

Abstract
Acute intermittent hypoxia (AIH) induces a form of spinal motor plasticity known as phrenic long-term facilitation (pLTF); pLTF is a prolonged increase in phrenic motor output after AIH has ended. In anesthetized rats, we demonstrate that pLTF requires activity of the novel PKC isoform, PKCθ, and that the relevant PKCθ is within phrenic motor neurons. Whereas spinal PKCθ inhibitors block pLTF, inhibitors targeting other PKC isoforms do not. PKCθ is highly expressed in phrenic motor neurons, and PKCθ knockdown with intrapleural siRNAs abolishes pLTF. Intrapleural siRNAs targeting PKCζ, an atypical PKC isoform expressed in phrenic motor neurons that underlies a distinct form of phrenic motor plasticity, does not affect pLTF. Thus, PKCθ plays a critical role in spinal AIH-induced respiratory motor plasticity, and the relevant PKCθ is localized within phrenic motor neurons. Intrapleural siRNA delivery has considerable potential as a therapeutic tool to selectively manipulate plasticity in vital respiratory motor neurons.
Keywords: intermittent hypoxia, motor neuron, plasticity, protein kinase C, spinal cord
Introduction
Our appreciation for spinal motor plasticity and its role in functional recovery from motor deficits caused by neural injury has increased in recent years (Wolpaw and Tennissen, 2001; Kokotilo et al., 2009; Dale-Nagle et al., 2010b). Prominent models of spinal plasticity include operant conditioning of the H reflex (Wolpaw et al., 1986), spinal instrumental learning (Grau et al., 2006), spontaneous phrenic motor recovery below a cervical hemisection (Goshgarian, 2009), and phrenic long-term facilitation (pLTF) following acute intermittent hypoxia (AIH) (Mitchell et al., 2001). Basic insights into spinal plasticity have guided preclinical trials demonstrating that intermittent hypoxia increases leg strength, walking time, and walking speed in patients with incomplete spinal injuries (Trumbower et al., 2012; Hayes et al., 2014).
The phrenic motor system is ideal to study spinal motor plasticity because rhythmic phrenic nerve activity is readily quantified and reflects a meaningful, automatic motor behavior that exhibits functional plasticity (Feldman et al., 2003; Mitchell and Johnson, 2003). The most frequently studied model of respiratory motor plasticity is pLTF, a prolonged increase in phrenic burst amplitude lasting hours following AIH (Hayashi et al., 1993; Bach and Mitchell, 1996). Studies of pLTF continue to guide therapies with intermittent hypoxia to restore motor function (Vinit et al., 2009; Lovett-Barr et al., 2012; Nichols et al., 2013), but we still have an incomplete understanding of the mechanisms giving rise to this important form of spinal plasticity.
Hypoxia activates medullary raphe neurons, increasing serotonin release within spinal respiratory motor nuclei (Kinkead et al., 2001). Episodic spinal serotonin Type 2 receptor activation is necessary (Fuller et al., 2001a; Baker-Herman and Mitchell, 2002) and sufficient (MacFarlane and Mitchell, 2009; MacFarlane et al., 2011) for pLTF, triggering new synthesis of BDNF. New BDNF synthesis is necessary, and spinal TrkB receptor activation is sufficient to elicit pLTF (Baker-Herman et al., 2004) via downstream ERK MAP kinase signaling (Hoffman et al., 2012). Molecular elements linking serotonin receptor activation to downstream signaling likely involve protein kinase C (PKC) because of its strong coupling to serotonin Type 2 receptors and its role in other forms of neuroplasticity (Hua et al., 1999; Sossin, 2007). Thus, we wished to determine whether spinal PKC activity is necessary for AIH-induced pLTF, and to identify which of the 15 known PKC mammalian isoforms is involved.
Another major goal of this study was to localize the plasticity to the critical spinal cellular elements, often hypothesized to be phrenic motor neurons (Mitchell et al., 2001; Feldman et al., 2003). To our knowledge, no studies have directly demonstrated that mammalian lower motor neurons are plastic. Motor plasticity known as sensorimotor long-term facilitation occurs in the sea slug, Aplysia, following tail stimulation or episodic serotonin receptor activation (Mauelshagen et al., 1998; Glanzman, 2009). Critical molecular events for this form of plasticity occur in the presynaptic sensory neuron (Kandel, 2001). However, protein synthesis, intracellular calcium elevation, and AMPA receptor insertion have been demonstrated in Aplysia postsynaptic motor neurons following induction of long-term facilitation (Cai et al., 2008; Glanzman, 2008). These studies suggest that motor neurons are malleable and lend credence to our hypothesis that mammalian spinal α motor neurons, which have long been thought of as of as “relays” between the CNS and muscles, are indeed highly plastic. Using spinal injections of differentially selective PKC inhibitors, immunofluorescence to confirm PKCθ expression in phrenic motor neurons, and RNA interference via intrapleural injections of siRNAs (Mantilla et al., 2013) targeting PKCθ mRNA within phrenic motor neurons, we test three hypotheses: (1) spinal PKC activity is necessary for pLTF; (2) the relevant PKC isoform is PKCθ; and (3) PKCθ is expressed within phrenic motor neurons. These studies provide the first evidence for a critical role of PKCθ in any form of motor plasticity and demonstrate that the relevant PKCθ is within the motor neurons per se.
Materials and Methods
Animals.
Experiments were performed on 3- to 4-month-old male Sprague Dawley rats (Harlan colony 211 or 218a) weighing 280–480 g. Rats were housed two per cage with food and water ad libitum and kept in a 12 h light/dark cycle. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison and conformed to policies found within the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Drugs
Pharmacological solutions.
Bisindolymalemide I, 1.4 mm (BIS, also known as GF109203X or Gö 6850; Tocris Biosciences), 40 mm Gö 6983 (Tocris Biosciences), 50 mm CID755673 (CID; Sigma-Aldrich), and 20 mm sotrastaurin (also known as AEB 071; Axon ligands) were dissolved in DMSO and stored at −20°C. Myristoylated peptide (2 μg/μl) mimicking the pseudosubstrate domain of PKCθ (theta inhibitory peptide, TIP; Calbiochem) dissolved in ACSF (in mm as follows: 120 NaCl, 3 KCl, 2 CaCl, 2 MgCl, 23 NaHCO3, 10 glucose; 95% O2/5% CO2) and stored at −20°C. NPC15437, 50 mm (NPC; Sigma-Aldrich) was stored in ACSF at 4°C. All PKC inhibitors were chosen based on published half-maximal inhibitory concentrations (Martiny-Baron et al., 1993; Gschwendt et al., 1996; Uberall et al., 1997; Saraiva et al., 2003; Felber et al., 2007; Sharlow et al., 2008; Evenou et al., 2009).
Intrathecal injections.
Drugs were delivered at doses consistent with spinal inhibition of PKC (Yashpal et al., 1995; Hua et al., 1999; Ferguson et al., 2008; Laferrière et al., 2011). Stock solutions were diluted in ACSF to their final concentration with a DMSO concentration between 0% and 20% (Gö 6983 20%; NPC 0%; CID 10%; BIS 20%; sotrastaurin 10%; TIP, 0%). In some cases, Gö 6983 in 100% DMSO or 100% DMSO alone (vehicle) was delivered intrathecally due to limited drug solubility. Limited dose–response curves are shown in Figure S1 (available at www.jneurosci.org as supplemental material). A 50 μl Hamilton syringe was loaded with drug solution and connected to silicone catheter (2 Fr Access Technologies; 0.3 mm inner diameter) inserted subdurally above C4 spinal segment. Injections were delivered 10–15 min before AIH (12 μl total, 2 μl/30 s). Vehicle-treated rats received ACSF with 0%–100% DMSO. There was no difference in pLTF when pretreated with 100% DMSO versus ACSF (47 ± 15% vs 44 ± 3%, respectively; p = 0.70).
RNA interference
siRNA delivery.
Pools of four 21-nucelotide duplexes of Accell-modified small interfering RNAs (siRNAs) targeting PKCθ (siPKCθ), PKCζ (siPKCζ), and nontargeting (NTsiRNA) were purchased from Dharmacon (Thermo Scientific). Accell-modified siRNA was chosen because this modification preferentially transfects neurons versus other cell types (Nakajima et al., 2012). Each siRNA pool was suspended in Dharmacon siRNA buffer to yield a concentration of 5 μm, aliquoted, and stored at −20°C. Before intrapleural injections, 20 μl of siRNA was added to 6 μl of 5× siRNA buffer (Dharmacon), 3.2 μl of Oligofectamine Transfection Reagent (Invitrogen) and 0.8 μl of RNAase free H2O (final siRNA concentration of 3.33 μm) and carefully mixed 20 min before injection, allowing the siRNA to complex with the transfection reagent.
