

Abstract
The age-dependent progression of tau pathology is a major characteristic of tauopathies, including Alzheimer's disease (AD), and plays an important role in the behavioral phenotypes of AD, including memory deficits. Despite extensive molecular and cellular studies on tau pathology, it remains to be determined how it alters the neural circuit functions underlying learning and memory in vivo. In rTg4510 mice, a Tau-P301L tauopathy model, hippocampal place fields that support spatial memories are abnormal at old age (7–9 months) when tau tangles and neurodegeneration are extensive. However, it is unclear how the abnormality in the hippocampal circuit function arises and progresses with the age-dependent progression of tau pathology. Here we show that in young (2–4 months of age) rTg4510 mice, place fields of hippocampal CA1 cells are largely normal, with only subtle differences from those of age-matched wild-type control mice. Second, high-frequency ripple oscillations of local field potentials in the hippocampal CA1 area are significantly reduced in young rTg4510 mice, and even further deteriorated in old rTg4510 mice. The ripple reduction is associated with less bursty firing and altered synchrony of CA1 cells. Together, the data indicate that deficits in ripples and neuronal synchronization occur before overt deficits in place fields in these mice. The results reveal a tau-pathology-induced progression of hippocampal functional changes in vivo.
Keywords: Alzheimer's, learning and memory, neurodegeneration, place cells, tau
Introduction
The age-dependent progression of tau pathology is a hallmark of tauopathies, including Alzheimer's disease (AD). Tau pathology in AD starts with tau protein hyperphosphorylation followed by the formation of neurofibrillary tangles and neurodegeneration, particularly in the neural circuits important for learning and memory such as the cortex and hippocampus (Grundke-Iqbal et al., 1986; Hardy et al., 1998; Wenk, 2003; Ashe and Zahs, 2010). Behaviorally, the most obvious manifestation of the disease is progressive memory loss (Carlesimo and Oscar-Berman, 1992; Salmon and Bondi, 2009). It is believed that tau pathology and subsequent neurodegeneration play an important role in the memory deficits of AD (Ghoshal et al., 2002; Ashe and Zahs, 2010). However, the critical link by which tau pathology and neurodegeneration lead to memory loss remains unclear.
Memory loss is likely linked to tau pathology through in vivo functional abnormalities of memory circuits including the hippocampus. Therefore, we focus on the functional changes of hippocampal neurons in freely moving animals of a tauopathy mouse model, the rTg4510 mouse (Tau mouse). In these mice, the overexpression of a mutated human tau gene (P301L) in the forebrain leads to the age-dependent progression of tau pathology that is similar to what is seen in tauopathies (Ramsden et al., 2005; Santacruz et al., 2005). These mice also show age-dependent memory deficits, with little impairment in spatial reference memory at early ages, but significant impairment at ≥4.5 months (Ramsden et al., 2005). We hypothesize that the progressive spatial memory deficit in Tau mice is accompanied by a progressive change in the functions of hippocampal neurons in vivo.
It is proposed that spatial memories are supported by hippocampal place cells, which fire action potentials only when an animal is at one or a few places (place fields) in an environment (O'Keefe and Dostrovsky, 1971; Wilson and McNaughton, 1993; Eichenbaum et al., 1999). Place cells are paced by oscillations of local field potentials (LFPs), including theta oscillations (6–12 Hz) occurring during running behavior (Buzsáki, 2002) and high-frequency (100–250 Hz) ripple oscillations during sleep (Buzsáki et al., 1992). When ripples occur, the firing of place cells is synchronized within a short period of time (Buzsáki et al., 1992; Csicsvari et al., 2000). Strong evidence exists for a role of ripples in sleep-dependent memory consolidation (Buzsáki, 1989; Wilson and McNaughton, 1994; Euston et al., 2007; Ji and Wilson, 2007; Girardeau et al., 2009; Ego-Stengel and Wilson 2010). To examine our hypothesis, in this study we ask how hippocampal place cells, LFP oscillations, and firing synchrony are altered in an age-dependent fashion in Tau mice. We showed previously that place cells in the CA1 area of the hippocampus are severely impaired in old (∼8 months) Tau mice (Cheng and Ji, 2013). By mainly focusing on young Tau mice at 2–4 months of age, here we aim to reveal the in vivo neurophysiological deficits that parallel the age-dependent progression of tau pathology and spatial memory deficits in Tau mice.
Materials and Methods
Animals
A total of 23 Tau mice and 15 wild-type (WT) littermates (Table 1) were used, which were generated from two mouse lines (Ramsden et al., 2005; Santacruz et al., 2005). The responder line carried a human tau gene with the P301L mutation downstream of a tetracycline-operon-responsive element, and was maintained on an FVB/N background. The accompanying activator line contained an open-frame Tet-Off gene downstream from a calcium-calmodulin kinase II (CamKII) promoter and was maintained on a 129Sv6 background. The resulting F1 progenies of the two mouse lines overexpressed P301L human tau in the forebrain and were the experimental subjects used for electrophysiological recordings. Two age groups of mice were used: 2–4 months and 7–9 months. Most mice were males, but a minor number of females (six Tau, four WT; Table 1) were used in the experiments due to the lengthy time of breeding. However, our previous work (Cheng and Ji, 2013) and the data in the present study (data not shown) revealed that male and female Tau mice displayed similar neurophysiological deficits, compared with their WT counterparts. Therefore, the recorded data from male and female mice of the same age group were combined in the analysis. The recording procedures used are similar to those published previously (Ji and Wilson, 2007; Cheng and Ji, 2013) and are briefly described below. The experimental protocol was approved by the Institutional Committee on Animal Care at Baylor College of Medicine and followed National Institutes of Health guidelines.
Table 1.
Animals used in this study and the number of CA1 cells analyzed from each animal under each experimental condition (familiar, novel, or sleep)
| Mouse # | Tau mice |
WT mice |
|||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Young |
Old |
Young |
Old |
||||||||||||||||||||||||||
| 1 | 2* | 3 | 4+ | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12+ | 13 | 14 | 2* | 15+ | 16 | 17+ | 18 | 19 | 20 | 21 | 22+ | 23 | 24 | 25 | 26 | 27+ | 28+ | |
| Familiar (active/total) | 7/10 | 20/28 | 24/27 | 6/12 | 9/14 | 11/31 | 4/13 | 6/18 | 9/14 | 8/14 | 3/3 | — | — | — | ∘ | ∙ | ∙ | ∙ | ∙ | 10/14 | 29/53 | 8/9 | 6/10 | 7/15 | — | ∙ | ∙ | ∙ | ∙ |
| Novel (active/total) | 3/5 | 11/19 | 17/33 | 4/11 | 7/11 | 6/19 | — | — | — | — | — | — | — | — | ∘ | ∙ | ∙ | ∙ | ∙ | 9/18 | 13/18 | 9/12 | 11/25 | — | — | ∙ | ∙ | ∙ | ∙ |
| Sleep | 8 | 31 | 17 | 8 | 16 | 13 | 16 | 30 | 12 | 10 | 29 | 12 | 3 | 15 | 18 | 31 | 29 | 5 | 13 | 52 | 13 | — | 9 | 3 | 5 | 14 | 23 | 27 | |
In addition, 10 animals (5 young Tau mice, 4 young WT mice, and 1 old WT mouse, including 3 females) used only for LFP analysis were not included in the table. One mouse (Mouse 6) was not used in the sleep LFP analysis (no SWS episodes recorded). —, No cells recorded; ∘, cells recorded but not relevant to this study; ∙, cells recorded and included in our previous study (Cheng and Ji, 2013), but not included in the present study.
+Female mouse;
*mouse analyzed at both young and old ages.
Surgery
Every mouse was surgically implanted with a hyperdrive containing eight or four tetrodes. Each mouse was anesthetized with 0.5–2% isoflurane and fixed with a stereotaxic device throughout the surgery. A craniotomy at the coordinates 2.0 mm antereoposterior and 1.5 mm mediolateral from Bregma was made. The hyperdrive cannulae containing the tetrodes were lowered to the exposure and cemented to the skull using stainless steel anchoring screws and dental acrylic. The analgesic ketoprofen (5 mg/kg) and saline (1 ml) were injected subcutaneously at the end of surgery before the animal recovered from anesthesia.
Tetrode recording
Tetrodes were slowly moved down to the CA1 pyramidal cell layer over 2–4 weeks following the surgery. The pyramidal cell layer was identified by the presence of bursty firing and sharp-wave ripples when animals were at rest (Buzsáki et al., 1992; Csicsvari et al., 2000). Spiking activity and LFPs were recorded by a Neuralynx Digital Lynx system while the animal either slept in its home cage for 1–3 h or performed the track-running tasks described below. Neuronal spikes were filtered between 0.6 and 6 kHz, identified using a preset threshold of 50–70 μV, and sampled at 32 kHz. LFPs were broadly filtered between 0.1 Hz and 9 kHz, sampled at 32 kHz, and digitally downsampled at 2 kHz. Data were stored in hard disks and analyzed offline. Spikes were sorted using the manual clustering program xclust (M. Wilson, Massachusetts Institute of Technology). The animal's position was tracked by two color diodes mounted over the animal's head. Position data were sampled at 33 Hz with a resolution of ∼0.2 cm.
