastic foci observed after week 4 may be associated with the increased multiplicity of adenocarcinomas. Also, other types of preneoplastic lesions such as mucin-depleted foci, as presented in rat colon carcinogenesis with DMH44, might partially contribute to the development of colon cancers in this model. Further studies on the early stage of carcinogenesis in this model are necessary, including comparative investigations of developing colonic dysplastic foci induced by BP/DSS or DSS alone from molecular biology and pathology points of view.
In conclusion, dysplastic foci accompanied by regenerating epithelial cells rapidly appeared in the colons of mice exposed to BP/DSS and DSS alone, with a marked decrease in the number of dysplastic foci with time, and thereafter showed a continued low multiplicity of dysplastic foci. There were no histological differences in the dysplastic foci between BP/DSS and DSS alone. However, in mice treated with BP/DSS, induction of colonic adenocarcinomas markedly increased in a time-dependent manner, but only a few adenocarcinomas were noted in mice treated with DSS alone. Aberrant nuclear/cytoplasmic expression of β-catenin was found in subsets of dysplastic foci and tumor cells, suggesting a close association between aberrant β-catenin accumulation and promotion/progression of colon carcinogenesis by BP and DSS. This BP/DSS model provides a short-term experimental bioassay system that can be used to investigate the development of colonic tumors and the mechanisms of action of specific genes or molecules, particularly in relation to inflammation.
Declaration of conflicting interest: The authors declare that they have no potential conflicts of interest with respect to the authorship and/or publication of this article. The authors received no financial support for the research and/or authorship of this article.
Acknowledgments
We thank E. Kawada, I. Mori, M. Nishikawa and M. Tanaka of Sunplanet Co., Ltd., for their skilful techniques. We are grateful to Dr. T. Imai of the National Cancer Center Research Institute for his useful advice on diagnosis of dysplastic and neoplastic lesions. We also thank Dr. K. Tsukidate for his helpful advice and Drs. David L. Hutto and S. Akare of Eisai Inc. for critical review of this manuscript.
References
- 1.Diggs DL, Huderson AC, Harris KL, Myers JN, Banks LD, Rekhadevi PV, Niaz MS, and Ramesh A. Polycyclic aromatic hydrocarbons and digestive tract cancers: a perspective. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 29: 324–357. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg DW, Giardina C, and Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 30: 183–196. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westbrook AM, Szakmary A, and Schiestl RH. Mechanisms of intestinal inflammation and development of associated cancers: lessons learned from mouse models. Mutat Res. 705: 40–59. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clapper ML, Cooper HS, and Chang W-CL. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 28: 1450–1459. 2007. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka T. Colorectal carcinogenesis: Review of human and experimental animal studies. J Carcinog. 8: 5 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mori H, Ohbayashi F, Hirono I, Shimada T, and Williams GM. Absence of genotoxicity of the carcinogenic sulfated polysaccharides carrageenan and dextran sulfate in mammalian DNA repair and bacterial mutagenicity assays. Nutr Cancer. 6: 92–97. 1984. [DOI] [PubMed] [Google Scholar]
- 7.Cooper HS, Murthy S, Kido K, Yoshitake H, and Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, β-catenin and p53 expression and the role of inflammation. Carcinogenesis. 21: 757–768. 2000. [DOI] [PubMed] [Google Scholar]