Intrapleural injections.
Bilateral intrapleural injections of siRNAs and cholera toxin B (CtB) fragment were performed as previously described (Mantilla et al., 2009, 2013). Rats were induced in a flow-through chamber with 5% inhaled isoflurane in 100% O2, placed on a surgical table, and maintained with a nose cone (2% isoflurane, 100% O2). For phrenic motor neuron back labeling, a 25 μl Hamilton syringe with attached sterile needle (6 mm) was loaded with 12.5 μl of CtB fragment (2 μg/μl in sterile H2O) per side. For intrapleural siRNA delivery, 30 μl was loaded into a 50 μl Hamilton syringe. The rib cage was palpated to locate the fifth intercostal space at the anterior axillary line, and the needle was inserted into the pleural space bilaterally to deliver the injectate. After injections, isoflurane was discontinued and respiratory chest movements were monitored for at least 15 min after discontinuing anesthesia. All rats recovered with minimal signs of distress.
Phrenic nerve recordings
Surgical preparation.
Surgical procedures for phrenic and hypoglossal nerve isolation were performed as described previously (Bach and Mitchell, 1996; Fuller et al., 2001a). Briefly, rats were induced in a closed chamber with isoflurane, placed on an experimental surgical table, and maintained with a nose cone (3.5% isoflurane, 50% O2). After performing a tracheotomy, rats were pump ventilated (Rodent Ventilator 683, Harvard Apparatus; tidal volume 2.2–2.7 ml; frequency 70–75 breaths/min) with 3.5% isoflurane in 50% O2 (balance N2). Small amounts of CO2 were added to the gas mixture to maintain end-tidal CO2 (Respironics Novametrix) at 42–45 mmHg. Tracheal pressure was monitored to prevent overinflation and to assure that the tracheostomy remained patent. Both vagus nerves were isolated in the mid-cervical region and cut bilaterally to prevent ventilator entrainment. A tail vein was catheterized for continuous delivery of intravenous fluids (∼1.5 ml/h of 75% lactated Ringer's solution, 10% HCO3, and 15% hetastarch; Hespan, 6% hetastarch in 0.9% NaCl). Intravenous fluid infusion was adjusted as needed to maintain acid-base status (−4.0 to 4.0 mEq/L base excess, ≤1.5 mEq/L change at 60 min) as determined via blood samples. Catheterization of the femoral artery was performed for blood gas sampling and blood pressure recording. The left hypoglossal and phrenic nerves were isolated using a dorsal approach, cut, desheathed, and covered with saline-soaked cotton. A C2 laminectomy was performed for intrathecal drug administration; the dura was exposed, cut dorsally, and a silicone catheter (2 Fr; Access Technologies) attached to a 50 μl Hamilton syringe was advanced under the dura to C4. Anesthesia was then slowly converted to urethane (1.8–2.0 mg/kg, i.v.) while isoflurane was slowly withdrawn. After sufficient time for isoflurane dissipation, rats were paralyzed with pancuronium bromide (2.5 mg/kg, i.v.). Body temperature was measured with rectal probe and kept at 37.5 ± 1°C using the heated surgical table. Blood pressure was monitored throughout the experiment to ensure physiological stability (baseline: 70–150 mmHg, ≤30 mmHg change at the end of an experiment). Depth of anesthesia was assessed by toe pinch responses in blood pressure and phrenic nerve activity. At the end of each protocol, a maximal CO2 response (end-tidal CO2> 90 mmHg) was assessed to verify adequate dynamic range for phrenic output. All rats included in the study had >30% hypoxic response during AIH and >50% maximal hypercapnic response at 60 min after AIH (or sham normoxia), signifying that neither chemosensory inputs nor phrenic motor output was saturated. All rats were sacrificed at the end of neurophysiological experiments with an intravenous urethane bolus.
Electrophysiology.
Desheathed phrenic and hypoglossal nerves were placed on bipolar silver electrodes and submerged in mineral oil to prevent dessication. Nerve activity was amplified (10,000×), bandpass filtered (300–10,000 Hz model 1800, A-M Systems), rectified and integrated with a continuous moving averager (time constant: 50 ms; CWE, MA-821 filter). The integrated signal was digitized and analyzed with a data acquisition system (WINDAQ,DATAQ Instruments).
One hour following conversion to urethane anesthesia, the CO2 apneic and recruitment thresholds for phrenic nerve activity were determined by adjusting inspired CO2 and/or ventilator frequency. Baseline CO2 was maintained at 2–3 mmHg above the recruitment threshold for the duration of an experiment. Baseline nerve activity was maintained for at least 20–30 min, and then a blood sample was drawn to measure arterial PCO2, PO2, and pH to assess “baseline” conditions. Some rats received intrathecal injections near the phrenic motor nucleus 10–15 min before beginning experimental paradigms. The rats were then exposed to either AIH or continuous baseline conditions (i.e., time control). AIH consisted of three 5 min episodes of 10%–12% inspired O2, separated by 5 min of baseline oxygen conditions. During hypoxic episodes, arterial PO2 was between 35 and 45 mmHg, and arterial PCO2 was maintained within 1.5 mmHg of baseline values. Following AIH, baseline conditions were reestablished. Blood gas analyses were performed at 15, 30, and 60 min after AIH; blood inclusion criteria included arterial PO2 > 180 mmHg, PaCO2 within 1.5 mmHg of baseline, and base excess within 1.5 mEq/L of baseline. All rats had blood values within this range. Additionally, rats that had short-term hypoxic responses of <50% increase from baseline were eliminated from the study (only one rat did not meet this criterion).
Rats treated as time controls received relevant pharmacological agents but did not receive AIH and were maintained at baseline conditions for an equivalent period. Blood samples were taken at similar times as AIH-treated rats.
Protein quantification
Tissue harvesting.
After electrophysiology experiments, a C2-C5 laminectomy was performed and rats were partially exsanguinated. The spinal cord was cut at the C3 (mid C3) and C6 rootlets (mid C6), and then placed on ice. After allowing the extracted spinal cord to cool for 1 min, a coronal cut was made separating dorsal and ventral cord. The ventral cord was placed in a Pyrex homogenizer (Corning, 1 ml PTFE Pestle Tissue Grinder) filled with 250 μl of homogenization buffer (150 mm NaCl, 1% Triton X-100, 0.5% Na deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0 with 1× Halt Phosphatase Inhibitor mixture, Thermo Scientific; and 1× Protease Inhibitor Mixture, Sigma-Aldrich #P8340) and homogenized by pumping 20 times. The sample was then spun for 10 min at 0.5 relative centrifugal force. The pellet was discarded; 12 μl of the supernatant was saved for bicinchoninic acid assay for protein. The remaining supernatant was transferred and mixed with an equal volume of 2× loading buffer (5% 2-mercaptoethanol in Laemmli buffer: 65.8 mm Tris-HCl, pH 6.8, 2.1% SDS, 26.3% (w/v) glycerol, 0.01% bromophenol blue). Both samples were stored at −20°C for protein analysis and immunoblotting. Right diaphragm samples were also taken from each rat and processed in the same manner as above, except that 500 μl of homogenization buffer was used in a 3 ml tissue grinder.
Immunoblotting.
Bicinchoninic acid protein assay was performed using the Thermo Scientific Pierce BCA Protein Assay Kit following manufacturer's instructions. Using the estimated protein concentration, each sample was diluted to 1 μg protein/μl with loading buffer before loading into 10% or 4%–15% Tris-HCl 18 well gels. The 25 μl/well of loading control (1:1 loading buffer and homogenization buffer) and 10–25 μl/well of a protein ladder (1:1:1 of loading buffer, Precision Plus Protein Dual Color Standard, Bio-Rad; and Biotinylated Protein Ladder, Cell Signaling Technology) were also loaded on each gel. The 25 μl (25 μg protein) of sample was loaded into each well and was run at 120 V for 1 h. Afterward, wet transfer to a 0.45 μm pore Immobilon PVDF membrane was performed on ice for 1 h at 100 V. After transfer, the blots were cut at 50 kDa to allow for concurrent separate probing of GAPDH for loading control (MW 37 kDa) and PKCθ (MW 80 kDa) or PKCζ (MW 75 kDa). Blots were blocked in 5% BSA (for PKCθ antibody) or 5% milk (for PKCζ antibody or GAPDH antibody) in Tris-buffered saline (20 mm Tris, 500 mm NaCl) with 0.05% Tween (TBST) for 1 h at room temperature. Antibodies were then added to the blocking solution, and the blots were incubated overnight at 4°C. Afterward, blots were washed 3 times (5 min) with TBST and probed with HRP-conjugated goat anti-rabbit antibodies (1:10000, Santa Cruz Biotechnology) in blocking solution for 1 h at room temperature. Blots were then washed 5 times (5 min) with TBST. Signal was resolved using Pierce Super Signal West Dura Extended Duration Substrate kit (Thermo Scientific). Chemiluminescence was detected with an AutoChemi Imaging system (UVP Bio-Imaging Systems) with Labworks 4.6. Densitometry was performed with ImageJ by following instructions found at http://lukemiller.org/index.php/2010/11/analyzing-gels-and-Western-blots-with-image-j/. All relative protein comparisons are made by normalizing to GAPDH signal for that well and then comparing the relative signal with the mean of the control values. Percentage change calculation: 100 − (sample PKC signal/sample GAPDH signal)/(mean NTsiRNA samples/GAPDH) × 100 Antibodies. Polyclonal antibodies to phospho-PKCθ (Cell Signaling Technology, #9377) were used at 1/1000 dilution in 5% BSA TBST to assess PKCθ protein expression with immunoblotting. We used the phospho-PKCθ antibody instead of other PKCθ antibodies because: (1) phospho-PKCθ was unaffected by AIH; (2) phospho-PKCθ labeling was significantly better than non-phospho-antibodies; and (3) the same phospho-PKCθ antibody has been used successfully in other studies to determine PKCθ expression (Jové et al., 2006; Wei et al., 2010).
