

Abstract
All synapses show activity-dependent changes in strength, which affect the fidelity of postsynaptic spiking. This is particularly important at auditory nerve synapses, where the presence and timing of spikes carry information about a sound's structure, which must be passed along for proper processing. However, it is not clear how synaptic plasticity influences spiking during ongoing activity. Under these conditions, conventional analyses erroneously suggest that synaptic plasticity has no influence on EPSC amplitude or spiking. Therefore, we developed new approaches to study how ongoing activity influences synaptic strength, using voltage- and current-clamp recordings from bushy cells in brain slices from mouse anteroventral cochlear nucleus. We applied identical trains of stimuli, except for one skipped stimulus, and found that EPSC amplitude was affected for 60 ms following a skipped stimulus. We further showed that the initial probability of release, calcium-dependent mechanisms of recovery, and desensitization all play a role even during ongoing activity. Current-clamp experiments indicated that these processes had a significant effect on postsynaptic spiking, as did the refractory period to a smaller extent. Thus short-term plasticity has real, important functional consequences.
Keywords: depression, endbulb, short-term plasticity, synapse
Introduction
Activity-dependent changes in synaptic strength have been evident since the very first recordings of synaptic potentials (Eccles et al., 1941; Feng, 1941). These include a number of processes, collectively referred to as short-term plasticity (STP), and have been a subject of intense interest particularly for revealing the mechanisms underlying neurotransmitter release and recovery (Magleby, 1987; Regehr and Stevens, 2001; Zucker and Regehr, 2002). STP appears to be a basic and defining property of synapses (Reyes et al., 1998), and likely contributes to neuronal processing in some systems (Fortune and Rose, 2001). However, it has also been unclear for a long time how STP acts in vivo, where many neurons are constantly active and synapses have limited time for recovery.
This is a particular issue in the auditory system, where spontaneous activity levels can be high (> 60 Hz), and increase further in the presence of sound, up to peak rates of 200–300 Hz (Kiang, 1965; Sachs and Abbas, 1974; Johnson, 1980). Auditory nerve synapses, called “endbulbs of Held,” show strong depression in vitro (Isaacson and Walmsley, 1996; Oleskevich et al., 2000; Wang and Manis, 2008; Yang and Xu-Friedman, 2008; Cao and Oertel, 2010), but these experiments are typically conducted with long recovery times between stimuli, so it is not clear how relevant STP is during ongoing activity. Indeed, recent experiments at endbulb synapses onto bushy cells (BCs) in the anteroventral cochlear nucleus (AVCN) suggest that STP is absent, and spiking depends primarily on the action potential refractory period and random fluctuations in EPSP amplitude (Kuenzel et al., 2011). A similar conflict is evident with the calyx of Held synapse in the medial nucleus of the trapezoid body (MNTB), which shows clear short-term depression in vitro (von Gersdorff and Borst, 2002; Wong et al., 2003; Müller et al., 2010; Grande and Wang, 2011), but the effects of this depression are not obvious in vivo (Lorteije et al., 2009). This has suggested that STP is absent or reduced in vivo, raising doubts about the relevance of STP to neuronal computation.
Yet, there are many differences between such in vivo and in vitro experiments. One major difference is the existence of ongoing activity in vivo, which could fundamentally alter the nature of STP or alternatively could render the conventional assays of STP inapplicable. We studied this issue at the endbulb of Held by applying conventional and novel approaches, which revealed that STP is active during ongoing activity. A new approach using skipped stimuli allowed us to quantitatively assess the importance of different aspects of STP and show that mechanisms important in rested synapses continue to influence synaptic strength even at steady state. We used dynamic clamp to show that these influences on synaptic strength have consequences on spiking. Thus, STP influences the transfer of information across synapses, even during ongoing activity similar to in vivo.