Behavioral apparatuses and tasks
For recording during track-running tasks, data were acquired while each animal was running back and forth (two trajectories) for food reward (condensed milk) on a familiar or novel narrow (6 cm wide) track. The familiar track was rectangular shaped and had a total length of ∼2 m, whereas the novel track was a ∼2 m long rectangular track for the majority of recorded mice and a ∼1.2 m long L-shaped track for the others. Each animal was trained on the familiar track starting about 1 week after surgery. The training lasted for at least 2 weeks before recordings began. Recordings started only after the animal was able to run back and forth at least 10 times on the track. On a given recording day, the animal went through two recording sessions, each ∼15 min long, on the familiar track, separated by a 10–15 min rest in an enclosed box. For a subset of mice (two Tau and two WT), familiar track recordings were repeated once every 3–5 d and lasted for up to 4 months. However, for each of these mice, only 1 d of spike data per age group (young or old) were included in the analysis. After at least 1 d of recording on the familiar track, the recording on the novel track took place in a novel room and followed a similar daily recording schedule. The animal had never been in the novel room or on the novel track before the first day of the recording to ensure novelty.
Electrolytic lesions, histology, and immunohistochemistry
After all recordings, the animal was killed using an overdose of pentobarbital (50 mg/kg). Electric current (30 μA, 5–10 s) was passed to each tetrode to create an electrolytic lesion at each recording site. The brain was perfused using 4% paraformaldehyde (PFA), dissected out, and stored in 4% PFA overnight before being cryoprotected in 30% sucrose at 4°C. To verify the tau pathology at different ages, a selected set of mouse brains (three old and three young WT and Tau animals) were later sectioned using a cryostat at varied thicknesses. In this case, two sets of 16 μm sections were stained with tau antibodies MC-1 and PHF-1 (Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY). Another set of 50 μm sections were stained with cresyl violet to identify the recording sites. For all other mice, the brains were sectioned at 50–100 μm and stained with cresyl violet only. Only the data acquired with recording sites verified at the CA1 pyramidal cell layer were used in the analysis (Fig. 1).
Figure 1.

Tau pathology in young and old Tau mice. A, cresyl violet staining of coronal sections from a 4-month-old WT mouse, 4-month-old Tau mouse, 8-month-old WT mouse, and 8-month-old Tau mouse. Arrows indicate recording sites. Note the thinner than normal CA1 pyramidal cell layer in the 8-month-old Tau mouse, but the nearly normal CA1 layer thickness in the 4-month-old Tau mouse. B, C, MC-1 and PHF-1 stained coronal sections of 4-month-old (B) and 8-month-old (C) WT and Tau mice. For each column, the boxed area in CA1 of the top picture is expanded at the bottom. Note the extensive staining (brownish black) of the CA1 pyramidal cells for both MC-1 and PHF-1 in the 8-month-old Tau mouse (C), but only MC-1 staining in the 4-month-old Tau mouse (B). Scale bars: black, −0.5 mm; red, −25 μm.
Data analysis
As described below, we quantified various properties of CA1 neurons and LFPs recorded from Tau and WT mice as they slept and as they ran on the familiar and novel tracks. Results are presented as mean ± SE or median and [10%, 90%] range values unless specified otherwise. Statistical comparisons between two groups of samples were made by using the t test (statistic presented as t(df)) or the nonparametric rank-sum test (statistic presented as W(N1, N2), where N1 and N2 are the number of samples in the two groups), respectively, unless specified otherwise. Two-way ANOVA was used for comparisons among the average LFP power spectral densities (PSDs) at different frequencies recorded from different groups of mice (statistic presented as F(number of groups − 1, df)). To examine the animal-to-animal variability within an age/genotype group for a given parameter, we computed a ratio of between-animal variation to within-animal variation (presented as F(number of animals − 1, df)) and its significance level as determined by the F test. Significance p values >1 × 10−5 are shown as actual values of the statistical tests.
Place field quantification.
For each running session, we defined two running trajectories on a track as the two paths from one end to the other in opposite running directions. A running lap was defined as a single run on one of the trajectories. Only those neurons active on at least one trajectory (mean firing rate, >0.5 Hz) with at least four laps were included in the analysis. Putative interneurons (mean firing rate, >7 Hz) were excluded. If a neuron was active on both trajectories, it was analyzed separately on each of the trajectories, and results were averaged. Each trajectory was linearized and spatially binned with a 2 cm bin size, with the reward sites at both ends of the trajectory excluded from analysis. A mean firing rate xi at each spatial bin i was defined as the total number of spikes divided by the total occupancy time (ti) across all laps on the trajectory in the session. The rate curve [x1, x2,…, xN], where N is the total number of bins, was smoothed using a Gaussian kernel with a σ of 2 bins. The spatial information (I) of the neuron on the trajectory in the session is defined as follows (Skaggs et al., 1993):
 |
where pi = is the occupancy probability, and r = is the mean firing rate.
Place fields on the trajectory were defined using the rate curve [x1, x2,…, xN]. Any peaks of the curve with a peak rate ≥1 Hz were identified as potential place fields. Place field boundaries for each peak were determined as the first and final bin across a threshold, which was 10% of the peak rate (Mehta et al., 2002). Neighboring place fields with gaps <6 cm were combined into a single field. Field length was defined as the distance between the boundaries. Those fields with lengths <6 or >100 cm were excluded from the analysis.
The firing stability of a place cell was computed to measure the similarity between its rate curves on the same trajectory across two recording sessions, if the cell was active on the trajectory during both sessions. Given two rate curves [x1, x2,…, xN] and [y1, y2,…, yN], we defined firing stability as their Pearson correlation:
 |
where |x and σx are the mean and SD of [x1, x2,…, xN], and |y and σy are the mean and SD of [y1, y2,…, yN], respectively.
Slow-wave sleep classification.
Similar to previous studies (Ji and Wilson, 2007), behavioral states were classified at a resolution of 1 s using a semiautomatic method. Briefly, the CA1 LFP was filtered through the delta band (1–4 Hz), theta band (6–12 Hz), and broad ripple band (100–250 Hz). The power of each filtered LFP was computed for every 1 s interval as the mean of all the squared LFP values within the interval. Three thresholds were then defined: one for movement speed, one for theta-to-delta power ratio, and one for broad ripple power. Because the overall LFP power differed across animals (for example, due to neurodegeneration), the latter two thresholds were manually defined for each mouse, relative to the baseline values when the animal was known to be awake. For this purpose, the LFP powers within different bands were graphed versus time for a time period including both slow-wave sleep (SWS) and waking states. A value that visually separated the two states was set as a threshold by a mouse click. SWS was then automatically detected from these thresholds for the entire recording period by a custom-made program. SWS was defined by no head movement (<2 cm/s), high ripple power (larger than the threshold), and low theta power (lower than the threshold). The detected SWS periods were then manually verified, similarly as in other classification methods (Robert et al., 1999; Gilmour et al., 2010). The thresholds were refined a few times to find values that corresponded best with manual verification in both Tau and WT mice. Further analyses related to sleep were only applied to the identified SWS episodes that were at least 5 s long.
LFP power analysis.
For a recording session, LFP segments during individual SWS episodes or during individual running laps were selected. For PSD analysis, a multitaper method was used to estimate the PSD at every 0.5 Hz for each segment, and PSDs were averaged across all the segments of a session. The mean and SE of PSDs at each frequency across all running/sleep sessions were computed for each genotype (Tau or WT) and age (young or old) group. To quantify power within a frequency band, the PSD of a session was integrated within the band. Theta power was integrated from 6–12 Hz. This definition of theta is a slightly higher frequency band than the conventional definition (Buzsáki, 2002), which we used because in our previous study we identified the peak theta frequency in our animals to be ∼9 Hz (Cheng and Ji, 2013). The ripple power was integrated from 120–180 Hz, because our PSD analysis revealed that this band was where the ripple power increased from the neighboring band (see Fig. 4). To examine how the theta and ripple powers were altered in Tau mice relative to the change in overall LFP, we also computed normalized theta and ripple powers. The normalized theta power of a session was defined as the theta power divided by the total power of the broad band 2–400 Hz. Ripple power was normalized by the power of a neighboring baseline frequency band (80–100 Hz), because normalizing to the broad band would produce tiny values since the power in ripple band was much smaller than those in lower-frequency bands. The mean and SE of the power (or normalized power) in the theta/ripple band were computed across all the sessions for each genotype (Tau or WT) and age group (young or old) of mice.
Figure 4.

Ripple power during SWS was reduced in young and old Tau mice. A–D, Raw and ripple-filtered (pass band, 100–250 Hz) LFP traces of a young WT mouse (A), a young Tau mouse (B), an old WT mouse (C), and an old Tau mouse (D). Scale bars apply to traces from all four mice. E, F, Average (mean and SE, which is half of the curve thickness) PSD of LFPs recorded from young (E) and old (F) WT (blue) and Tau (red) mice. G, H, Same as E and F, but with the PSD normalized by the power of the baseline frequency band of 80–100 Hz. I, Average (mean and SE) ripple power within the frequency band of 120–180 Hz in young and old WT and Tau mice. J, Same as E, but with ripple power normalized by the power of baseline frequency band of 80–100 Hz. ***p < 0.001.
Theta phase precession.