Polyclonal antibodies to atypical PKC (Santa Cruz Biotechnology, PKCζ/ι C-20) were used at 1/1000 dilution in 5% milk to probe for PKCζ (MW ∼75 kDa). GAPDH antibody (Cell Signaling Technology, 14C10, #2118) was used in 5% milk at 1/50,000 to assess GAPDH protein expression. Polyclonal rabbit anti-phospho-PKCθ (1/250, Cell Signaling Technology, #9377), polyclonal donkey anti-CtB (1/5000, EMD Millipore), monoclonal mouse anti-rabbit GFAP (1/500, EMD Millipore), and monoclonal mouse anti-rabbit CD11b (1/200, Serotec), conjugated goat anti-donkey AlexaFluor-495 (1/1000, Invitrogen), and conjugated goat anti-rabbit AlexaFluor-488 (1/500, Invitrogen) were used in immunofluorescence treatments.
Immunofluorescence
Tissue preparation and staining.
Following electrophysiology experiments, nine rats were killed and perfused transcardially with 300–400 ml of chilled 0.01 M PBS followed by 300–400 ml of chilled 4% PFA (pH, 7.4).The brain and spinal cord were harvested and immersion fixed in 4% PFA for 8 h (4°C). The tissues were then transferred to 20% sucrose followed by 30% sucrose until the tissues were dehydrated. The 40 μm transverse slices were cut using a microtome (SM2000R Leica) from C3-C5 and placed into an antifreeze solution of ethylene glycol:glycerol:01 M PBS (3:3:4) until stained. Free-floating sections were washed with 0.05 M Tris-buffered saline with 0.1% Triton-X (TBS-TX) and blocked with 1.0% BSA for 1 h. The tissues were labeled with rabbit anti-P-PKCθ (1:250, Cell Signaling Technology), mouse anti-GFAP (1:500, EMD Millipore), mouse anti-CD11b (1:200 Serotec), and donkey anti-CtB (1:5000, EMD Millipore) at room temperature for 16 h. Slices were washed with TBS-TX and subsequently stained with conjugated donkey anti goat AlexaFluor-647 (1:1000, Invitrogen), conjugated donkey anti-rabbit AlexaFluor-488 (1:500, Invitrogen), and donkey anti-mouse AlexaFluor-695 (1:1000, Invitrogen) for 2 h at room temperature. Tissue slices were washed a final time with TBS-TX and mounted on charged slides with an anti-fade solution (Prolong Gold, Invitrogen).
Imaging.
Slices were imaged at 20× and 40× with a Nikon C1 laser scanning confocal microscope with λ strobing in the Nikon EZ-C1 Gold (version 3.80) confocal imaging software. For z-stacks, 2 μm step increments were used.
Statistical analysis
Integrated phrenic burst amplitude and frequency were analyzed in 60 s bins before, during, and 15, 30, and 60 min after AIH or at similar points in time controls (no AIH). Integrated nerve burst amplitudes were expressed as a percentage change from baseline. Two-way ANOVA with a repeated-measures design (Sigmaplot 12) was used to compare integrated nerve burst amplitudes and frequency (as well as arterial PCO2, PO2, pH, and base excess) to detect significant treatment (AIH vs time control; drug treatment) and time effects. Individual comparisons between groups and times were made via post hoc analysis using Fisher's LSD test. Values of p < 0.05 were considered statistically significant.
Because no significant differences were found among the different time control experiments (NPC, n = 3; BIS, n = 3; sotrastaurin, n = 2; TIP, n = 4 vehicle, n = 3; p = 0.59), all time control experiments were grouped for comparison with AIH-treated rats. Similarly, because differences were not observed between time control experiments with intrapleural siPKCθ (n = 3), siPKCζ (n = 3), and NTsiRNA (n = 4) (p = 0.43), all intrapleural siRNA injection time controls were grouped.
Results
pLTF requires spinal PKC activity
To test the hypothesis that spinal PKC activation is necessary for pLTF, and to begin identification of the specific PKC isoform(s) involved, six PKC inhibitors with different pharmacological profiles were delivered intrathecally over the phrenic motor nucleus at C4 before AIH; reported half-maximal inhibitory concentration (IC50) values for each inhibitor and PKC isoform are shown in Table 1. Short-term hypoxic phrenic responses were unaltered by any spinal PKC inhibitor (data not shown, p > 0.05), consistent with prior studies using intrathecal pharmacology (MacFarlane and Mitchell, 2009).
Table 1.
Reported half-maximal inhibitory concentration (IC50) of each tested PKC inhibitor for each mammalian PKC isoforma
| Isoform/inhibitor | Gö 6983 (nm) | NPC | CID (μm) | BIS (nm) | Sotrastaurin (nm) |
|---|---|---|---|---|---|
| α | 7 | + | >>10 | 8 | 1 |
| β | 7 | + | >10 | 20 | 0.6 |
| γ | 6 | + | - | 2–6 | - |
| δ | 10 | + | >>7 | 0.2 | 2 |
| ε | - | + | - | 0.1 | 3 |
| η | - | + | - | - | 2 |
| μ (PKD) | 20 | - | 0.2 | 2 | - |
| θ | - | - | - | 20 | 0.2 |
| ι/λ | - | - | - | - | - |
| ζ | 60 | + | - | 6 | - |
aClassical (α, β, and γ), novel (δ, ζ, η, μ, θ), and atypical PKC isoforms (ι/λ, ζ) are shown. All IC50 values were obtained from published studies (see Materials and Methods). For NPC, no published IC50 values are available, but known inhibitory activity is denoted with a plus sign (+). Hyphen (-) indicates no known activity at the particular PKC isoform.
Time-compressed phrenic neurograms (Fig. 1A) illustrate integrated phrenic nerve burst amplitude before, during, and after AIH. In control rats (Fig. 1A, top, vehicle), phrenic nerve burst amplitude was significantly elevated above baseline at 30 min (29 ± 4%, p < 0.05) and 60 min (47 ± 4%, p < 0.001) after AIH, an effect not observed in time controls (i.e., no AIH; 8 ± 4% at 60 min, p > 0.05; Fig. 1A, bottom); persistent elevation of phrenic nerve burst amplitude represents pLTF.
Figure 1.

Spinal inhibition of PKCθ, but not other isoforms, blocks pLTF. A, Representative neurograms after AIH from rats given intrathecal injections (12 μl) of PKC inhibitors. ↓ indicates intrathecal delivery of inhibitor. Dotted line indicates baseline amplitude. Gray represents increases from baseline. B, Averaged percentage increase from baseline for each group at 60 min after AIH. Delivered drug doses: vehicle, n = 20; 1–2 mm Gö 6983, n = 8; 5 mm NPC-15437 (NPC), n = 6; 280 μm BIS, n = 5; 2–20 μm sotrastaurin, n = 5; myristolated pseudosubstrate peptide targeting PKCθ (theta inhibitory peptide, TIP, 2 μg/μl), n = 5. C, Average pLTF following intrathecal NPC or TIP. #p < 0.05 versus time controls (no AIH). *p < 0.05 versus vehicle.
pLTF does not require classical PKC isoforms
Gö 6983 is a broad-spectrum PKC active-site inhibitor that potently inhibits all classical PKC isoforms (Table 1) (Gschwendt et al., 1996). Rats pretreated with intrathecal Gö 6983 exhibited significant pLTF at 60 min (46 ± 9%; Fig. 1B) versus time controls (8 ± 4%, p < 0.05), which was similar to vehicle pretreated rats (100% DMSO; 44 ± 15%, p ≥ 0.28). There was no dose effect of Gö 6983 on pLTF in the range studied (Fig. 2; p > 0.05).