Materials and Methods
All procedures were approved by the Institutional Animal Care and Use Committee at the University at Buffalo. Brain slices were prepared from the cochlear nucleus of CBA/CaJ mice of either sex aged P15 to 30 (average age, 17.4 d postnatal). Slices were cut at a slight angle from sagittal, to best preserve straight projections of the auditory nerve, using an Integraslice 7500 MM (Campden Instruments) at 170–200 μm thickness. Slices were cut into low-sodium, ice-cold cutting solution containing the following (in mm): 76 NaCl, 26 NaHCO3, 75 sucrose, 1.25 NaH2PO4, 2.5 KCl, 25 glucose, 7 MgCl2, 0.5 CaCl2, bubbled with 95% O2 and 5% CO2 (pH 7.8, 305 mOsm), and incubated at 30°C for 20 min in standard recording solution containing the following (in mm): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 20 glucose, 1 MgCl2, 1.5 CaCl2, 4 Na l-lactate, 2 Na-pyruvate, and 0.4 Na l-ascorbate bubbled with 95% O2 and 5% CO2 (pH 7.4, 310 mOsm). In some experiments, slices were instead cut into recording solution and incubated at 30°C for 15 min in solution containing the following (in mm): 93 NMDG, 93 HCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na l-ascorbate, 3 Na-pyruvate, 10 MgCl2, and 0.5 CaCl2. After that, slices were incubated in recording solution. For fiber-stimulation experiments in Figure 7, 5 μm CPP was also included to block NMDA receptors. Patch electrodes contained the following (in mm): (voltage clamp) 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, 1 QX-314, pH 7.3, 311 mOsm, or (current/dynamic clamp) 130 KMeSO3, 10 NaCl, 2 MgCl2, 0.16 CaCl2, 0.5 EGTA, 10 HEPES, 4 Na2ATP, 0.4 NaGTP, 14 phosphocreatine di(tris), pH 7.3, 305 mOsm.
Figure 7.
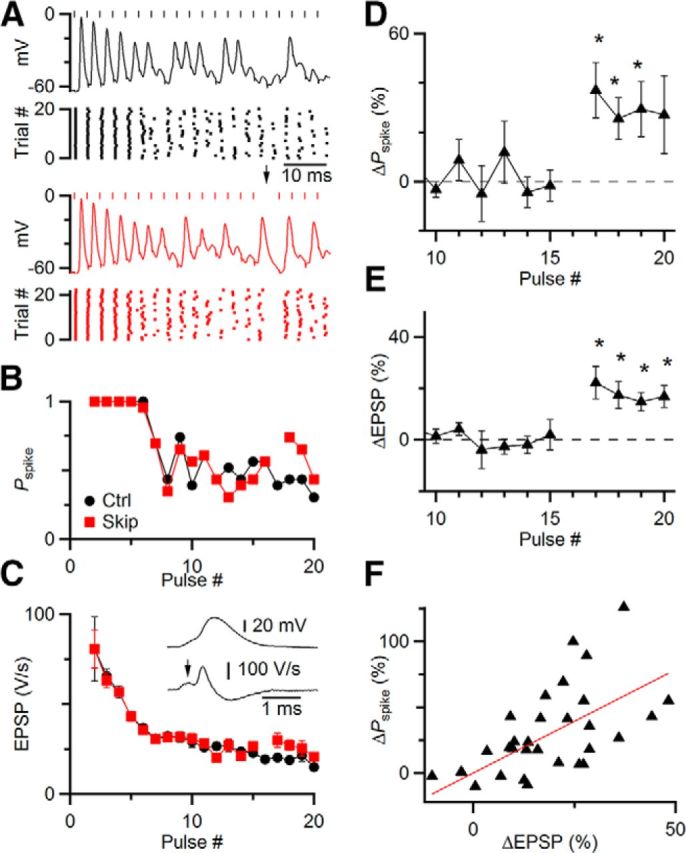
Skipping stimuli has functional consequences, increasing the probability of spiking (Pspike). A–C, Representative current-clamp experiment. A single endbulb was stimulated at 333 Hz in regular trains in the presence of 5 μm CPP, in control (A, top) and with one skipped stimulus (A, bottom). Rasters show spike timing over many trials. Pspike (B) and EPSP amplitude (C) declined over the train but increased immediately after the skipped stimulus. C, Inset, Method of estimating EPSP amplitude from the derivative of membrane potential. D–F, Average results from 8 similar experiments, showing a significant increase in Pspike and EPSP amplitude following a skipped stimulus. Asterisks indicate significant increases in D and E (p < 0.05). F, The increase in Pspike was strongly correlated with the increase in EPSP for the 4 pulses following the skipped stimulus in 8 experiments (r = 0.42).