Theta phase was determined by using the Hilbert transform on LFP traces filtered within the theta band. For a given place field of a CA1 cell on a trajectory, the theta phases of all the spikes within the field boundary were circularly correlated with their linearized positions (circular correlation) to quantify the overall quality of phase precession [see the formula in the study by Ravassard et al. (2013)]. In addition, following the traditional method (O'Keefe and Recce, 1993), we also computed the linear correlation and the linear regression slope between the theta phases and the positions. In this case, all the phase values (in degrees) were shifted by a value from 0° to 360° at every degree, and their linear correlation with the positions was computed for each shift. The phase values at the shift that yielded the most negative correlation were linearly regressed with the positions. The correlation value and the regression slope at this shift were taken as the linear correlation and phase precession slope, respectively, for the place field.
Ripple event detection and quantification.
In addition to examining total power within the ripple band, individual ripple events were detected via CA1 LFPs recorded while mice were in SWS, as in previous studies (Csicsvari et al., 2000; Lee and Wilson, 2002; Suh et al., 2013). LFPs were first filtered through the broad ripple band of 100–250 Hz. A ripple peak threshold, defined as 6 SDs of the filtered LFP trace, was used to identify the potential ripple events (Csicsvari et al., 2000; Ji and Wilson, 2007). Another threshold, defined as 2.5 SDs of the filtered LFP trace, was used to define the start and end times of ripple events. Neighboring ripple events with a gap smaller than 30 ms were combined into a single ripple event (Siapas and Wilson, 1998). Only events with durations between 30 and 400 ms were included in further analyses. Ripple amplitude (the absolute value of the most negative trough) and ripple duration were quantified for each ripple event and averaged across all the events in a session.
Ripple-associated sharp waves.
We computed a ripple-peak-triggered LFP average to quantify ripple-associated sharp waves. For a given ripple event, we determined the time of its most negative trough. The trough times of all ripple events in a recording session were then used as trigger times. The raw LFP trace without filtering in the session was broken into 200 ms long segments, each centered at a trigger time (time lag 0). Such LFP segments for all ripple events in the session were stacked together with their centers aligned at trigger time lag 0 and a mean (and SE) LFP curve (vs the trigger time lag within [−100, 100] ms) was then averaged across the stacked segments. The peak amplitude of the LFP average curve was computed as a quantification of the sharp waves in the session.
Burst index.
A bursty spike was defined as the one that trailed an earlier spike by <10 ms. Burst index was the number of bursty spikes in a spike train divided by the total number of spikes.
Pairwise spike cross-correlation.
Similar to previous studies (Sirota et al., 2003; Wierzynski et al., 2009), we computed a normalized spike count cross-correlation, which is insensitive to firing rate, for each pair of cells active during a SWS session (mean rate, >0.5 Hz). Given two spike trains made of M, N spikes, P = [P1, P2,…, PM] and Q = [Q1, Q2,…, QN], and a time bin size B, for each time lag ΔT and for each spike Pi (i = 1, 2,…, M) in P, we counted the number of spikes in Q that were within [Pi + ΔT − B/2 Pi + ΔT + B/2]. The spike counts were summed across i = 1, 2,…, M for each time lag ΔT. The summed spike count C was then normalized by the spike count that would be expected if the two spike trains were random Poisson processes, which was m = MN/B. The cross-correlation between the two cells at time lag ΔT was (C − m)/. The bin size B was set to 10 ms. The mean value of the central time lag bins with [−20, 20] ms was taken as the correlation between the two cells during SWS. The cross-correlations for all the pairs within an age/genotype group were then averaged.
Results
To examine how in vivo hippocampal neurophysiological activities are altered in an age-dependent manner in Tau mice, we recorded LFPs and/or firing activities of CA1 neurons from 18 Tau mice and 10 WT littermates at 2–4 months of age (young mice) and compared them with those from 5 Tau and 5 WT mice at 7–9 months of age (old mice). These two age groups were studied because they represent an early stage and a mature stage, respectively, of pathology and spatial memory impairment (Ramsden et al., 2005; Santacruz et al., 2005). The recordings were made while mice were actively running on a track or were in SWS.
Tau pathology in young and old Tau mice
We verified the recording sites in CA1 by electrolytic lesions and histology and verified tau pathology of young and old Tau mice by immunohistochemistry, performed after the recording was completed (Fig. 1). In agreement with previous reports (Ramsden et al., 2005; Santacruz et al., 2005), the hippocampi of old Tau mice were severely degenerated compared with those of old WT mice, whereas the hippocampi of young Tau mice were relatively normal. We measured the thickness of the CA1 pyramidal layer and the thickness of the cortex above CA1. The mean thickness of the CA1 pyramidal layer in old Tau mice was only 48% of that of old WT mice (Tau, 26 ± 1 μm, N = 7; WT, 53 ± 1 μm, N = 7; t(12) = 23.6; p < 1 × 10−5, t test), whereas the mean thickness of the cortex in Tau mice was 77% of that of old WT mice (Tau, 567 ± 6 μm, N = 7; WT, 737 ± 22 μm, N = 6; t(11) = 8.1; p < 1 × 10−5). In young Tau mice, the thickness of the CA1 pyramidal layer was 88% of that of WT mice (Tau, 49 ± 2 μm, N = 9; WT, 56 ± 3 μm, N = 7), but the change was not statistically significant (t(14) = 1.8; p = 0.095). Similarly, the cortical thickness of young Tau mice was 90% of that of WT mice (Tau, 740 ± 48 μm, N = 9; WT, 826 ± 26 μm, N = 7), but the difference was not significant (t(14) = 1.5; p = 0.17). In a sample group of the recorded Tau mice for each age group (three young and three old Tau mice), we also performed immunohistochemical staining against abnormal tau using MC-1 and PHF-1 antibodies. MC-1 recognizes the tau protein with abnormal conformation at an early stage of tau pathology, while PHF-1 is believed to label a more advanced stage of tau pathology with intracellular neurofibrillary tangles (Ramsden et al., 2005). We found positive staining against MC-1 in both young and old Tau mice, whereas the staining against PHF-1 was only obvious in old Tau mice (Fig. 1B,C). The staining was similar across all the animals within the same age group. These results are consistent with previous findings that abnormal tau protein was abundant but neurodegeneration was not significant in young Tau mice, whereas both were prominent in old Tau mice (Ramsden et al., 2005; Santacruz et al., 2005).
Largely normal place fields with subtle abnormalities in young Tau mice
We first asked how hippocampal place cells were affected by early stage tau pathology in young Tau mice. We recorded from CA1 cells in 11 young Tau mice and 5 young WT mice while animals were running two trajectories (back and forth) on a familiar track for two sessions per day, each ∼15 min long with a 10–15 min break in between. On average, the Tau mice ran 13.0 ± 0.8 (mean ± SE) laps per trajectory per session, whereas WT mice ran 13.2 ± 1.9 laps (t(60) = −0.13; p = 0.90, t test). The running speed was also comparable between Tau and WT mice (Tau, 22 ± 2 cm/s; WT, 21 ± 3 cm/s; t(60) = 0.37; p = 0.71). A total of 184 CA1 cells from young Tau mice (Tau cells) and 101 from young WT mice (WT cells) were recorded on the familiar track (Table 1). Among these, 107 (58%) Tau cells and 60 (59%) WT cells were determined to be putative principal cells that were active on at least one trajectory during at least one session. There was no significant difference in the median firing rate of the track-active Tau cells (median, 1.2 Hz; [10%, 90%] range, [0.63, 2.7] Hz) compared to WT cells (median, 1.3; [10%, 90%] range, [0.64, 2.6] Hz; W(107,60) = 9050; p = 0.84, rank-sum test).
As expected, WT cells fired reliably at well-defined place fields, which were stable between the two sessions on familiar trajectories (Fig. 2A). Tau cells also showed place fields (Fig. 2B) that appeared comparable to WT. To evaluate just how similar Tau place fields were to their WT controls, we first examined place specificity of WT and Tau cells. We quantified specificity using two parameters: spatial information, which measures how much information (in bits per spike) a cell's firing tells about the animal's location on a trajectory, and place field length, a finer measure which is the length of an individual place field on the trajectory. Our results show that Tau cells had similar median spatial information on familiar trajectories to WT cells (Fig. 2C; Tau, median, 1.2 bits/spike; [10%, 90%] range, [0.34, 2.2] bits/spike; WT, 1.1 bits/spike, [0.48, 2.4] bits/spike; W(107,60) = 9076; p = 0.77). However, the median field length of Tau cells was slightly (24%) but significantly greater than that of WT cells (Fig. 2D; Tau, median, 42 cm; [10%, 90%] range, [28, 58] cm; WT, 33 cm, [24, 56] cm; W(107,58) = 9933; p = 0.00033). We then examined the stability of the place fields of Tau and WT cells across the two running sessions. The stability of Tau cells was significantly lower than that of WT cells (Fig. 2E; Tau, median, 0.67 cm; [10%, 90%] range, [0.25, 0.94] cm; WT, 0.79 cm, [0.45 0.96] cm; W(73,38) = 3741; p = 0.03). In addition, we also computed the average values of these parameters for each mouse and compared them between young Tau and WT mice using each mouse average instead of each cell as a sample. The results were similar. The median spatial information was similar between Tau and WT mice (Tau, median, 1.3; Tau, median, 0.67 bits/spike; [10%, 90%] range, [0.82, 1.7] bits/spike; WT, 1.4 bits/spike, [0.57, 1.7] bits/spike; W(11,5) = 95; p = 0.91). The median field length in Tau mice was slightly greater than that in WT mice (Tau, median, 42 cm; [10%, 90%] range, [32, 52] cm; WT, 33 cm, [26, 45] cm) with a trend toward a significant difference (W(11,5) = 108; p = 0.11). The median stability in Tau mice was lower than that in WT mice (Tau, median, 0.69 cm; [10%, 90%] range, [0.28, 0.76] cm; WT, 0.78 cm, [0.69, 0.85] cm), again with a trend toward a significant difference (W(11,5) = 77; p = 0.069). Finally, to examine how these parameters varied from animal to animal within WT and Tau mice, we computed a ratio of between-animal variance to within-animal variance (F) for each parameter and each genotype. We found significant animal-to-animal variations only within the Tau mice in the place field length (F(10,96) = 3.5; p = 0.0006) and stability (F(10,62) = 3.4; p = 0.0013), suggesting the heterogeneous nature of this group. These results indicate that CA1 place cells in young Tau mice had comparable place specificity on familiar trajectories to those in young WT mice at a gross level, but exhibited subtle differences in the quality of their place fields as measured by field length and stability.