- 8.Okayasu I, Yamada M, Mikami T, Yoshida T, Kanno J, and Ohkusa T. Dysplasia and carcinoma development in a repeated dextran sulfate sodium-induced colitis model. J Gastroenterol Hepatol. 17: 1078–1083. 2002. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki R, Kohno H, Sugie S, and Tanaka T. Sequential observations on the occurrence of preneoplastic and neoplastic lesions in mouse colon treated with azoxymethane and dextran sodium sulfate. Cancer Sci. 95: 721–727. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki R, Kohno H, Sugie S, Nakagama H, and Tanaka T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis. 27: 162–169. 2006. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, and Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 94: 965–973. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imai T, Fukuta K, Hasumura M, Cho Y-M, Ota Y, Takami S, Nakagama H, and Hirose M. Significance of inflammation-associated regenerative mucosa characterized by Paneth cell metaplasia and β-catenin accumulation for the onset of colorectal carcinogenesis in rats initiated with 1,2-dimethylhydrazine. Carcinogenesis. 28: 2199–2206. 2007. [DOI] [PubMed] [Google Scholar]
- 13.Onose J, Imai T, Hasumura M, Ueda M, and Hirose M. Rapid induction of colorectal tumors in rats initiated with 1,2-dimethylhydrazine followed by dextran sodium sulfate treatment. Cancer Lett. 198: 145–152. 2003. [DOI] [PubMed] [Google Scholar]
- 14.Wang J-G, Wang D-F, Lv B-J, and Si J-M. A novel mouse model for colitis-associated colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sulfate sodium. World J Gastroenterol. 10: 2958–2962. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakura A, Seki Y, Sonoda J, Hosokawa S, Aoki T, Suganuma A, Kerns WD, and Tsukidate K. Rapid induction of colonic adenocarcinoma in mice exposed to benzo[a]pyrene and dextran sulfate sodium. Food Chem Toxicol. 49: 2997–3001. 2011. [DOI] [PubMed] [Google Scholar]
- 16.Hakura A, Tsutsui Y, Sonoda J, Kai J, Imade T, Shimada M, Sugihara Y, and Mikami T. Comparison between in vivo mutagenicity and carcinogenicity in multiple organs by benzo[a]pyrene in the lacZ transgenic mouse (Muta Mouse). Mutat Res. 398: 123–130. 1998. [DOI] [PubMed] [Google Scholar]
- 17.Hakura A, Tsutsui Y, Sonoda J, Mikami T, Tsukidate K, Sagami F, and Kerns WD. Multiple organ mutation in the lacZ transgenic mouse (Muta Mouse) 6 months after oral treatment (5 days) with benzo[a]pyrene. Mutat Res. 426: 71–77. 1999. [DOI] [PubMed] [Google Scholar]
- 18.Benford D, Dinovi M, and Setzer RW. Application of the margin-of-exposure (MoE) approach to substances in food that are genotoxic and carcinogenic e.g.: benzo[a]pyrene and polycyclic aromatic hydrocarbons. Food Chem Toxicol. 48(Suppl 1): S42–S48. 2010. [DOI] [PubMed] [Google Scholar]
- 19.Culp SJ, Gaylor DW, Sheldon WG, Goldstein LS, and Beland FA. A comparison of the tumors induced by coal tar and benzo[a]pyrene in a 2-year bioassay. Carcinogenesis. 19: 117–124. 1998. [DOI] [PubMed] [Google Scholar]
- 20.Hakura A, and Sonoda J. Benzo[a]pyrene and colonic cancer. In: Pyrene: Chemical properties, biochemistry applications and toxic effects. Ruzicka ZP, and Kral T (eds). Nova Science Publishers. New York. 43–78. 2013. [Google Scholar]
- 21.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr Eval Carcinog Risks Hum. 92: 1–853. 2010. [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips DH. Polycyclic aromatic hydrocarbons in the diet. Mutat Res. 443: 139–147. 1999. [DOI] [PubMed] [Google Scholar]
- 23.Aoki K, Aoki M, Sugai M, Harada N, Miyoshi H, Tsukamoto T, Mizoshita T, Tatematsu M, Seno H, Chiba T, Oshima M, Hsieh C-L, and Taketo MM. Chromosomal instability by β-catenin/TCF transcription in APC or β-catenin mutant cells. Oncogene. 26: 3511–3520. 2007. [DOI] [PubMed] [Google Scholar]
- 24.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, and Clevers H. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 111: 241–250. 2002. [DOI] [PubMed] [Google Scholar]
- 25.Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC, Sommers SC, and Yardley JH. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum Pathol. 14: 931–968. 1983. [DOI] [PubMed] [Google Scholar]
- 26.Pascal RR. Dysplasia and early carcinoma in inflammatory bowel disease and colorectal adenomas. Hum Pathol. 25: 1160–1171. 1994. [DOI] [PubMed] [Google Scholar]