Figure 2.

Dose–responses of intrathecal PKC inhibitors on pLTF. Each group received 12 μl intrathecal injections of PKC inhibitor of shown concentrations over the phrenic motor nucleus. A, Gö 6983 had no effect on pLTF at any dose. Amplitude of pLTF at 60 min after AIH in rats given injections of 0.1–2 mm Gö 6983 (n = 7) or 10 mm Gö 6983 (n = 2) compared with rats given vehicle (100% DMSO, n = 7) injections. B, NPC had no effect on pLTF at any dose. Amplitude of pLTF at 60 min after AIH in rats given injections of 100 μm NPC15437 (n = 2) or 5–40 mm NPC15437 (n = 7) compared with rats given vehicle (ACSF, n = 3) injections (p > 0.05). C, CID755673 had no effect on pLTF at any dose tested. Amplitude of pLTF at 60 min after AIH in rats given injections of 10 μm (n = 2), 50 μm (n = 2), or 0.2–5 mm CID755673 (n = 3) compared with rats given vehicle (10–20% DMSO in ACSF, n = 3) injections (p > 0.05). D, Sotrastaurin significantly attenuated pLTF. Amplitude of pLTF at 60 min after AIH in rats given injections of 0.2 μm (n = 2), 2 μm (n = 4), or 20 μm sotrastaurin (n = 2) compared with rats given vehicle (10% DMSO in ACSF, n = 2) injections. *Significant difference versus grouped time controls (rats given vehicle or drug with no AIH, n = 13). #Significant difference versus grouped vehicle AIH rats (0%–20% DMSO in ACSF, 100% DMSO with AIH, n = 20).
pLTF requires a novel PKC isoform
BIS is a broad-spectrum PKC inhibitor that potently inhibits novel and classical PKC isoforms (Table 1) (Martiny-Baron et al., 1993; Gschwendt et al., 1996; Uberall et al., 1997). Intrathecal BIS (280 μm) significantly attenuated pLTF (22 ± 6%) versus vehicle controls (46 ± 4%, p < 0.001; Fig. 1B).
Because Gö 6983 and BIS are active-site inhibitors, we searched for PKC inhibitors acting by a distinct mechanism. NPC binds PKC regulatory subunits, blocking the binding of its activator, diacylglycerol. Based on in vitro studies, NPC inhibits most PKC isoforms, with some notable exceptions, such as PKCμ and PKCθ (Table 1) (Saraiva et al., 2003; Felber et al., 2007). Surprisingly, rats given 5 mm NPC exhibit qualitatively normal pLTF (Fig. 1A, trace, B; 54 ± 12%), which is indistinguishable from vehicle controls (47 ± 4%, p = 0.40; Fig. 1B). Because NPC does not inhibit pLTF at any dose tested (0.1–40 mm, p = 0.61; Fig. 2), NPC likely does not inhibit the relevant PKC isoform.
Since NPC has no known inhibitory effects on PKCμ or PKCθ, we tested inhibitors for these PKC isoforms (Table 1). In rats given the PKCμ inhibitor CID755673, pLTF was indistinguishable from vehicle-treated rats at doses ranging 50 μm to 5 mm (40 ± 13%, p = 0.26; Figs. 1B, 2). However, a broad-spectrum PKC inhibitor that potently inhibits PKCθ, sotrastaurin, attenuated pLTF at doses ≥2 μm (17 ± 4%, p < 0.001 vs vehicle; Figs. 1B, 2). Because BIS also inhibits PKCθ, we hypothesized that spinal PKCθ is the critical PKC isoform for pLTF.
pLTF does not require atypical PKC isoforms
PKCζ activity is expressed in phrenic motor neurons and required for phrenic motor facilitation induced by inactivity (Strey et al., 2012). In this same study, PKCζ inhibition with the myristolated peptide targeting PKCζ had no effect on pLTF, demonstrating that PKCζ and PKMζ are not required for pLTF. Our results fit with this conclusion because Gö 6983, an inhibitor of PKCζ, had no effect on pLTF (Figs. 1B, 2A).
pLTF requires PKCθ activity
Because BIS and sotrastaurin inhibit many PKC isoforms other than PKCθ, we sought to selectively block PKCθ activity with a myristolated peptide mimicking the pseudosubstrate region of PKCθ (TIP). TIP binds to the catalytic domain of PKCθ, blocking substrate phosphorylation (Harris et al., 1996; Kilian et al., 2004). Intrathecal TIP (0.86 mm) abolished pLTF (10 ± 7%) versus baseline (p = 0.17; Fig. 1A) or time controls (8 ± 4%, p = 0.74; Fig. 1B). Furthermore, TIP significantly decreased pLTF versus vehicle (47 ± 4%, p < 0.001; Fig. 1C) and NPC-treated rats (54 ± 12%, p = 0.001; Fig. 1C), suggesting that PKCθ activity per se is necessary for pLTF.
pLTF in NPC-treated rats was not significantly different from pLTF in vehicle-treated rats at any time point (16 ± 6% at 15 min, 31 ± 8% at 30 min, 54 ± 12% at 60 min, p = 0.994; Fig. 1C), yet NPC inhibits all PKC isoforms aside from PKCθ and PKCμ (also known as protein kinase D). Because, the PKCμ inhibitor had no effect on pLTF, we found no evidence that any PKC isoform other than PKCθ is necessary for AIH-induced pLTF.
PKCθ is highly expressed in phrenic motor neurons
Because spinal PKCθ activity appears to be critical for pLTF, we sought to determine cellular expression patterns within the phrenic motor nucleus. Strong PKCθ labeling (green; Fig. 3A,C) was observed in labeled phrenic motor neurons (blue; CtB-positive cells; Fig. 3B,D) and in other, unidentified neurons (higher magnification, Fig. 3F) in transverse C4-C5 spinal sections. PKCθ immunolabeling was negligible in microglia (red; CD11b-positive cells; Fig. 3B) or astrocytes (red; GFAP-positive cells; Fig. 3D), suggesting that PKCθ is predominantly expressed in ventral cervical spinal neurons. High-power images of phrenic motor neurons clearly demonstrate immunoreactive PKCθ protein (Fig. 3E) colocalized in identified phrenic motor neurons (Fig. 3F).
Figure 3.
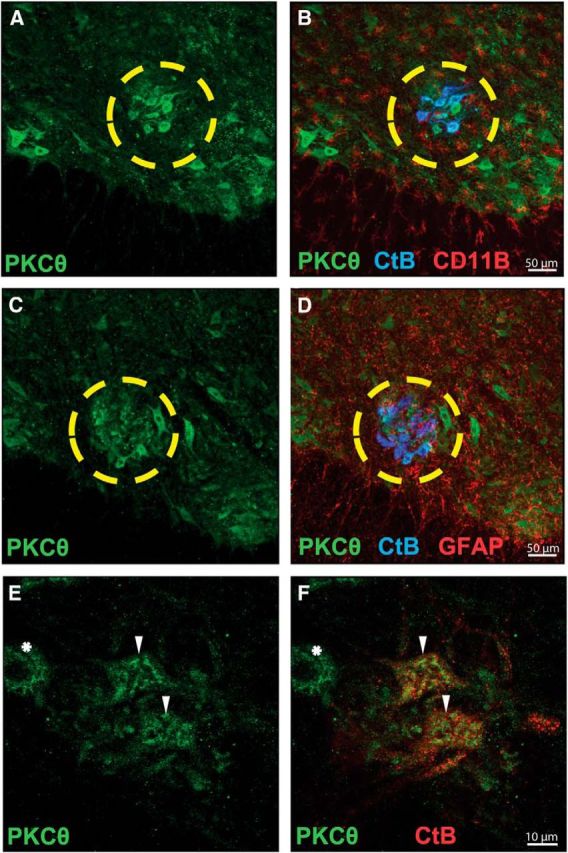
PKCθ is highly expressed in identified (CtB-positive) phrenic motor neurons. A, C, Representative confocal fluorescence images of ventral C4-C5 spinal sections from rats fluorescently labeled for PKCθ (Cell Signaling Technology, #9377, green). B, Colocalization of phrenic motor neurons (CtB, blue) and PKCθ fluorescence (green) is evident; in contrast, no colocalization could be appreciated between microglia (CD11b, red) and PKCθ (green) fluorescence. D, Colabeling for phrenic motor neurons (CtB, blue) and astrocytes (GFAP, red) reveals no expression of PKCθ in astrocytes. The yellow circle outlines the phrenic motor nucleus. E, Higher-magnification images of phrenic motor nucleus demonstrate PKCθ immunofluorescence (green). F, PKCθ immunofluorescence is present in phrenic motor neurons (CtB-positive, red; ▾), but also in unidentified putative neurons near the phrenic motor nucleus (CtB-negative, *).