Figure 2.

Place fields on familiar and novel tracks in young Tau mice were largely normal. A, B, Example place cells recorded from a WT (A) and a Tau (B) mouse during two sessions in which the mice ran along a trajectory on a familiar track. In each panel, the top spike raster plot shows the linearized position of the animal at the time of each spike during each lap of a session, and the bottom curve shows the average firing rate of the cell across all the laps of the session. Arrows indicate running direction. Asterisks indicate place fields. C–E, Distributions of spatial information (C), place field length (D), and stability (E) of WT and Tau place cells on the familiar track. Counts are normalized by total number of cells. F–H, Same as C–E, but for place cells on a novel track.
We next studied CA1 place cells on a novel track where six Tau and four WT mice ran two trajectories (back and forth) for two sessions on their first day. The findings are similar to those on the familiar track. Tau mice traversed a similar number of laps per trajectory per session as WT mice (Tau, 8.0 ± 0.7; WT, 7.7 ± 1.3; t(38) = 0.27; p = 0.79), but with a faster speed (Tau, 14.0 ± 1.3 cm/s; WT, 7.6 ± 0.8 cm/s; t(38) = 3.6; p = 0.00091). A total of 48 Tau cells and 42 WT cells were active on at least one trajectory during at least one session. Despite the apparent difference in the running speed between Tau and WT mice, the firing rates of Tau and WT cells were similar during the running of novel trajectories (Tau, median, 1.0 Hz; [10%, 90%] range, [0.5, 2.5] Hz; WT, 1.1 Hz, [0.6, 2.8] Hz; W(48,42) = 2084; p = 0.41). The median spatial information of Tau cells on the novel trajectories was not significantly different from that of WT cells (Fig. 2F; Tau, 1.1 bits/spike, [0.5, 1.9] bits/spike; WT, 1.1 bits/spike, [0.6, 1.8] bits/spike; W(48,42) = 2112; p = 0.94), but the median length of identified place fields of Tau cells was greater than that of WT cells (Fig. 2G; Tau, 43 cm, [33, 65] cm; WT, 34 cm, [26, 45] cm; W(47,33) = 2456; p < 1 × 10−5). In addition, the median stability of Tau cells across the two novel track sessions was lower than that of WT cells (Fig. 2H; Tau, 0.65 cm, [0.17, 0.87] cm; WT, 0.79 cm, [0.19, 0.97] cm; W(34,29) = 921; p = 0.022). Using animal averages instead of cells as samples produced similar results. The median spatial information was similar between Tau and WT mice (Tau, 1.2 bits/spike, [0.9, 1.6] bits/spike; WT, 1.3 bits/spike, [0.9, 1.9] bits/spike; W(6,4) = 33; p = 0.99). The median field length in Tau mice was slightly but significantly greater than that in WT mice (Tau, 46 cm, [40, 49] cm; WT, 36 cm, [31, 40] cm; W(6,4) = 44; p = 0.019). The median stability in Tau mice was lower than that in WT mice (Tau, 0.57 cm, [0.42, 0.76] cm; WT, 0.77 cm, [0.50, 0.83] cm), with a trend toward a significant difference (W(6,4) = 24; p = 0.067). Finally, unlike the case of the familiar track, we did not find any significant animal-to-animal variations in these parameters on the novel track within the Tau or WT mouse group (p ≥ 0.13).
These data suggest that CA1 cells of young Tau mice can form place fields in both familiar and novel environments, as the firing of Tau cells contained a similar amount of spatial information as that of WT cells. However, there could be fine differences in the quality of their place fields; namely, our data suggest that place fields in Tau cells were slightly larger and were less stable across sessions. Our previous work, however, showed that CA1 cells in old Tau mice do not have stable firing locations even on a lap-by-lap basis, resulting in firing activities that contain very little spatial information (Cheng and Ji, 2013). Together, these results indicate a progressive deterioration of place fields as tau pathology matures with age.
Largely normal theta oscillations and theta phase precession in young Tau mice
The largely normal place fields in CA1 cells of young Tau mice shown here were recorded during track running. A prominent phenomenon in the hippocampus at the LFP level during track running is the theta oscillation. Given the importance of theta oscillations in shaping place fields (Buzsáki, 2002; Mehta et al., 2002; Harris et al., 2003), we examined theta oscillations in young Tau and WT mice while the mice ran on a familiar track. Theta oscillations in young Tau and WT mice appeared quite similar (Fig. 3A,B). We quantified theta oscillations by computing a PSD curve around the theta band (3–15 Hz) and the overall theta power (integration of PSD between 6 and 12 Hz) for each LFP trace. The averaged PSD curve among young Tau mice was slightly below that of young WT mice (Fig. 3C; F(1,824) = 8.8; p = 0.0031, ANOVA), meaning the average power in young Tau mice was slightly lower than in their WT controls. In old Tau mice, the total LFP power was significantly reduced from that of old WT mice, but the proportion of theta relative to the total LFP power was not (Cheng and Ji, 2013). The slight change in the PSD curve of young Tau mice could be due to a change in the total LFP power caused by the tau pathology. To account for this possibility, we computed the total power (integration of PSD between a broad band of 2–400 Hz) for each LFP trace and then normalized the PSD curve by the total power. The average total LFP power of young Tau mice was slightly reduced, but not significantly different from that of young WT mice (Tau, 0.011 ± 0.001 mV2; WT, 0.012 ± 0.001 mV2; t(32) = −0.92; p = 0.37). When normalized by the total power, the average PSD curves were similar between young Tau and WT mice (Fig. 3D; F(1,824) = 0.61; p = 0.44). The overall theta power in young Tau mice was slightly lower than, but not significantly different from, that in young WT mice (Fig. 3E; Tau, 0.0030 ± 0.0003 mV2; WT, 0.0038 ± 0.0006 mV2; t(32) = −1.3; p = 0.22). The normalized overall theta power relative to total power in young Tau mice was similar to that in young WT mice (Fig. 3F; Tau, 0.27 ± 0.01; WT, 0.29 ± 0.02; t(32) = −0.83; p = 0.41). LFPs recorded while running on the novel track showed similar results (data not shown). These results indicate that although absolute theta power may be slightly lower, CA1 LFPs in young Tau mice contain a normal amount of theta oscillations.
Figure 3.

Theta power was normal in young Tau mice during track running. A, B, Raw and theta-filtered (pass band, 6–12 Hz) LFP traces of a young WT mouse (A) and a young Tau (B) mouse. Calibrations apply to traces in both A and B. C, Average (mean and SE) PSD of LFPs recorded from young WT (blue) and Tau (red) mice. D, Same as C, but with PSDs normalized by total power of LFPs. E, Average (mean and SE) theta power of WT and Tau mice. F, Same as E, but normalized by total power. G, Spike theta phases versus spike positions for a place cell recorded from a young WT mouse and a cell recorded from a young Tau mouse. The phases (top) are plotted with three cycles to show phase precession. The firing rate curve of each cell (bottom) is also shown. Lines indicate linear regression results. H–J, Mean circular correlation (H), mean linear correlation (I), and mean slope of regression (J) between spike phases and spike positions across all cells recorded from young WT and Tau mice. ***p < 0.0001.