Recordings were made at 32°C using a Multiclamp 700B controlled by an Instrutech ITC-18 digital interface through Wavemetrics Igor running custom-written routines. Patch electrodes had resistances of 1.5–2.5 MΩ, yielding series resistances of 5–10 MΩ, which were compensated to 70%. Individual auditory nerve fibers were stimulated in the neuropil using a 3–5 μm electrode connected to a stimulus isolator (WPI, A365), passing 14 μA of current for 0.2 ms. Dynamic clamp was performed using the ITC-18 interface at 50 kHz. Individual EPSCs were derived by convolving a unitary EPSC with amplitudes based on a stochastic model of neurotransmitter release (Yang and Xu-Friedman, 2013). Briefly, this model tracks vesicle release, depletion, and recovery at a set of release sites using a binomial distribution. The model also includes desensitization and activity-dependent recovery. The parameters used to derive conductances were as follows: P0 = 0.4, k0 = 0.5/s, kmax = 7/s, τD = 10 ms, KD = 0.05, τS = 5 ms, KS = 1 NV, NS = 60, NV = 3. The conductance amplitudes for each cell were scaled according to its conductance threshold. Threshold was measured with 0.5 nS resolution by applying AMPA-like conductances, and finding the minimum conductance amplitude that drove spiking with an adaptive procedure. The initial conductance of dynamic-clamp trains was scaled to 5–6 times this value. Fiber stimulation trials were separated by 30 s, and dynamic-clamp trials were separated by 0.2 s. Spike probability was calculated by counting the number of spikes elicited by a given stimulus pulse on each trial, and normalizing by the number of trials. All statistical tests are paired Student's t tests, except where otherwise noted.
Results
We first applied standard approaches to characterize short-term plasticity during ongoing activity. We made whole-cell recordings from BCs in brain slices of mice and stimulated individual auditory nerve fibers in long Poisson-distributed trains. Under these conditions, the EPSCs, mediated by AMPA receptors, showed considerable variability in amplitude (Fig. 1A). This variability could result from stochastic processes, such as the randomness of neurotransmitter release, as well as deterministic processes, such as the various forms of STP. One simple test for STP is to examine how one EPSC affects the subsequent EPSC. Because endbulb EPSCs usually depress in vitro, we would predict that particularly large EPSCs should deplete neurotransmitter and cause the next EPSC to be smaller than the average, as occurs in synapses at rest (Debanne et al., 1996). Small EPSCs should preserve enough transmitter that the next can be larger than average. Thus, a plot of one EPSC (EPSCi) against the following EPSC (ESPCi+1) is expected to be negatively correlated. However, there was only a very weak correlation, and in the opposite direction from expected (slope = 0.09 ± 0.05; Fig. 1B). Similar results were found in 5 cells, with no significant correlation (r = 0.05 ± 0.05, p = 0.1, t test; Fig. 1B, inset).
Figure 1.
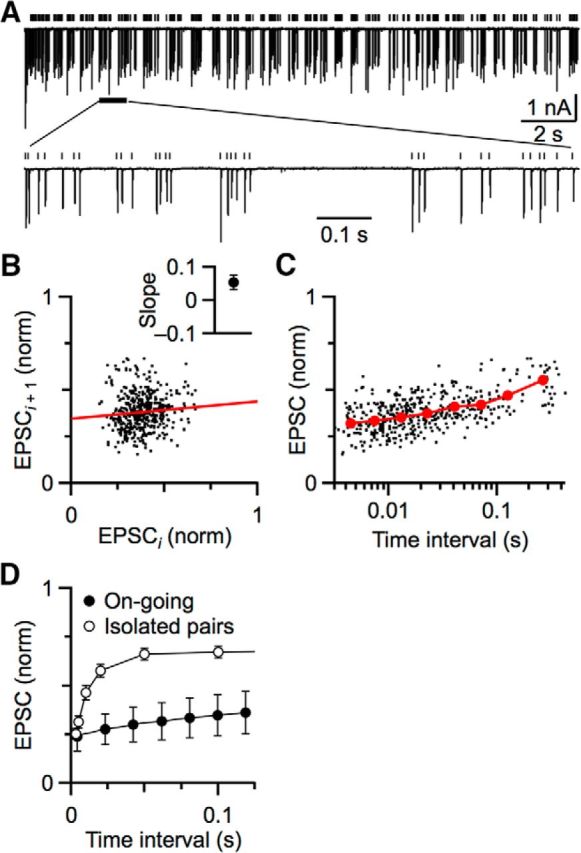
Simple analyses of EPSC amplitudes at the endbulb synapse fail to reveal short-term plasticity during ongoing activity. A, EPSCs were evoked by stimulating single auditory nerve fibers for 20 s at 25 Hz. Bottom, Portion of the data on an expanded timescale. Vertical lines indicate the timing of stimulations. B, Lack of correlation between adjacent EPSCs. Each EPSC in the train in A is designated as EPSCi, and the subsequent EPSC as EPSCi + 1. The slope of the fit (0.09 ± 0.05) indicates no correlation. Inset, Average slope from 5 cells, which is not significantly different from 0 (p = 0.1). C, Weak effect of previous interval on subsequent EPSC amplitude. Each dot is one EPSC from the train plotted against its previous interval. Red circles represent binned averages of 440 EPSCs. D, Average relationship between EPSC and previous interval during ongoing activity from 5 experiments (closed circles). For comparison, we show the same relationship when synapses were activated in pairs of stimuli of various intervals, with paired stimuli well isolated in time from each other (open circles). Data are from Yang and Xu-Friedman (2009). These “rested” synapses show faster and greater recovery.