- 27.Ward JM. Morphogenesis of chemically induced neoplasms of the colon and small intestine in rats. Lab Invest. 30: 505–513. 1974. [PubMed] [Google Scholar]
- 28.Weinberg RA. The biology of cancer. Garland Science. New York. 2007. [Google Scholar]
- 29.Yamada Y, and Mori H. Multistep carcinogenesis of the colon in Apc(Min/+) mouse. Cancer Sci. 98: 6–10. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brittan M, and Wright NA. Stem cell in gastrointestinal structure and neoplastic development. Gut. 53: 899–910. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leedham SJ, Schier S, Thliveris AT, Halberg RB, Newton MA, and Wright NA. From gene mutations to tumours—stem cells in gastrointestinal carcinogenesis. Cell Prolif. 38: 387–405. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka T, Suzuki R, Kohno H, Sugie S, Takahashi M, and Wakabayashi K. Colonic adenocarcinomas rapidly induced by the combined treatment with 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and dextran sodium sulfate in male ICR mice possess β-catenin gene mutations and increases immunoreactivity for β-catenin, cyclooxygenase-2 and inducible nitric oxide synthase. Carcinogenesis. 26: 229–238. 2005. [DOI] [PubMed] [Google Scholar]
- 33.van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, Thiele A, van den Born M, Begthel H, Brabletz T, Taketo MM, and Clevers H. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 7: 381–386. 2005. [DOI] [PubMed] [Google Scholar]
- 34.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, and Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 469: 415–418. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobaek-Larsen M, Thorup I, Diederichsen A, Fenger C, and Hoitinga MR. Review of colorectal cancer and its metastases in rodent models: comparative aspects with those in humans. Comp Med. 50: 16–26. 2000. [PubMed] [Google Scholar]
- 36.Heijstek MW, Kranenburg O, and Borel Rinkes IH. Mouse models of colorectal cancer and liver metastases. Dig Surg. 22: 16–25. 2005. [DOI] [PubMed] [Google Scholar]
- 37.Harris DL, Washington MK, Hood DB, Roberts LJ, 2nd , and Ramesh A. Dietary fat-influenced development of colon neoplasia in Apc Min mice exposed to benzo(a)pyrene. Toxicol Pathol. 37: 938–946. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mori H, Yamada Y, Kuno T, and Hirose Y. Aberrant crypt foci and β-catenin accumulated crypts; significance and roles for colorectal carcinogenesis. Mutat Res. 566: 191–208. 2004. [DOI] [PubMed] [Google Scholar]
- 39.Ochiai M, Ushigome M, Fujiwara K, Ubagai T, Kawamori T, Sugimura T, Nagao M, and Nakagama H. Characterization of dysplastic aberrant crypt foci in the rat colon induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Am J Pathol. 163: 1607–1614. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzui M, Morioka T, and Yoshimi N. Colon preneoplastic lesions in animal models. J Toxicol Pathol. 26: 335–341. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamada Y, and Mori H. Pre-cancerous lesions for colorectal cancers in rodents: a new concept. Carcinogenesis. 24: 1015–1019. 2003. [DOI] [PubMed] [Google Scholar]
- 42.Yamada Y, Yoshimi N, Hirose Y, Kawabata K, Matsunaga K, Shimizu M, Hara A, and Mori H. Frequent β-catenin gene mutations and accumulations of the protein in the putative preneoplastic lesions lacking macroscopic aberrant crypt foci appearance, in rat colon carcinogenesis. Cancer Res. 60: 3323–3327. 2000. [PubMed] [Google Scholar]
- 43.Yamada Y, Oyama T, Hirose Y, Hara A, Sugie S, Yoshida K, Yoshimi N, and Mori H. β-Catenin mutation is selected during malignant transformation in colon carcinogenesis. Carcinogenesis. 24: 91–97. 2003. [DOI] [PubMed] [Google Scholar]
- 44.Yoshimi N, Morioka T, Kinjo T, Inamine M, Kaneshiro T, Shimizu T, Suzui M, Yamada Y, and Mori H. Histological and immunohistochemical observations of mucin-depleted foci (MDF) stained with Alcian blue, in rat colon carcinogenesis induced with 1,2-dimethylhydrazine dihydrochloride. Cancer Sci. 95: 792–797. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]