The phases of a CA1 place cell's spikes relative to the theta oscillation systematically shift earlier as the animal travels through its place field, a phenomenon known as phase precession (O'Keefe and Recce, 1993). We examined whether theta phase precession was altered in young Tau mice. Since CA1 cells in old Tau mice do not show stable place fields (Cheng and Ji, 2013), we did not analyze phase precession in old mice. CA1 cells in both Tau and WT young mice displayed phase precession (Fig. 3G). Following a previous study (Ravassard et al., 2013), we used a circular correlation between the theta phases and the positions of the spikes within a place field to quantify the overall quality of phase precession in the field. The average circular correlations across place fields in Tau and WT mice were similar (Tau, 0.27 ± 0.01; WT, 0.27 ± 0.02; t(154) = 0.18; p = 0.86; Fig. 3H). To examine phase precession at a finer level, we also computed the linear correlation between spike phases and positions, and the slope of phase decrease within a place field (O'Keefe and Recce, 1993). There was a small (21%) but significant reduction in the absolute value of the linear correlation in young Tau mice (Fig. 3I; −0.36 ± 0.01) compared with that of young WT mice (−0.45 ± 0.02; t(139) = 4.7; p < 1 × 10−5). Similarly, there was a 31% reduction in the absolute slope of phase precession in young Tau mice (Fig. 3J; Tau, −5.4 ± 0.4°/cm; WT, −7.8 ± 0.6°/cm; t(139) = 3.6; p = 0.00039), which may be related to the larger place fields in young Tau mice (Fig. 2D). Using animal averages instead of place fields as samples produced similar results. The mean circular correlation was similar between Tau and WT mice (Tau, 0.26 ± 0.02; WT, 0.29 ± 0.04; t(14) = 0.99; p = 0.34). The mean absolute linear correlation in Tau mice was slightly but significantly smaller than that in WT mice (Tau, −0.36 ± 0.02; WT, −0.45 ± 0.04; t(14) = 2.4; p = 0.032). The mean (absolute) slope of phase precession in Tau mice was lower than that in WT mice (Tau, −5.9 ± 0.7°/cm; WT, −8.0 ± 1.0°/cm), with a trend toward a significant difference (t(14) = 1.7; p = 0.10). We also examined the animal-to-animal variability of these parameters in young Tau and WT mice. A significant animal-to-animal variation was found in the precession slope within the young Tau mice (F(9,77) = 3.6; p = 0.0008), but not in other parameters (p ≥ 0.078). The analysis indicates that CA1 cells in young Tau mice showed qualitatively similar phase precession to those in young WT mice, but there were signs of subtle quantitative differences.
Reduced ripples in young and old Tau mice
The largely normal place fields in young Tau mice suggest that CA1 cells in these mice can encode spatial memories. We next examined the neurophysiological features that are currently thought to be involved in memory consolidation. One phenomenon implicated in stabilizing newly formed memories is the LFP ripple event during resting and sleep (Buzsáki, 1989; Girardeau et al., 2009; Ego-Stengel and Wilson, 2010). Therefore, we asked whether there were any ripple deficits in young Tau mice and how such deficits might progress in old Tau mice. We recorded LFPs in the CA1 pyramidal layer while animals were in SWS, a sleep stage when ripple events occur most often. A total of 24 SWS sessions from 18 young Tau mice, 14 sessions from 10 young WT mice, 5 sessions from 5 old Tau mice, and 5 sessions from 5 old WT mice, with one to three sessions per mouse, were analyzed.
We found that ripples in young Tau mice had smaller amplitudes than those in WT mice, and ripple amplitudes in old Tau mice were even more reduced, as shown by the raw and filtered (100–250 Hz) LFP traces during SWS (Fig. 4A–D). We quantified this observation by first computing the PSD of each raw LFP around the ripple band. Comparing the average PSD curves across all SWS sessions of young Tau and WT mice revealed lower power in young Tau mice than in young WT mice across a large range of frequencies (Fig. 4E; F(1,16316) = 2486; p < 1 × 10−5; ANOVA). This difference was further amplified between old Tau and WT mice (Fig. 4F; F(1,3968) = 3193; p < 1 × 10−5).
Our PSD analysis reveals that most ripple power was concentrated in the 120–180 Hz band in young and old WT mice, as shown by a peak within this frequency range in their PSD curves (Fig. 4E,F). In old Tau mice, this peak was almost absent (Fig. 4F). In young Tau mice, the peak within 120–180 Hz appeared to be less prominent than in young WT mice, particularly when examined in relation to the proceeding and lagging frequency bands (80–100 and 200–280 Hz). To confirm this observation, we normalized the PSDs by the total power within a baseline band of 80–100 Hz. Aligning the normalized PSD curves of Tau and WT mice clearly showed a smaller peak in young Tau mice (Fig. 4G; F(1,16316) = 1223; p < 1 × 10−5) and the almost disappearance of the peak in old Tau mice (Fig. 4H; F(1,3968) = 864; p < 1 × 10−5).
We then computed the absolute and normalized (relative to the power within the baseline band 80–100 Hz) ripple power within 120–180 Hz. There were significant differences in both absolute and normalized ripple power between WT and Tau mice within both the young and old age groups (Fig. 4I,J). Specifically, there was a 54% reduction in ripple power in young Tau mice compared to young WT mice (Tau, 0.0011 ± 0.0002 mV2; WT, 0.0024 ± 0.0003 mV2; t(36) = 3.5; p = 5.0 × 10−4). This reduction increased to 92% in old Tau mice compared to old WT mice (Tau, 0.00019 ± 0.00005 mV2; WT, 0.0024 ± 0.0004 mV2; t(8) = 5.1; p = 9.4 × 10−4). When measured using normalized ripple power, the reduction was 34% in young Tau mice versus young WT mice (Tau, 2.3 ± 0.1; WT, 3.5 ± 0.3; t(36) = 4.1; p = 2.3 × 10−4), and 59% in old Tau mice versus old WT mice (Tau, 1.5 ± 0.2; WT, 3.7 ± 0.2; t(8) = 8.2; p = 3.5 × 10−5). The results also reveal that the ripple power was stable across age groups in WT mice, as measured by both total ripple power (t(17) = −0.026; p = 0.98) and normalized ripple power (t(17) = −0.39; p = 0.70). In contrast, the ripple power decreased with age, as measured by both total ripple power (t(27) = 2.2; p = 0.036) and normalized ripple power (t(27) = 2.3; p = 0.032) in young versus old Tau mice.
The power analysis in the ripple band revealed a deficit in the high-frequency ripple oscillations in Tau mice. To understand what exact aspects of the ripples were altered, we next analyzed individual ripple events, defined as short-lasting (30–150 ms) bouts of high-amplitude ripple oscillations. We detected ripple events within each SWS session and quantified the amplitude and duration for each ripple event. Because many ripple events (up to thousands) were detected per SWS session, comparing all ripple events in Tau mice with those in WT mice from the same age group could yield statistically significant, yet tiny, differences due to the large number of samples. Therefore, we computed the average values of these parameters across all the ripple events in a session, resulting in one parameter value per session that we could use to compare Tau and WT mice in each age group. We also computed the occurrence rate (number of ripple events per second) for each SWS session. The median amplitude of ripple events in young Tau mice was reduced by 38% compared to young WT mice (Fig. 5A; Tau, median, 0.18 mV; [10%, 90%] range, [0.10, 0.24] mV; WT, 0.29 mV, [0.21, 0.36] mV; W(24,14) = 339; p = 1.0 × 10−4). The reduction was further increased to 90% in old Tau mice, compared to old WT mice (Tau, 0.082 mV, [0.067, 0.12] mV; WT, 0.36 mV, [0.20, 0.39] mV; W(5,5) = 15; p = 0.0079). However, there was no difference in ripple event duration either between young Tau and WT mice (Fig. 5B; Tau, 60 ms, [56, 69] ms; WT, 61 ms, [57, 72] ms; W(24,14) = 434; p = 0.31) or between old Tau and WT mice (Tau, 59 ms, [54, 67] ms; WT, 59 ms, [55, 73] ms; W(5,5) = 25; p = 0.69). Ripple events occurred at a similar rate in young Tau and WT mice (Fig. 5C; Tau, 0.71 events/s, [0.57, 1.1] events/s; WT, 0.84 events/s, [0.66, 1.0] events/s; W(24,14) = 424; p = 0.19), but occurred less frequently in old Tau mice than in old WT mice (Tau, 0.36 events/s, [0.083, 0.49] events/s; WT, 0.80 events/s, [0.46 0.87] events/s; W(5,5) = 16; p = 0.016). The results indicate that ripple events in Tau mice had smaller amplitude but similar durations, compared with their age-matched WT counterparts.
Figure 5.
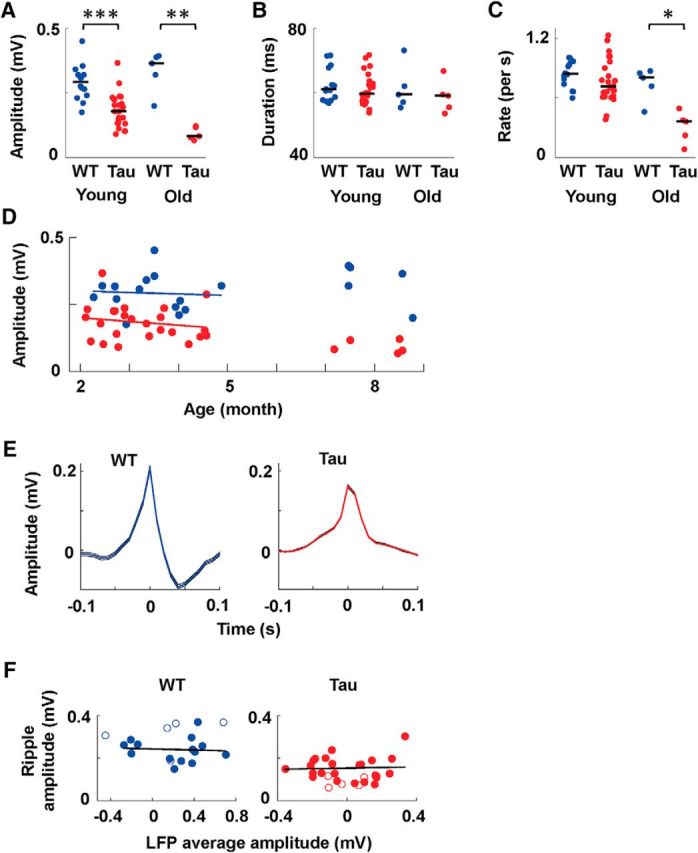
Identified ripple events during SWS were smaller in young and old Tau mice. A–C, Average amplitude (A), average duration (B), and average occurrence rate (C) of ripples during slow-wave sleep sessions for young and old WT (blue) and Tau (red) mice. Each dot is the average value of all ripples recorded in a session. Black lines are median values. D, Average ripple amplitudes of all sessions (blue, WT mice; red, Tau mice) are plotted against animal age at time of recording. Each dot represents a session. Red and blue lines are linear regressions for the red and blue dots, respectively, within the young age group (<5 months). *p < 0.05; **p < 0.01; ***p < 0.001. E, Ripple-triggered LFP averages of two example SWS sessions, one from a young WT mouse and one from a young Tau mouse. F, Ripple amplitudes plotted versus the peak amplitudes of ripple-triggered LFP averages for individual SWS sessions recorded from WT and Tau mice. Solid dots, Young mice; open circles, old mice; lines, linear regressions for the young WT and young Tau mice.