We also used a second approach to detect STP, taking into account the interval between stimuli. We would predict that, for two closely spaced stimuli, there would be little time for recovery, so the synapse should depress, whereas with longer pauses, sufficient recovery could take place that the EPSC would be larger. When we plotted EPSC amplitude against previous interval, some recovery was visible at long times (Fig. 1C), but it was slow and limited. Similar results were found for 5 cells (Fig. 1D, closed symbols), with an average τ of recovery of 90.6 ± 6.5 ms. By contrast, as we and others have shown before, recovery of an endbulb stimulated in simple pairs is greater and faster (Fig. 1D, open symbols), with average recovery τfast of 10 ± 1 ms and τslow of 2.0 ± 0.9 s (Yang and Xu-Friedman, 2009). Furthermore, high activity activates calcium-dependent forms of recovery, which appear to speed recovery from vesicle depletion (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998). At the endbulb, this form of recovery has a τ of 45 ± 4 ms (Yang and Xu-Friedman, 2008). Thus, the analysis approaches used so far show little evidence of these well-described mechanisms during ongoing activity, suggesting that STP may be different.
However, different analyses seem to indicate that deterministic processes do operate during ongoing activity. In hippocampal slices, applying the same stimulus train multiple times triggered virtually identical field potentials each time (Dobrunz and Stevens, 1999), suggesting that deterministic processes underlie the changes in field potential. It is not clear whether individual synapses are so consistent. We tested this by stimulating single auditory nerve fibers repeatedly with the same random train. EPSC amplitudes showed the effects of both stochastic and deterministic processes. An example experiment is shown in Figure 2A. Each trial showed different amplitudes for a given EPSC in the train (Fig. 2A, colored markers). We have shown before that this variability can have a large influence on the function of the synapse (Yang and Xu-Friedman, 2013). In addition, EPSC amplitude showed predictable trends from one trial to the next, where on most trials, the EPSC showed similar increases and decreases. These deterministic changes appear to indicate the presence of STP.
Figure 2.

Deterministic processes underlie EPSC amplitude. A, A representative experiment showing that EPSC amplitudes are highly repeatable on multiple presentations. The same random train was repeated 6 times. Colored bars indicate EPSC amplitudes from each trial. Black trace represents the average EPSC. There was considerable variability from trial to trial, but there were also consistent trends across trials. B, C, EPSC amplitudes from different trials are highly correlated. B, EPSCs at the same position in the train in A, but on different trials, are plotted against each other and fit with a line. Red dots represent the first 10 EPSCs of the train. Black dots represent the subsequent EPSCs, which are at steady state. In this representative experiment, the EPSCs on different trials were highly correlated, with a slope of 0.95 ± 0.01 for all EPSCs (red line), and 0.96 ± 0.01 excluding the first 10 EPSCs (black line). C, Similar results were seen in four experiments. For clarity, just the linear fits to the steady-state EPSCs are shown. Inset, Average slope (all EPSCs: 0.94 ± 0.05, open symbol; steady-state EPSCs: 0.94 ± 0.06, closed symbol). This indicates that EPSC amplitude during ongoing activity is subject to both random and deterministic processes.