PKCθ knockdown within phrenic motor neurons blocks pLTF
Intrapleural siRNAs are retrogradely transported to phrenic motor neurons, where they knock down target mRNA and protein levels (Mantilla et al., 2013). We delivered three daily intrapleural injections of either nontargeting siRNAs (NTsiRNA), or siRNAs targeting PKCθ (siPKCθ) or the ζ isoform of atypical PKC (siPKCζ). PKCζ is expressed in phrenic motor neurons (Guenther et al., 2010) but is not required for pLTF (Strey et al., 2012). On the other hand, PKCζ is necessary for a distinct form of phrenic motor plasticity known as inactivity induced phrenic motor facilitation (Strey et al., 2012). Rats given NTsiRNA had normal pLTF following AIH (48 ± 12%, p < 0.001 vs time controls; Fig. 4A, top), demonstrating that nonspecific effects of the injection procedures, transfection reagent or small, double-stranded RNAs had no impact on pLTF. In contrast, siPKCθ blocked pLTF (11 ± 13%, p = 0.24 vs time controls, p < 0.001 vs NTsiRNA; Fig. 4A, middle), confirming that PKCθ is essential for pLTF and that the relevant PKC is located within phrenic motor neurons per se. pLTF was unaffected in rats given siPKCζ (57 ± 15%, p < 0.001 vs time control, p = 0.27 vs NTsiRNA; Fig. 4A, bottom), demonstrating that neither knockdown of this atypical PKC isoform nor nonspecific siRNA effects could account for loss of pLTF after siPKCθ delivery. In summary (Fig. 4B), pLTF was normal in rats treated with NTsiRNA or siPKCζ, whereas siPKCθ completely blocked pLTF.
Figure 4.
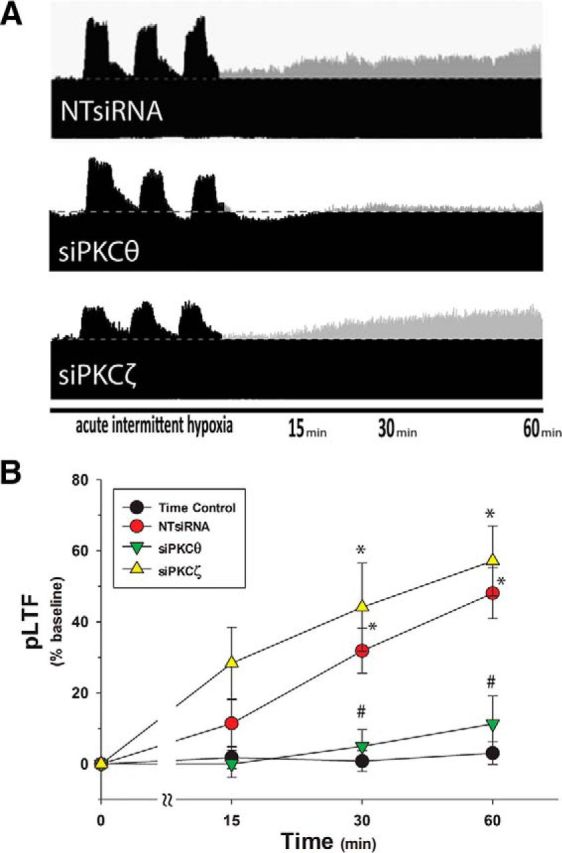
Intrapleural siRNAs targeting PKCθ block pLTF. A, Representative phrenic neurograms after AIH from rats given three, daily intrapleural injections of nontargeting siRNAs (NTsiRNA, n = 7, top), PKCθ targeting siRNAs (siPKCθ, n = 7, middle), or PKCζ targeting siRNAs (siPKCζ, n = 5, bottom). Horizontal dotted line indicates baseline amplitude. Gray represents increases from baseline. B, Summary data demonstrate significant pLTF at 30 and 60 min after AIH in NTsiRNA and siPKCζ groups, but not with siPKCθ. Rats given intrapleural NTsiRNA (n = 4), siPKCθ (n = 3), or siPKCζ (n = 3) without AIH (time controls, n = 10) did not exhibit significant facilitation at 60 min. *p < 0.05 versus time controls. #p < 0.05 versus siPKCζ or NTsiRNA.
Short-term hypoxic phrenic responses were unaltered by siPKCθ (83 ± 9%) versus NTsiRNA (83 ± 7%, p = 0.70; Fig 5). Rats treated with siPKCζ appeared to have enhanced short-term hypoxic phrenic responses versus siPKCθ (120 ± 24%, p = 0.03; Fig 5); however, this effect was not significant versus NTsiRNA (p = 0.52).
Figure 5.

Hypoxic phrenic responses are unaltered by intrapleural PKCθ siRNA. Phrenic nerve amplitude (% baseline) during short-term hypoxia after pretreatment with intrapleural injections of nontargeting siRNA (NTsiRNA, n = 7), PKCθ siRNA (siPKCθ, n = 7), or PKCζ siRNA (siPKCζ, n = 5). Time control rats (no AIH, n = 10) are shown for comparison. *p < 0.05, significant difference versus time controls. #p < 0.05, significant difference versus intrapleural siPKCθ+AIH. &p < 0.05, significant difference versus intrapleural NTsiRNA+AIH.
Hypoglossal motor output exhibits AIH-induced LTF, although it is more variable than pLTF, depending on age, sex, and rat colony (Fuller et al., 2000, 2001b; Behan et al., 2002; Baker-Herman and Strey, 2011). There was no evidence for unintended (indirect) brainstem effects from intrapleural siRNAs because hypoglossal LTF was unaffected by any of the intrapleural siRNAs tested (Fig. 6; siPKCθ and siPKCζ, p > 0.05 vs NTsiRNA, p < 0.05 vs time controls). Because there is no direct route for intrapleural siRNAs to reach hypoglossal motor neurons, these data suggest that siRNAs do not have indirect effects, such as trans-synaptic transport to brainstem premotor neurons, which provide the respiratory-related synaptic inputs to hypoglossal motor neurons (Yamada et al., 1988; Woch et al., 2000).
Figure 6.

Hypoglossal LTF is unaffected by intrapleural siRNA. Hypoglossal nerve amplitude (% baseline) following AIH exposure after pretreatment with intrapleural injections of nontargeting siRNA (NTsiRNA, n = 5), PKCθ siRNA (siPKCθ, n = 3), or PKCζ siRNA (siPKCζ, n = 3). All groups were significantly elevated compared with time control rats (NTsiRNA, n = 3; siPKCθ, n = 2; or siPKCζ, n = 3 + no AIH, n = 8). *p < 0.05, significant difference versus time control.
PKCθ knockdown in the ventral cervical spinal cord was confirmed with immunoblots of ventral spinal homogenates from the C3 to C5 spinal segments (Fig. 7A). Intrapleural siPKCθs significantly decreased PKCθ protein levels (−49 ± 18%, p = 0.03) versus rats given NTsiRNAs (Fig. 7B). As expected, siPKCζ (Fig. 7C) had no significant effect on PKCθ protein levels (−13 ± 15%, p = 0.528 vs NTsiRNA; Fig. 7D). Thus, intrapleural siPKCθs selectively target and knock down ventral spinal PKCθ, whereas siPKCζs had no off-target effects on PKCθ. These measurements confirm that intrapleural siPKCθs selectively reduce PKCθ expression within the ventral cervical cord encompassing the phrenic motor nucleus and strengthen the argument that PKCθ is the PKC isoform necessary for pLTF.
Figure 7.

Intrapleural siPKCθs decrease PKCθ protein expression in ventral cervical spinal homogenates (C3-C5). A, Representative immunoblot of ventral C3-C5 spinal cord from rats given three, daily intrapleural injections of nontargeting siRNAs (NTsiRNA, n = 6) or PKCθ mRNA targeting siRNAs (siPKCθ, n = 6). The immunoblot was cut (dotted line) and probed for PKCθ and GAPDH (as a loading control). B, Semiquantitative analysis of optical density (normalized to GAPDH) demonstrates significant PKCθ knockdown after intrapleural siPKCθs (vs NTsiRNA). C, Representative immunoblots for NTsiRNA (n = 4) or PKCζ mRNA targeting siRNAs (siPKCζ, n = 3). The immunoblot was cut (dotted line) and probed for PKCθ and GAPDH (as a loading control). D, Semiquantitative analysis of optical density (normalized to GAPDH) reveals no significant change in PKCθ protein after intrapleural siPKCζ (vs NTsiRNA). *p < 0.05 versus NTsiRNA.