This examination of ripple events also confirms our power spectrum result that ripples were stable in WT mice between the two age groups. There was no significant difference in either ripple amplitude (W(14,5) = 124; p = 0.16) or ripple occurrence rate (W(14,5) = 151; p = 0.34) between young and old WT mice. In contrast, there was an age-dependent decline in both ripple amplitude (W(24,5) = 412; p = 0.0029) and ripple occurrence rate (W(24,5) = 418; p = 9.0 × 10−4) in Tau mice. Nevertheless, even for young mice, ripple events were already smaller than those in WT mice. In our experiments, the young age group was somewhat heterogeneous, containing mice across an age range of 2–4 months. Is it possible that ripples in Tau mice were normal in the youngest mice of this group and then gradually declined with age? To examine this, we plotted ripple amplitude versus animal age for each individual session (Fig. 5D). Regression analysis within this age group indicates that the ripple amplitude in both Tau and WT mice did not significantly depend on age (Tau, r = −0.17, N = 24, p = 0.42; WT, r = −0.059, N = 14, p = 0.84; Pearson's r). The lack of age dependence of ripple amplitude in young Tau mice suggests that the ripple deficit was already present in the youngest Tau mice (∼2 months) we could record. Around this early age, previous studies have established that while neuronal loss is likely absent, abundant abnormal tau protein is already present in Tau mice (Ramsden et al., 2005; Santacruz et al., 2005). Therefore, our results suggest that abnormal tau protein is involved in the ripple deficit in Tau mice.
High-frequency ripple oscillations are often accompanied by sharp waves (Csicsvari et al., 2000), which are presumably generated by the synchronous drive of CA1 from CA3 pyramidal neurons and could be a trigger for memory consolidation during ripples. We then examined whether sharp waves in Tau mice were altered. For each SWS session, we computed the ripple-peak-triggered LFP average. The peak amplitude of the LFP average was used to quantify the sharp waves underlying ripples (Fig. 5E). In our experiments, the recordings were made in the CA1 pyramidal cell layer, where sharp waves are relatively flat and where they change polarity from depth positive to depth negative (Ylinen et al., 1995; Csicsvari et al., 1999). Consistent with this, although the peak amplitudes in WT and Tau mice were either positive or negative, all were small (Fig. 5F; all but two sessions within [−0.5, 0.5] mV). The mean absolute value of the peak amplitudes in Tau mice was significantly smaller than that in WT mice for both the young (Tau, 0.16 ± 0.02 mV; WT, 0.33 ± 0.04 mV; t(36) = −4.5; p = 5.4 × 10−5) and old (Tau, 0.08 ± 0.01 mV; WT, 0.34 ± 0.10 mV; t(8) = −2.5; p = 0.038) age groups. The smaller peak amplitude suggests an impairment in sharp waves in Tau mice. However, given the sensitivity of sharp waves to the precise recording location within the CA1 cell layer, we cannot rule out the possibility that the change in peak amplitude could be due to a subtle difference between Tau and WT mice in the recording locations, which would be too small to detect using electrolytic lesions and histology. If so, this raises the concern that the smaller ripples in Tau mice could also be a result of recording location differences. To address this concern, we asked how much sharp-wave amplitude affected ripple amplitude within an age/genotype group. As seen in Figure 5F, we found no correlation in either the young Tau (r = −0.053; p = 0.81; Pearson's r) or young WT (r = 0.064; p = 0.83) mice (including old mouse data in the analysis produced a very similar result). Also apparent from Figure 5F is that the ripple amplitudes in Tau mice were consistently smaller than those of WT mice no matter what the corresponding sharp wave amplitudes were. Therefore, even if subtle differences in recording location were responsible for the sharp-wave change in Tau mice, they did not cause the ripple deficit, as long as the recording sites were in the CA1 pyramidal cell layer. In conclusion, our data provide supporting evidence that, in addition to ripples, the sharp waves were reduced in Tau mice and that the ripple reduction was unlikely a direct result of subtle differences in precise recording locations.
Altered firing patterns of CA1 cells in young and old Tau mice
Ripples are associated with synchronized firing of CA1 neurons (Buzsáki et al., 1992; Csicsvari et al., 2000); therefore, we next investigated the consequences of ripple reduction on firing patterns of CA1 neurons during SWS in Tau mice. A total of 205 putative pyramidal cells recorded from 13 young Tau mice, 90 cells from 5 young WT mice, 98 cells from 5 old Tau mice, and 69 cells from 4 old WT mice were included in the analysis (Table 1). The median firing rate of the cells did not significantly differ between either young Tau and WT mice (Tau, median, 0.45 Hz; [10%, 90%] range, [0.11, 1.7] Hz; WT, 0.50 Hz, [0.15, 1.5] Hz; W(205,90) = 29487; p = 0.21) or between old Tau and WT mice (Tau, 0.65 Hz, [0.10, 2.4] Hz; WT, 0.61 Hz, [0.17, 1.8] Hz; W(98,69) = 8632; p = 0.19). Because individual CA1 neurons tend to burst when ripple events occur, we first examined whether the burstiness of CA1 cells during SWS was altered in Tau mice, as quantified by a burst index. We found that the median burst index of CA1 cells in Tau mice was significantly lower than that of WT mice for both the young (Fig. 6A; Tau, 0.11, [0.0, 0.26]; WT, 0.25, [0.15, 0.37]; W(205,90) = 23601; p < 1 × 10−5) and old (Fig. 6B; Tau, 0.071, [0.024, 0.19]; WT, 0.17, [0.11, 0.26]; W(98,69) = 5782; p < 1 × 10−5) groups. The ratio of between-animal variance to within-animal variance revealed a significant animal-to-animal variation in the burst index within the young Tau mice (F(12,188) = 9.7; p < 1 × 10−5), but not within other groups (p ≥ 0.20). Even with this variability, comparing the burst index using mouse averages as samples showed reduced burst index in both the young (Tau, 0.11, [0.07, 0.27]; WT, 0.25, [0.24, 0.31]; W(5,13) = 72; p = 0.014) and old (Tau, 0.08, [0.07, 0.10]; WT, 0.17, [0.14, 0.21]; W(4,5) = 30; p = 0.016) Tau mice.
Figure 6.

A, B, Distributions of burst index for neurons recorded from young (A) and old (B) WT and Tau mice during SWS. Each plot is a histogram normalized by the total number of neurons.
We next analyzed the firing synchrony of CA1 cells at the population level by computing pairwise cross-correlations among the active cells within SWS sessions. Similar to previous studies, we used a normalized spike-count correlation that was insensitive to firing rate (Sirota et al., 2003; Wierzynski et al., 2009). We analyzed 276 pairs in young Tau, 147 pairs in young WT, 300 pairs in old Tau, and 144 pairs of cells in old WT mice. The average cross-correlograms of all four age/genotype groups displayed a prominent peak at time lag 0, indicating the existence of firing synchrony in all groups (Fig. 7A,B). However, the peaks in Tau mice were smaller than those in WT mice for both the young and old age groups. We took the mean of the cross-correlation values around the peak (within the [−20, 20] ms time lags) as the correlation of a cell pair (Wilson and McNaughton, 1994). The average correlation of young Tau mice was significantly lower than that of young WT mice (Tau, 3.7 ± 0.3; WT, 8.3 ± 0.9; t(421) = −5.4; p < 1 × 10−5). The same was true for old Tau and WT mice (Tau, 2.2 ± 0.2; WT, 5.1 ± 0.4; t(442) = −6.8; p < 1 × 10−5). We also made the comparisons using mouse averages as samples. In this case, the average correlation of Tau mice was also significantly lower than that of WT mice for both the young (Tau, 4.6 ± 0.8; WT, 8.8 ± 0.8; t(14) = −3.2; p = 0.006) and the old groups (Tau, 1.5 ± 0.6; WT, 4.7 ± 0.7; t(7) = −3.3; p = 0.013). In addition, we found a significant animal-to-animal variation in the correlation within the young (F(10,264) = 2.9; p = 0.0022) and old (F(4,295) = 2.9; p = 0.023) Tau mice, but not within the WT groups (p ≥ 0.15). This analysis reveals that firing synchrony during SWS was still present in Tau mice, but significantly reduced.
Figure 7.