To quantify the contribution of the deterministic component to EPSC amplitude, we measured EPSC amplitude for 6 trials of a representative cell. We refer to EPSCi on trial j as EPSCij. We plotted the amplitude on each trial j against the corresponding EPSCi in each of the other 5 trials k (EPSCik, j ≠ k; Fig. 2B) and normalized to the overall average of the initial EPSC amplitude. The EPSC amplitudes were strongly correlated between trials whether all EPSCs were included in the analysis (r = 0.80), or the first 10 EPSCs (Fig. 2B, red dots) were excluded to focus on the steady state (r = 0.71). The slope was very close to unity both including (0.95 ± 0.01; Fig. 2B, red line) and excluding the first 10 EPSCs (0.96 ± 0.01; Fig. 2B, black line). Similar results were found in 4 cells, with highly significant correlation coefficients of 0.83 ± 0.10 when including and 0.74 ± 0.09 when excluding the first 10 EPSCs. Average slopes were 0.94 ± 0.05 including and 0.94 ± 0.06 excluding the first 10 EPSCs (Fig. 2C; p ≪ 0.001).
The variance in EPSC amplitude could result from two sources: stochastic variability resulting from the probabilistic nature of neurotransmitter release and deterministic variability resulting from activity-dependent processes of STP. To estimate the contribution of STP to EPSC variance, we used the correlation coefficients from the linear regression. The value of r2 equals 1 − SSresidual/SStotal, where SSresidual is the residual sum of squares difference between the linear fit and the data, and SStotal is the total variance about the mean. Thus, r2 represents the fraction of the variance in EPSC amplitude accounted for by the fit (Glantz, 1997). For these experiments, r2 was 55 ± 15%. In other words, 55% of the variance in EPSC amplitude was deterministic, leaving 45% of the EPSC variance resulting from stochastic variability. Thus, a major determinant of EPSC amplitude during ongoing activity is STP.
This correlation approach is valuable for revealing the contribution of deterministic processes to EPSC amplitude, but it does not provide insight into the underlying mechanisms that lead to the moment-by-moment EPSC amplitude. We have already shown that the analysis approaches in Figure 1 are inadequate, perhaps because they only treat a single previous EPSC. We considered the impact of a longer history of activity by summing the normalized EPSC amplitudes over a window of 200 ms preceding each EPSCi. An example is shown in Figure 3A, where the single EPSC indicated by the arrow is preceded by 4 EPSCs over the previous 200 ms (Fig. 3A, red bar), whose amplitudes are summed. This analysis showed a clear, significant correlation between activity level and EPSC amplitude (slope = −0.05, r = −0.50; Fig. 3B). To test the specificity of this approach, we shifted the window earlier by 25 ms (blue bar) and 100 ms (magenta bar), and saw progressively shallower slopes and weaker correlations (25 ms: slope = −0.03 and r = −0.32; Fig. 3C; 100 ms: slope = −0.01 and r = −0.11; Fig. 3D). We obtained similar results for 6 cells (Fig. 3E), with significant correlations for windows ending 0 (p < 0.001), 25 ms (p < 0.01), and 100 ms before the EPSC (p < 0.05). Thus, the amplitude of an EPSC is influenced by the immediately preceding activity.
Figure 3.

Relating EPSC amplitude to longer history of activity. A, Illustration of the analysis method. EPSCs are collected during a long random train. For each EPSC in the train (e.g., arrow), EPSC amplitudes are summed over a preceding 200 ms window (colored bars). The window position was shifted from 0 to 100 ms before the EPSC. B–D, Correlation of EPSC amplitude with previous activity, for windows shifted 0 (B), 25 (C), and 100 ms (D) before the EPSC. There was a strong negative correlation for the 0 ms shift, which weakened when the window was shifted earlier. E, Average from 6 experiments, showing that recent activity does correlate with EPSC amplitude. Correlations differed significantly from 0 for shifts of 0 (p < 0.001), 25 (p < 0.01), and 100 ms (p < 0.05).