Discussion
Here we demonstrate that a single novel PKC isoform, PKCθ, is necessary for an important form of spinal motor plasticity, AIH-induced pLTF. The role of PKCθ in pLTF was demonstrated with the following: (1) spinal application of drugs targeting multiple PKC isoforms, but with distinct affinities; (2) a cell-permeable peptide targeting the catalytic site of PKCθ; and (3) targeted PKCθ knockdown within phrenic motor neurons via RNA interference. No other known mammalian PKC isoform was required for AIH-induced pLTF.
Although PKCθ is highly expressed in phrenic motor neurons and is also found in unidentified neurons of the ventral cervical spinal cord, there was minimal expression in adjacent glia. Functional, localization of the relevant PKCθ was achieved with intrapleural siRNAs targeting PKCθ mRNA; intrapleural injections are accessible to respiratory motor neuron terminals accessing the pleural space, and are retrogradely transported to phrenic motor neuron cell bodies (Mantilla et al., 2009, 2013). Although we cannot completely rule out transcellular siRNA exchange as claimed in vitro (Valadi et al., 2007; Mittelbrunn and Sánchez-Madrid, 2012; Montecalvo et al., 2012), the combination of restricted PKCθ localization within neurons and the profound effect of intrapleural siPKCθ on pLTF suggest, at a minimum, that the relevant PKCθ is within the phrenic motor network. Collectively, our data provide the first evidence that motor neuron PKCθ is required for any form of spinal plasticity.
Spinal PKCθ activity is required for pLTF
Using available reagents, we demonstrated that a single spinal PKC isoform (PKCθ) is required for AIH-induced pLTF. Although mechanisms giving rise to AIH-induced pLTF have been studied extensively in recent years (Dale-Nagle et al., 2010a, 2010b; Devinney et al., 2013), this is the first confirmation of the role played by PKC. PKCθ activation likely occurs downstream from spinal serotonin Type 2 (5-HT2) receptor activation because spinal Gq protein-coupled 5-HT2 receptor activation is both necessary and sufficient for pLTF (Fuller et al., 2001a; MacFarlane and Mitchell, 2009), and these receptors are highly expressed in phrenic motor neurons (Basura et al., 2001; MacFarlane et al., 2011). 5-HT2 receptors activate phospholipase C to produce diacylglycerol and inositol triphosphate (Pandey et al., 1995). Diacylglycerol binds to the regulatory domain (C1) of PKC, activating the catalytic domain to enable phosphorylation of downstream targets (Steinberg, 2008). Thus, we suggest that PKCθ activity is most likely induced by 5-HT2 receptor activation and subsequent diacylglycerol production within phrenic motor neurons.
PKCθ differs from all other novel and classical PKC isoforms because it binds diacylglycerol predominantly at the C1B versus the C1A region of the regulatory domain (Melowic et al., 2007; Steinberg, 2008). Moreover, the C1B region of PKCθ may be unable to bind NPC because structural studies demonstrate narrowing of the phorbol ester-binding surface of C1B in PKCθ versus the relatively similar PKCδ C1B region, which has 80% sequence homology (Rahman et al., 2013). This difference may explain how NPC, which binds the diacylglycerol binding site of most PKC isoforms (Sullivan et al., 1991), had no effect on pLTF. Most likely, NPC fails to block PKCθ because it cannot bind the distinct C1B-diacylglycerol binding-site of PKCθ. This key difference in PKCθ regulatory domain structure enabled us to use NPC as a pharmacological tool to rule out other conventional and novel PKC isoforms. Because inhibitors that have no known PKCθ activity (Gö 6983, CID, NPC) had no effect on pLTF and collectively cover all other known conventional and novel PKC isoforms aside from PKCθ, it is highly unlikely that any other PKC isoforms are required for pLTF. Thus, our pharmacological studies support the idea that PKCθ is uniquely required for pLTF.
Cellular localization of pLTF: targeting spinal neurons with siRNAs
Because 5-HT2 receptors are abundantly expressed on phrenic neurons (Basura et al., 2001; Fuller et al., 2005), and many molecules associated with pLTF (5-HT2 receptors, BDNF, TrkB, and phospho-ERK) are upregulated in presumptive phrenic motor neurons following repetitive exposure to acute intermittent hypoxia (Lovett-Barr et al., 2012; Satriotomo et al., 2012), we previously hypothesized that pLTF arises from cellular mechanisms within phrenic motor neurons per se (Fuller et al., 2000; Mitchell et al., 2001; Feldman et al., 2003; Mahamed and Mitchell, 2008; Dale-Nagle et al., 2010b). However, no studies to date provide direct experimental evidence to support this hypothesis. Here, we used a novel approach, intrapleural siRNA injections and phrenic nerve recordings, to specifically target phrenic motor neurons (Mantilla et al., 2013). Although the observation that siPKCθ knockdown within phrenic motor neurons abolishes pLTF is consistent with the idea that the relevant PKCθ for pLTF is within phrenic motor neurons per se, we cannot completely rule out the possibility of transcellular siRNA exchange. Indeed, some claim that micro-RNAs are exchanged between cells in cell culture conditions (Valadi et al., 2007; Mittelbrunn and Sánchez-Madrid, 2012; Montecalvo et al., 2012), suggesting the possibility (not certainty) of transcellular siRNA exchange in vivo. However, intrapleural siPKCθs are unlikely to affect astrocytes or microglia in the phrenic motor nucleus because their PKCθ expression is minimal (Fig. 2B,D). siPKCθs could possibly jump synapses, conceivably affecting PKCθ expression in spinal interneurons (Lane et al., 2008; Lane, 2011) or presynaptic modulatory neurons, such as the raphe serotonergic neurons that initiate AIH-induced pLTF (Mitchell et al., 2001; Mahamed and Mitchell, 2008; Dale-Nagle et al., 2010b). However, because intrapleural siPKCθ did not alter hypoglossal LTF or the short-term hypoxic phrenic response, our evidence contradicts this idea. Finally, intrapleural siRNAs targeting the receptor tyrosine kinase, TrkB, knockdown TrkB protein within the phrenic motor nucleus, but not in nearby regions containing phrenic interneurons (D.P.F., E.A.D., G.S.M., unpublished observations). Thus, collective evidence argues against the possibility that intrapleural siPKCθ blocked pLTF via indirect, trans-synaptic effects in premotor neurons.
Intrapleural siRNAs most likely affect other respiratory motor pools that contact the pleural space, such as intercostal motor neurons; indeed, these motor pools retrogradely transport CtB after intrapleural injections (Mantilla et al., 2009). Because intercostal motor neurons have no direct connections to phrenic motor neurons, there is no reason to believe that intercostal motor neurons will affect pLTF. On the other hand, we suspect that intrapleural siPKCθ would block intercostal LTF (Fregosi and Mitchell, 1994), unless there are unknown mechanistic differences between these motor pools. There is some possibility that intrapleural siRNAs are retrogradely transported by the vagus nerve to the dorsal motor nucleus of the vagus or the nucleus ambiguous. Although we cannot rule out PKCθ knockdown in these areas, it highly unlikely such knockdown (if it occurs) would influence pLTF.
Motor neurons are plastic
Historically, motor neurons have been thought of as simple relays linking the CNS to muscles. Accumulating evidence has suggested that motor neurons are dynamic, including studies on plasticity in the model organism, Aplysia (Glanzman, 2008). Moreover, spinal motor neurons have extensive dendritic trees, which are influenced by developmental experiences, such as gravitational changes (Inglis et al., 2000). In the respiratory system, we know of at least five distinct cellular mechanisms giving rise to phrenic motor facilitation (Dale-Nagle et al., 2010a). Here, we provide evidence that AIH elicits plasticity within motor neurons (and/or their immediate neural network), thereby inducing pLTF. Further, we demonstrate that a single PKC isoform (of 15) is necessary for pLTF, and the relevant PKC is within phrenic motor neurons per se. Further studies aimed at understanding spinal motor neuron plasticity will enable a better understanding of how motor systems adapt to the ever-changing demands of life, such as normal development and aging, gaining or losing weight, changes in physiological conditions, or the onset of disease (Mitchell and Johnson, 2003).