Firing synchrony among CA1 neurons during SWS was reduced in young and old Tau mice. A, B, Average pairwise cross-correlograms during SWS for young (A) and old (B) WT and Tau mice. Thin lines are the mean ± SE. Note the smaller peaks in the average cross-correlograms of Tau mice, compared to those of age-matched WT controls. C, Mean pairwise correlation, defined as the average cross-correlation value within the [−20, 20] ms time lag, for cell pairs recorded from young and old WT and Tau mice. ***p < 0.00001.
Discussion
To understand how neurophysiological features related to memory processing are affected by tau pathology in vivo, we examined hippocampal CA1 place cells and LFPs in freely behaving Tau and WT mice. We found that place cells in young Tau mice are largely normal, yet upon closer inspection they may be less stable and have slightly larger place fields than in WT controls. These subtle differences are in sharp contrast to the dramatic decrease in the spatial specificity of CA1 cells in old Tau mice (Cheng and Ji, 2013). At the LFP level, theta oscillations remain prominent in both young and old Tau mice (Cheng and Ji, 2013). However, we found that both ripple power and individual ripple events during SWS are reduced in young Tau mice, a deficit that becomes even greater in old Tau mice. The ripple reduction is accompanied by less burstiness of CA1 cells and reduced firing synchrony of CA1 cells during SWS. These results reveal an age-related progression of functional deficits in hippocampal neurons of freely behaving animals with ongoing tauopathy.
These neurophysiological deficits in the hippocampus in vivo occur in parallel with behavioral deficits in young and old Tau mice. Previous studies have found little to no spatial memory deficit in 2.5-month-old Tau mice, with memory impairments becoming apparent at ≥4.5 months of age, as measured by the Morris water maze task (Ramsden et al., 2005; Santacruz et al., 2005). We found that place fields can form in young Tau mice and contain a similar amount of spatial information to age-matched WT controls, yet CA1 cells in old Tau mice display much less distinct place fields on novel and familiar tracks (Cheng and Ji, 2013). Nevertheless, there are subtle, quantitative differences between place fields of young Tau and WT mice; namely, place fields of young Tau mice are slightly larger and less stable across sessions, differences that may be too subtle to impact a behavioral test such as the Morris water maze. In contrast, we found that ripples and firing synchrony in the young Tau mice are already altered and that the ripple reduction increases as Tau mice age. The appearance of relatively normal memory performance at the same age that we observe relatively normal place fields in young Tau mice, despite reduced ripples, suggests that memory impairment at the behavioral level appears to be more associated with the degree of place field abnormality, consistent with a previous study on an Aβ-related model of AD (Cacucci et al., 2008), than that of ripples. On the other hand, the same results support the notion that significant functional changes in the hippocampal network already start to occur in young Tau mice, before the manifestation of overt behavioral impairment at later ages, which are likely produced by both the cell loss and the dysfunction of existing neurons described in our studies.
Our findings suggest that the neural processes that contribute to memory encoding and consolidation are impaired in Tau mice. Current theories propose that new spatial memories are encoded by the place fields of populations of place cells (cell assemblies) that are functionally linked together during novel experiences, likely via mechanisms related to synaptic plasticity (Wilson and McNaughton, 1993; Eichenbaum et al., 1999; Harris et al., 2003; Cacucci et al., 2007; Resnik et al., 2012). Synchronized firing of the cell assemblies stabilizes newly formed memory codes during ripple events, which induces further synaptic plasticity for memory consolidation (Buzsáki, 1989). In young Tau mice, ripples and firing synchrony are altered, suggesting that the memory codes may be improperly consolidated and consequently less stable. However, our data also show that place fields are largely normal in young Tau mice, meaning their hippocampal cells can form relatively normal memory codes, which could support the normal spatial memory performance seen previously at this age (Ramsden et al., 2005; Santacruz et al., 2005). In old Tau mice, new specific place fields, and consequently, new cell assemblies, fail to form in novel experiences (Cheng and Ji, 2013), suggesting an impairment in memory encoding. Our results thus raise the possibility of abnormal memory consolidation early on, which results in unstable memory codes, followed by an eventual loss of the ability to form new memory codes altogether. How much this process contributes to the decline in behavioral performance, however, is difficult to speculate, because the behavioral impairment could also be produced by neurodegeneration itself in old Tau mice.
Looking beyond behavior, the escalating deficits in place fields and ripples also parallel the progression of tau pathology and neurodegeneration. What pathological features—abnormal tau protein, tau neurofibrillary tangles, or neuronal loss—are responsible for the observed functional deficits? Our results show that ripples are significantly reduced in young Tau mice, starting as early as ∼2 months, an age when abnormal tau protein is abundant but neuronal loss seems minimal, and mature tangles are yet to occur in the hippocampus (Ramsden et al., 2005). Although we cannot rule out the possible subtle neurodegeneration in young Tau mice, our observations suggest that the ripple deficit can be produced by abnormal tau protein. On the other hand, place fields are subtly altered in young Tau mice, but severely impaired in old Tau mice when mature tangles are prolific and neuronal loss is severe (Ramsden et al., 2005; Santacruz et al., 2005). Therefore, abnormal place fields are more likely linked to tau tangles and/or neurodegeneration. Several lines of evidence suggest that tau tangles may not be detrimental at the cellular and behavioral levels. Halting neurodegeneration without clearing neurofibrillary tangles seems to improve the memory performance of old Tau mice (Santacruz et al., 2005). Cortical neurons with tau tangles in old Tau mice are functionally normal in vivo (Kuchibhotla et al., 2014). Finally, the morphological and electrophysiological features of tau tangle positive and negative neurons in cortical slices of old Tau mice are indistinguishable (Rocher et al., 2010). Thus, it is likely that the place field deficit observed in old Tau mice is mainly caused by the severe neurodegeneration present at this age. Together, our data support the idea that overt place field impairment may require apparent neurodegeneration, whereas abnormal tau protein may be sufficient to cause the ripple deficit.
Interestingly, our findings of ripple and place field deficits in vivo also parallel the results of a previous in vitro electrophysiological study on cortical neurons of Tau mice (Crimins et al., 2012). In this study, a few electrophysiological parameters, including resting membrane potential and evoked firing rate, are abnormal in cortical neurons of Tau mice at young ages (<4 months), and these abnormalities persist in Tau mice at old ages (>8 months). In contrast, atrophic-like morphological changes are only robustly observed in cortical neurons of old Tau mice, independent of the presence of neurofibrillary tangles. In our study, although we do not find a consistent change in the firing rates of CA1 neurons, deficits in ripples and burstiness occur early in young Tau mice and persist in old Tau mice. Robust deficits in place fields of the recorded CA1 neurons are only observed in old Tau mice. Based on these parallel in vitro and in vivo findings, we speculate that the ripple deficit is due to changes in the intrinsic properties of individual neurons, possibly resulting from abnormal tau protein, and the place field deficit is due to the circuit-level change that occurs as a result of neurodegeneration. Although the ripple deficit was observed at the earliest age of our recording, it is unlikely produced by abnormal development in Tau mice. Previous studies have shown that the development of Tau mice is normal (Ramsden et al., 2005). In addition, the abnormal tau protein is expressed via a CamKII promoter, which is activated only after birth (Mayford et al., 1996).
How can abnormal tau protein produce the ripple deficit? We know that ripples are triggered by CA3 input but are most prominent within the CA1 pyramidal layer (Csicsvari et al., 2000; Behrens et al., 2005). Unfortunately, how CA1 neurons are synchronized during ripples is not well understood. Previous studies show that precise interactions among local CA1 pyramidal neurons and inhibitory interneurons are crucial in ripple generation (Ylinen et al., 1995; Rácz et al., 2009; Stark et al., 2014). It is possible that the presence of abnormal tau may alter the properties of CA1 pyramidal neurons and impair their precise firing patterns. Second, the abnormal tau could also alter inhibition in the hippocampal circuits. Since the CamKII promoter is rarely expressed in GABAergic interneurons (Mayford et al., 1996; Sík et al., 1998), abnormal tau should be absent from interneurons. However, there could be adaptations in interneurons to the altered intrinsic properties of the abnormal tau-bearing pyramidal neurons (Rocher et al., 2010; Crimins et al., 2012). Finally, there is evidence for the involvement of gap junctions in ripple generation (Maier et al., 2002, 2003; but see Buhl et al., 2003). Another possibility, then, is that abnormal tau may change the gap junction coupling in the CA1.
We observed that significant ripple deficits occur earlier than overt place field abnormality in Tau mice. This brings into question what the relationship is between ripples and place fields. If ripples are involved in the consolidation of spatial codes as current theories suggest, how can place fields in familiar environments be normal if ripples are reduced in young Tau mice? The answer could lie in the fact that place fields in familiar environments do not just result from previously formed memories, but contain a response to the sensory cues in the environment, probably to an even greater degree (Muller and Kubie, 1987; Siegel et al., 2008). The largely normal place fields suggest that young Tau mice are able to generate a spatial representation from environmental cues. As long as the cues are stable, this representation is maintained. Therefore, seemingly normal place fields can coexist with reduced ripples in the CA1 of Tau mice.
Footnotes
This work supported by NINDS Grants NS075726 (D.J.) and NS048884 (H.-C. L.).
The authors declare no competing financial interests.