We wanted to determine the effect of an individual release event on subsequent EPSCs. The standard approaches of Figure 1 are unable to distinguish such an effect, which makes it impossible to probe individual mechanisms of STP during ongoing activity. We reasoned that the influence of one EPSC could be assessed by how its absence changed subsequent EPSCs. For each cell, we generated a Poisson-distributed stimulus train of >20–30 stimuli and applied it repeatedly, skipping one stimulation on alternate trials. One advantage of the approach of skipping one stimulus is that it would be compatible with any pattern of stimulation, such as more naturalistic random trains. The stimulus skipped was always pulse #16, at which point the EPSC appeared to have reached a steady-state level of depression (Fig. 4A, arrow). EPSCs from trials with the identical stimulus pattern were then averaged together (Fig. 4A). Over most of the train, the average EPSC amplitudes were virtually identical, reflecting the consistent presence of STP (Fig. 4B). However, after the skipped stimulus, the subsequent few EPSCs differed. In the example of Figure 4B, the following three EPSCs were elevated compared with control trials, indicating that the EPSC contributed to depression during control trials. For the EPSC immediately following the skipped stimulus, the EPSC increased by 60%, and this decayed back to no change over the following three pulses (Fig. 4B, bottom).
Figure 4.
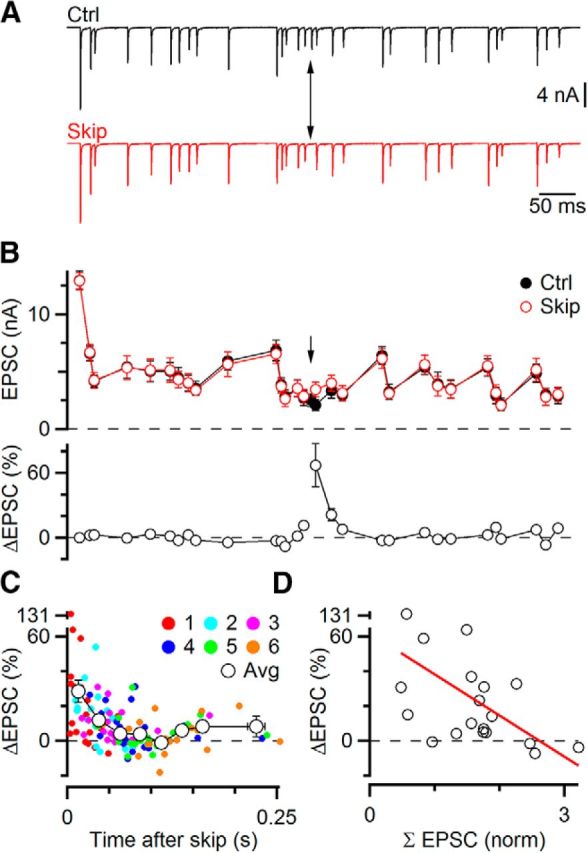
Using skipped stimuli to study influence of one EPSC on subsequent EPSCs. A, Representative experiment. A Poisson-distributed random stimulus train was applied repeatedly, skipping a single stimulus (arrow) on alternate trials. Traces represent averages of 15 individual trials. B, Quantification of the experiment in A. EPSC amplitudes were highly similar for both trains, except immediately after the skipped stimulus (arrow, top). The EPSC amplitude showed a large percentage increase (bottom, EPSCiskip/EPSCictrl − 100%) that subsequently decayed back to 0%. This indicates that the skipped stimulus normally triggered short-term depression. C, Effect of a single EPSC on subsequent six EPSCs from 19 similar experiments. Open circles represent averages of multiple points, binned by time after the skipped stimulus. D, Variability in the effect of the skipped stimulus depends in part on a longer history of activity. EPSC amplitudes were normalized to the first EPSC in the train and summed over the 200 ms preceding the EPSC immediately after the skipped stimulus. There was a strong negative correlation with the percentage increase in the EPSC (r = −0.52). This indicates that earlier activity influenced whether the skipped EPSC caused depression or not.
We did similar experiments in 12 cells. In 7 cells, two different Poisson trains were applied, for a total of 19 distinct patterns. We plotted the relative increase of EPSCs for the 6 pulses following the skipped stimulus and averaged the points together in 8 groups depending on their time after the skipped stimulus (Fig. 4C). The average EPSC increase was greatest immediately after the skipped pulse (28 ± 6%) and remained significant up to 60 ms after (p < 0.05). There was no significant increase in EPSC amplitude 80 ms after the skipped stimulus (p = 0.13).