Clinical significance
Motor plasticity also has therapeutic implications in clinical disorders that compromise both respiratory and nonrespiratory motor function. For example, intermittent hypoxia induces recovery of breathing capacity (Lovett-Barr et al., 2012) as well as rat forelimb (Lovett-Barr et al., 2012) and human leg function (Trumbower et al., 2012) after chronic, incomplete spinal injuries. AIH-induced plasticity may also prove useful in preserving or restoring breathing capacity in patients with motor neuron disease, such as amyotrophic lateral sclerosis (Nichols et al., 2013). Manipulating key molecules, such as PKCθ, may enable us to harness motor plasticity to restore motor function, without the concerns and inconveniences involved with hypoxic exposures (Almendros et al., 2014). Moreover, understanding the role of PKCθ in plasticity will guide our use of specific PKCθ inhibitors, which are currently being developed for immunosuppression (Zhang et al., 2013). Appreciating the role of PKCθ in plasticity may enable us to predict unintended effects of these agents.
Targeted RNA interference in respiratory motor pools with intrapleural siRNAs may lead to simple and effective therapies in patients with compromised breathing capacity, such as occurs in amyotrophic lateral sclerosis or spinal cord injury. For example, intrapleural siRNAs targeting molecules that restrain motor plasticity might be used to enhance motor output, thereby preserving/restoring some of the lost capacity. In ventilator-dependent patients, increased breathing capacity might enable unassisted ventilation, improving quality of life (Criner, 2002; Martin, 2002). Because intrapleural siRNAs profoundly affect phrenic motor plasticity, and the technique is relatively noninvasive and well tolerated by rodents, we provide proof of principle that this technique could be useful to implement gene therapy in respiratory motor neurons.
Footnotes
This work was supported by National Institutes of Health Grants T32 GM007507, T32 GM86926, and GM007507 to M.J.D., HL007654 to M.J.D. and E.A.D., and HL080209, HL111598, and HL69064; the Francis Family Foundation to A.G.H.; and the American Physiological Society UGSRF to T.J.P. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Bradley Wathen for excellent technical assistance and Erica Rosenbaum for technical advice.
The authors declare no competing financial interests.
References
- Almendros I, Wang Y, Gozal D. The polymorphic and contradictory aspects of intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2014;307:L129–L140. doi: 10.1152/ajplung.00089.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Strey KA. Similarities and differences in mechanisms of phrenic and hypoglossal motor facilitation. Respir Physiol Neurobiol. 2011;179:48–56. doi: 10.1016/j.resp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Basura GJ, Zhou SY, Walker PD, Goshgarian HG. Distribution of serotonin 2A and 2C receptor mRNA expression in the cervical ventral horn and phrenic motoneurons following spinal cord hemisection. Exp Neurol. 2001;169:255–263. doi: 10.1006/exnr.2001.7682. [DOI] [PubMed] [Google Scholar]
- Behan M, Zabka AG, Mitchell GS. Age and gender effects on serotonin-dependent plasticity in respiratory motor control. Respir Physiol Neurobiol. 2002;131:65–77. doi: 10.1016/S1569-9048(02)00038-1. [DOI] [PubMed] [Google Scholar]
- Cai D, Chen S, Glanzman DL. Postsynaptic regulation of long-term facilitation in Aplysia. Curr Biol. 2008;18:920–925. doi: 10.1016/j.cub.2008.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criner GJ. Care of the patient requiring invasive mechanical ventilation. Respir Care Clin North Am. 2002;8:575–592. doi: 10.1016/S1078-5337(02)00064-3. [DOI] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Mitchell GS. Multiple pathways to long-lasting phrenic motor facilitation. Adv Exp Med Biol. 2010a;669:225–230. doi: 10.1007/978-1-4419-5692-7_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Hoffman MS, MacFarlane PM, Satriotomo I, Lovett-Barr MR, Vinit S, Mitchell GS. Spinal plasticity following intermittent hypoxia: implications for spinal injury. Ann N Y Acad Sci. 2010b;1198:252–259. doi: 10.1111/j.1749-6632.2010.05499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinney MJ, Huxtable AG, Nichols NL, Mitchell GS. Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann N Y Acad Sci. 2013;1279:143–153. doi: 10.1111/nyas.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evenou JP, Wagner J, Zenke G, Brinkmann V, Wagner K, Kovarik J, Welzenbach KA, Weitz-Schmidt G, Guntermann C, Towbin H, Cottens S, Kaminski S, Letschka T, Lutz-Nicoladoni C, Gruber T, Hermann-Kleiter N, Thuille N, Baier G. The potent protein kinase C-selective inhibitor AEB071 (sotrastaurin) represents a new class of immunosuppressive agents affecting early T-cell activation. J Pharmacol Exp Ther. 2009;330:792–801. doi: 10.1124/jpet.109.153205. [DOI] [PubMed] [Google Scholar]
- Felber M, Sonnemann J, Beck JF. Inhibition of novel protein kinase C-epsilon augments TRAIL-induced cell death in A549 lung cancer cells. Pathol Oncol Res. 2007;13:295–301. doi: 10.1007/BF02940308. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Bolding KA, Huie JR, Hook MA, Santillano DR, Miranda RC, Grau JW. Group I metabotropic glutamate receptors control metaplasticity of spinal cord learning through a protein kinase C-dependent mechanism. J Neurosci. 2008;28:11939–11949. doi: 10.1523/JNEUROSCI.3098-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregosi RF, Mitchell GS. Long-term facilitation of inspiratory intercostal nerve activity following carotid sinus nerve stimulation in cats. J Physiol. 1994;477:469–479. doi: 10.1113/jphysiol.1994.sp020208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller D, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic motor output. Respir Physiol. 2000;121:135–146. doi: 10.1016/S0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001a;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Baker TL, Behan M, Mitchell GS. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001b;4:175–181. doi: 10.1152/physiolgenomics.2001.4.3.175. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Baker-Herman TL, Golder FJ, Doperalski NJ, Watters JJ, Mitchell GS. Cervical spinal cord injury upregulates ventral spinal 5-HT2A receptors. J Neurotrauma. 2005;22:203–213. doi: 10.1089/neu.2005.22.203. [DOI] [PubMed] [Google Scholar]
- Glanzman DL. New tricks for an old slug: the critical role of postsynaptic mechanisms in learning and memory in Aplysia. Prog Brain Res. 2008;169:277–292. doi: 10.1016/S0079-6123(07)00017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzman DL. Habituation in Aplysia: the Cheshire cat of neurobiology. Neurobiol Learn Mem. 2009;92:147–154. doi: 10.1016/j.nlm.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Goshgarian HG. The crossed phrenic phenomenon and recovery of function following spinal cord injury. Respir Physiol Neurobiol. 2009;169:85–93. doi: 10.1016/j.resp.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau JW, Crown ED, Ferguson AR, Washburn SN, Hook MA, Miranda RC. Instrumental learning within the spinal cord: underlying mechanisms and implications for recovery after injury. Behav Cogn Neurosci Rev. 2006;5:191–239. doi: 10.1177/1534582306289738. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors: differentiation from protein kinase C isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- Guenther CH, Vinit S, Windelborn JA, Behan M, Mitchell GS. Atypical protein kinase C expression in phrenic motor neurons of the rat. Neuroscience. 2010;169:787–793. doi: 10.1016/j.neuroscience.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TE, Persaud SJ, Saermark T, Jones PM. A myristoylated pseudosubstrate peptide inhibitor of protein kinase C: effects on glucose- and carbachol-induced insulin secretion. Mol Cell Endocrinol. 1996;121:133–141. doi: 10.1016/0303-7207(96)03858-0. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Coles SK, Bach KB, Mitchell GS, McCrimmon DR. Time-dependent phrenic nerve responses to carotid afferent activation: intact vs. decerebellate rats. Am J Physiol. 1993;265:R811–R819. doi: 10.1152/ajpregu.1993.265.4.R811. [DOI] [PubMed] [Google Scholar]
- Hayes HB, Jayaraman A, Herrmann M, Mitchell GS, Rymer WZ, Trumbower RD. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology. 2014;82:104–113. doi: 10.1212/01.WNL.0000437416.34298.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MS, Nichols NL, Macfarlane PM, Mitchell GS. Phrenic long-term facilitation after acute intermittent hypoxia requires spinal ERK activation but not TrkB synthesis. J Appl Physiol. 2012;113:1184–1193. doi: 10.1152/japplphysiol.00098.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua XY, Chen P, Yaksh TL. Inhibition of spinal protein kinase C reduces nerve injury-induced tactile allodynia in neuropathic rats. Neurosci Lett. 1999;276:99–102. doi: 10.1016/S0304-3940(99)00818-6. [DOI] [PubMed] [Google Scholar]