References
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens CJ, van den Boom LP, de Hoz L, Friedman A, Heinemann U. Induction of sharp wave-ripple complexes in vitro and reorganization of hippocampal networks. Nat Neurosci. 2005;8:1560–1567. doi: 10.1038/nn1571. [DOI] [PubMed] [Google Scholar]
- Buhl DL, Harris KD, Hormuzdi SG, Monyer H, Buzsáki G. Selective impairment of hippocampal gamma oscillations in connexin-36 knock-out mouse in vivo. J Neurosci. 2003;23:1013–1018. doi: 10.1523/JNEUROSCI.23-03-01013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. Two-stage model of memory trace formation: a role for “noisy” brain states. Neurosci. 1989;31:551–570. doi: 10.1016/0306-4522(89)90423-5. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/S0896-6273(02)00586-X. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Horvath Z, Urioste R, Hetke J, Wise K. High-frequency network oscillation in the hippocampus. Science. 1992;256:1025–1027. doi: 10.1126/science.1589772. [DOI] [PubMed] [Google Scholar]
- Cacucci F, Wills TJ, Lever C, Giese KP, O'Keefe J. Experience-dependent increase in CA1 place cell spatial information, but not spatial reproducibility, is dependent on the autophosphorylation of the alpha-isoform of the calcium/calmodulin-dependent protein kinase II. J Neurosci. 2007;27:7854–7859. doi: 10.1523/JNEUROSCI.1704-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacucci F, Yi M, Wills TJ, Chapman P, O'Keefe J. Place cell firing correlates with memory deficits and amyloid plaque burden in Tg2576 Alzheimer mouse model. Proc Natl Acad Sci U S A. 2008;105:7863–7868. doi: 10.1073/pnas.0802908105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlesimo GA, Oscar-Berman M. Memory deficits in Alzheimer's patients: a comprehensive review. Neuropsy Rev. 1992;3:119–169. doi: 10.1007/BF01108841. [DOI] [PubMed] [Google Scholar]
- Cheng J, Ji D. Rigid firing sequences undermine spatial memory codes in a neurodegenerative mouse model. Elife. 2013;2:e00647. doi: 10.7554/eLife.00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimins JL, Rocher AB, Luebke JI. Electrophysiological changes precede morphological changes to frontal cortical pyramidal neurons in the rTg4510 mouse model of progressive tauopathy. Acta Neuropathologica. 2012;124:777–795. doi: 10.1007/s00401-012-1038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurkó A, Mamiya A, Buzsáki G. Fast network oscillations in the hippocampal CA1 region of the behaving rat. J Neurosci. 1999;19:RC20. doi: 10.1523/JNEUROSCI.19-16-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Mamiya A, Buzsáki G. Ensemble patterns of hippocampal CA3-CA1 neurons during sharp wave-associated population events. Neuron. 2000;28:585–594. doi: 10.1016/S0896-6273(00)00135-5. [DOI] [PubMed] [Google Scholar]
- Ego-Stengel V, Wilson MA. Disruption of ripple-associated hippocampal activity during rest impairs spatial learning in the rat. Hippocampus. 2010;20:1–10. doi: 10.1002/hipo.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenbaum H, Dudchenko P, Wood E, Shapiro M, Tanila H. The hippocampus, memory, and place cells: is it spatial memory or a memory space? Neuron. 1999;23:209–226. doi: 10.1016/S0896-6273(00)80773-4. [DOI] [PubMed] [Google Scholar]
- Euston DR, Tatsuno M, McNaughton BL. Fast-forward playback of recent memory sequences in prefrontal cortex during sleep. Science. 2007;318:1147–1150. doi: 10.1126/science.1148979. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, García-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp Neurol. 2002;177:475–493. doi: 10.1006/exnr.2002.8014. [DOI] [PubMed] [Google Scholar]
- Gilmour TP, Fang J, Guan Z, Subramanian T. Manual rat sleep classification in principal component space. Neurosci Lett. 2010;469:97–101. doi: 10.1016/j.neulet.2009.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB. Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci. 2009;12:1222–1223. doi: 10.1038/nn.2384. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- Harris KD, Csicsvari J, Hirase H, Dragoi G, Buzsáki G. Organization of cell assemblies in the hippocampus. Nature. 2003;424:552–556. doi: 10.1038/nature01834. [DOI] [PubMed] [Google Scholar]
- Ji D, Wilson MA. Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat Neurosci. 2007;10:100–107. doi: 10.1038/nn1825. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Wegmann S, Kopeikina KJ, Hawkes J, Rudinskiy N, Andermann ML, Spires-Jones TL, Bacskai BJ, Hyman BT. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci U S A. 2014;111:510–514. doi: 10.1073/pnas.1318807111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Wilson MA. Memory of sequential experience in the hippocampus during slow wave sleep. Neuron. 2002;36:1183–1194. doi: 10.1016/S0896-6273(02)01096-6. [DOI] [PubMed] [Google Scholar]
- Maier N, Güldenagel M, Söhl G, Siegmund H, Willecke K, Draguhn A. Reduction of high-frequency network oscillations (ripples) and pathological network discharges in hippocampal slices from connexin 36-deficient mice. J Physiol. 2002;541:521–528. doi: 10.1113/jphysiol.2002.017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier N, Nimmrich V, Draguhn A. Cellular and network mechanisms underlying spontaneous sharp wave-ripple complexes in mouse hippocampal slices. J Physiol. 2003;550:873–887. doi: 10.1113/jphysiol.2003.044602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Mehta MR, Lee AK, Wilson MA. Role of experience and oscillations in transforming a rate code into a temporal code. Nature. 2002;417:741–746. doi: 10.1038/nature00807. [DOI] [PubMed] [Google Scholar]
- Muller RU, Kubie JL. The effects of changes in the environment on the spatial firing of hippocampal complex-spike cells. J Neurosci. 1987;7:1951–1968. doi: 10.1523/JNEUROSCI.07-07-01951.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Recce ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus. 1993;3:317–330. doi: 10.1002/hipo.450030307. [DOI] [PubMed] [Google Scholar]
- Rácz A, Ponomarenko AA, Fuchs EC, Monyer H. Augmented hippocampal ripple oscillations in mice with reduced fast excitation onto parvalbumin-positive cells. J Neurosci. 2009;29:2563–2568. doi: 10.1523/JNEUROSCI.5036-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravassard P, Kees A, Willers B, Ho D, Aharoni D, Cushman J, Aghajan ZM, Mehta MR. Multisensory control of hippocampal spatiotemporal selectivity. Science. 2013;340:1342–1346. doi: 10.1126/science.1232655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik E, McFarland JM, Sprengel R, Sakmann B, Mehta MR. The effects of GluA1 deletion on the hippocampal population code for position. J Neurosci. 2012;32:8952–8968. doi: 10.1523/JNEUROSCI.6460-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Guilpin C, Limoge A. Automated sleep staging systems in rats. J Neurosci Methods. 1999;88:111–122. doi: 10.1016/S0165-0270(99)00027-8. [DOI] [PubMed] [Google Scholar]
- Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, Luebke JI. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2010;223:385–393. doi: 10.1016/j.expneurol.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon DP, Bondi MW. Neuropsychological assessment of dementia. Ann Rev Psych. 2009;60:257–282. doi: 10.1146/annurev.psych.57.102904.190024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siapas AG, Wilson MA. Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron. 1998;21:1123–1128. doi: 10.1016/S0896-6273(00)80629-7. [DOI] [PubMed] [Google Scholar]
- Siegel JJ, Neunuebel JP, Knierim JJ. Dominance of the proximal coordinate frame in determining the locations of hippocampal place cell activity during navigation. J Neurophysiol. 2008;99:60–76. doi: 10.1152/jn.00731.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sík A, Hájos N, Gulácsi A, Mody I, Freund TF. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci U S A. 1998;95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirota A, Csicsvari J, Buhl D, Buzsáki G. Communication between neocortex and hippocampus during sleep in rodents. Proc Natl Acad Sci U S A. 2003;100:2065–2069. doi: 10.1073/pnas.0437938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaggs WE, McNaughton BL, Gothard KM, Markus EJ. An information-theoretic approach to deciphering the hippocampal code. In: Hanson SJ, Cowan JD, Giles CJ, editors. Advances in neural information processing systems. San Mateo, CA: Morgan Kaufmann; 1993. pp. 1030–1037. [Google Scholar]
- Stark E, Roux L, Eichler R, Senzai Y, Royer S, Buzsáki G. Pyramidal cell-interneuron interactions underlie hippocampal ripple oscillations. Neuron. 2014;83:467–480. doi: 10.1016/j.neuron.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh J, Foster DJ, Davoudi H, Wilson MA, Tonegawa S. Impaired hippocampal ripple-associated replay in a mouse model of schizophrenia. Neuron. 2013;80:484–493. doi: 10.1016/j.neuron.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL. Neuropathologic changes in Alzheimer's disease. J Clinic Psychiarty. 2003;64(Suppl 9):7–10. [PubMed] [Google Scholar]
- Wierzynski CM, Lubenov EV, Gu M, Siapas AG. State-dependent spike-timing relationships between hippocampal and prefrontal circuits during sleep. Neuron. 2009;61:587–596. doi: 10.1016/j.neuron.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Dynamics of the hippocampal ensemble code for space. Science. 1993;261:1055–1058. doi: 10.1126/science.8351520. [DOI] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- Ylinen A, Bragin A, Nádasdy Z, Jandó G, Szabó I, Sik A, Nuzsaki G. Sharp wave-associated high-frequency oscillation (200 Hz) in the intact hippocampus: network and intracellular mechanisms. J Neurosci. 1995;15:30–46. doi: 10.1523/JNEUROSCI.15-01-00030.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]