- Inglis FM, Zuckerman KE, Kalb RG. Experience-dependent development of spinal motor neurons. Neuron. 2000;26:299–305. doi: 10.1016/S0896-6273(00)81164-2. [DOI] [PubMed] [Google Scholar]
- Jové M, Planavila A, Sánchez RM, Merlos M, Laguna JC, Vázquez-Carrera M. Palmitate induces tumor necrosis factor-alpha expression in C2C12 skeletal muscle cells by a mechanism involving protein kinase C and nuclear factor-kappaB activation. Endocrinology. 2006;147:552–561. doi: 10.1210/en.2005-0440. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kilian K, Dernedde J, Mueller EC, Bahr I, Tauber R. The interaction of protein kinase C isozymes alpha, iota, and theta with the cytoplasmic domain of L-selectin is modulated by phosphorylation of the receptor. J Biol Chem. 2004;279:34472–34480. doi: 10.1074/jbc.M405916200. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Bach KB, Johnson SM, Hodgeman BA, Mitchell GS. Plasticity in respiratory motor control: intermittent hypoxia and hypercapnia activate opposing serotonergic and noradrenergic modulatory systems. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:207–218. doi: 10.1016/S1095-6433(01)00393-2. [DOI] [PubMed] [Google Scholar]
- Kokotilo KJ, Eng JJ, Curt A. Reorganization and preservation of motor control of the brain in spinal cord injury: a systematic review. J Neurotrauma. 2009;26:2113–2126. doi: 10.1089/neu.2008.0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laferrière A, Pitcher MH, Haldane A, Huang Y, Cornea V, Kumar N, Sacktor TC, Cervero F, Coderre TJ. PKMζ is essential for spinal plasticity underlying the maintenance of persistent pain. Mol Pain. 2011;7:99. doi: 10.1186/1744-8069-7-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA. Spinal respiratory motoneurons and interneurons. Respir Physiol Neurobiol. 2011;179:3–13. doi: 10.1016/j.resp.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Lane MA, White TE, Coutts MA, Jones AL, Sandhu MS, Bloom DC, Bolser DC, Yates BJ, Fuller DD, Reier PJ. Cervical prephrenic interneurons in the normal and lesioned spinal cord of the adult rat. J Comp Neurol. 2008;511:692–709. doi: 10.1002/cne.21864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J Neurosci. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587:5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Vinit S, Mitchell GS. Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience. 2011;178:45–55. doi: 10.1016/j.neuroscience.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Respiratory long-term facilitation: too much or too little of a good thing? Adv Exp Med Biol. 2008;605:224–227. doi: 10.1007/978-0-387-73693-8_39. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Gransee HM, Zhan WZ, Sieck GC. Motoneuron BDNF/TrkB signaling enhances functional recovery after cervical spinal cord injury. Exp Neurol. 2013;247:101–109. doi: 10.1016/j.expneurol.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin UJ. Whole-body rehabilitation in long-term ventilation. Respir Care Clin North Am. 2002;8:593–609. doi: 10.1016/S1078-5337(02)00041-2. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marmé D, Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Mauelshagen J, Sherff CM, Carew TJ. Differential induction of long-term synaptic facilitation by spaced and massed applications of serotonin at sensory neuron synapses of Aplysia californica. Learn Mem. 1998;5:246–256. doi: 10.1101/lm.5.3.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melowic HR, Stahelin RV, Blatner NR, Tian W, Hayashi K, Altman A, Cho W. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Cθ. J Biol Chem. 2007;282:21467–21476. doi: 10.1074/jbc.M700119200. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mittelbrunn M, Sánchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol. 2012;13:328–335. doi: 10.1038/nrm3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, Baty CJ, Gibson GA, Erdos G, Wang Z, Milosevic J, Tkacheva OA, Divito SJ, Jordan R, Lyons-Weiler J, Watkins SC, Morelli AE. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756–766. doi: 10.1182/blood-2011-02-338004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Kubo T, Semi Y, Itakura M, Kuwamura M, Izawa T, Azuma YT, Takeuchi T. A rapid, targeted, neuron-selective, in vivo knockdown following a single intracerebroventricular injection of a novel chemically modified siRNA in the adult rat brain. J Biotechnol. 2012;157:326–333. doi: 10.1016/j.jbiotec.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2013;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Davis JM, Pandey GN. Phosphoinositide system-linked serotonin receptor subtypes and their pharmacological properties and clinical correlates. J Psychiatry Neurosci. 1995;20:215–225. [PMC free article] [PubMed] [Google Scholar]
- Rahman GM, Shanker S, Lewin NE, Kedei N, Hill CS, Prasad BV, Blumberg PM, Das J. Identification of the activator-binding residues in the second cysteine-rich regulatory domain of protein kinase Cθ (PKCθ) Biochem J. 2013;451:33–44. doi: 10.1042/BJ20121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva L, Fresco P, Pinto E, Gonçalves J. Isoform-selectivity of PKC inhibitors acting at the regulatory and catalytic domain of mammalian PKC-alpha, -betaI, -δ, -eta and -zeta. J Enzyme Inhib Med Chem. 2003;18:475–483. doi: 10.1080/14756360310001603158. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Dale EA, Dahlberg JM, Mitchell GS. Repetitive acute intermittent hypoxia increases expression of proteins associated with plasticity in the phrenic motor nucleus. Exp Neurol. 2012;237:103–115. doi: 10.1016/j.expneurol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS, Wang QJ. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–33526. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossin WS. Isoform specificity of protein kinase Cs in synaptic plasticity. Learn Mem. 2007;14:236–246. doi: 10.1101/lm.469707. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strey KA, Nichols NL, Baertsch NA, Broytman O, Baker-Herman TL. Spinal atypical protein kinase C activity is necessary to stabilize inactivity-induced phrenic motor facilitation. J Neurosci. 2012;32:16510–16520. doi: 10.1523/JNEUROSCI.2631-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JP, Connor JR, Tiffany C, Shearer BG, Burch RM. NPC 15437 interacts with the C1 domain of protein kinase C: an analysis using mutant PKC constructs. FEBS Lett. 1991;285:120–123. doi: 10.1016/0014-5793(91)80739-P. [DOI] [PubMed] [Google Scholar]
- Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil Neural Repair. 2012;26:163–172. doi: 10.1177/1545968311412055. [DOI] [PubMed] [Google Scholar]
- Uberall F, Giselbrecht S, Hellbert K, Fresser F, Bauer B, Gschwendt M, Grunicke HH, Baier G. Conventional PKC-alpha, novel PKC-epsilon and PKC-theta, but not atypical PKC-λ are MARCKS kinases in intact NIH 3T3 fibroblasts. J Biol Chem. 1997;272:4072–4078. doi: 10.1074/jbc.272.7.4072. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- Vinit S, Lovett-Barr MR, Mitchell GS. Intermittent hypoxia induces functional recovery following cervical spinal injury. Respir Physiol Neurobiol. 2009;169:210–217. doi: 10.1016/j.resp.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei XY, Liu JP, Zhao CH, Ju G, Wong-Riley MT, Liu YY. Expressions of 5-HT/5-HT(2A) receptors and phospho-protein kinase C theta in the pre-Bötzinger complex in normal and chronic intermittent hypoxic rats. Neuroscience. 2010;168:61–73. doi: 10.1016/j.neuroscience.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Woch G, Ogawa H, Davies RO, Kubin L. Behavior of hypoglossal inspiratory premotor neurons during the carbachol-induced, REM sleep-like suppression of upper airway motoneurons. Exp Brain Res. 2000;130:508–520. doi: 10.1007/s002210050053. [DOI] [PubMed] [Google Scholar]
- Wolpaw JR, Tennissen AM. Activity-dependent spinal cord plasticity in health and disease. Annu Rev Neurosci. 2001;24:807–843. doi: 10.1146/annurev.neuro.24.1.807. [DOI] [PubMed] [Google Scholar]
- Wolpaw JR, O'Keefe JA, Noonan PA, Sanders MG. Adaptive plasticity in primate spinal stretch reflex: persistence. J Neurophysiol. 1986;55:272–279. doi: 10.1152/jn.1986.55.2.272. [DOI] [PubMed] [Google Scholar]
- Yamada H, Ezure K, Manabe M. Efferent projections of inspiratory neurons of the ventral respiratory group: a dual labeling study in the rat. Brain Res. 1988;455:283–294. doi: 10.1016/0006-8993(88)90087-X. [DOI] [PubMed] [Google Scholar]
- Yashpal K, Pitcher GM, Parent A, Quirion R, Coderre TJ. Noxious thermal and chemical stimulation induce increases in 3H-phorbol 12,13-dibutyrate binding in spinal cord dorsal horn as well as persistent pain and hyperalgesia, which is reduced by inhibition of protein kinase C. J Neurosci. 1995;15:3263–3272. doi: 10.1523/JNEUROSCI.15-05-03263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EY, Kong KF, Altman A. The yin and yang of protein kinase C-theta (PKCθ): a novel drug target for selective immunosuppression. Adv Pharmacol. 2013;66:267–312. doi: 10.1016/B978-0-12-404717-4.00006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]