There was considerable variability in the increase for the EPSC immediately after the skipped stimulus in different cells. One possible explanation for this was that the amount of depression relieved depended on more than just the single skipped stimulus, and the longer history of activity was different for each pattern. To test this, we used the same approach as in Figure 3, summing normalized EPSC amplitudes over the 200 ms before the skipped EPSC. There was a significant negative correlation between the increase in EPSC amplitude and the summed previous activity (slope = −23.6%, r = −0.52, p < 0.05; Fig. 4D). In other words, a small summed EPSC was reflective of little previous activity, and the skipped EPSC played a large role in setting the amplitude of the subsequent EPSC. A large summed EPSC was reflective of high previous activity, and skipping one EPSC had little additional effect. The correlation between previous activity and effect of the skipped stimulus is consistent with activity causing depression, where the amount of depression is context-dependent. This also likely explains why the analyses in Figure 1 fail to reveal much depression because the larger history of activity is critical. In sum, the skipped stimulus approach reveals that STP is present during ongoing activity.
Another advantage of the skipped stimulus approach is that it allows us to probe the importance of different synaptic properties in STP during ongoing activity. The first major question we had was how the initial probability of release (Pr) influences EPSC amplitude during ongoing activity. Synapses with high versus low Pr show depression or facilitation (Zucker and Regehr, 2002). Synapses that undergo changes in Pr, through the actions of neuromodulators or changes in bath calcium concentration, show large initial differences in EPSC amplitude and STP but become more similar in amplitude during prolonged activity (Brenowitz and Trussell, 2001). We have observed similar effects at the endbulb when Pr was manipulated by activating modulatory receptors, or by genetically ablating proteins involved in synaptic transmission (Strenzke et al., 2009; Chanda and Xu-Friedman, 2010b).
We found similar effects when we modulated Pr by changing bath calcium concentrations. A representative cell is shown in Figure 5A, B. When Pr was decreased by lowering bath calcium to 1 mm, the initial EPSC amplitude (data not shown) decreased by nearly 60%, but EPSC amplitudes later in the train were relatively unaffected (Fig. 5A, right, red vs black traces). When Pr was increased by raising bath calcium to 2 mm, the initial EPSC increased by 13% (data not shown), and the steady-state EPSC changed little (Fig. 5A, right, blue vs black traces). We found similar effects in multiple cells. Initial EPSC amplitude increased to 136 ± 15% of control in 2 mm Ca2+ (p < 0.05, N = 10 cells) and decreased to 58 ± 11% of control in 1 mm Ca2+ (p < 0.05, N = 6 cells). By contrast, the 5 EPSCs just preceding the skip (i.e., at steady state) had small and nonsignificant changes in amplitude after switching from 1.5 mm Ca2+ to 1 mm Ca2+ (81 ± 12%, p = 0.19, N = 6 cells) or 2 mm Ca2+ (103 ± 7%, p = 0.71, N = 10 cells). This raises the question how Pr influences EPSC amplitude during ongoing activity.
Figure 5.

Using skipped stimuli to study the influence of initial probability of release (Pr) on EPSC amplitude during ongoing activity. Pr was manipulated by changing the bath calcium concentration. A, Example experiment testing different calcium concentrations in a single cell. Long trains of EPSCs were applied (top traces, 50 Hz Poisson-distributed), of which only EPSCs 15–18 are shown, with stimulus 16 skipped on alternate trials (middle traces). Bottom traces, overlaid, Average EPSCs from control and skipped trials on an expanded timescale. Right, EPSCs from 1.0 mm (red), 1.5 mm (black), and 2 mm (blue) Ca2+ are overlaid with small offsets for comparison. B, The percentage change in EPSC amplitude between skipped and control trials from A. C, D, Results from 9 similar experiments. Percentage changes after the skipped stimulus are averaged by time (C) or based on the pulse following the skipped stimulus (D). Both show a large initial increase, which then decays back to 0. Increases were significant for 1.5 and 2 mm Ca2+ for the first 40 ms (C) or 2 pulses (D) after the skipped stimulus (*p < 0.05 for both calcium concentrations). Changes were not significant for 1 mm Ca2+. This indicates that the dominant form of plasticity is depression in 1.5 and 2 mm Ca2+.
To test this, we examined the EPSCs following the skipped stimulus. For the representative cell in Figure 5A, there were very different effects of the skipped stimulus in different bath calcium concentration despite the weak effects on steady-state EPSC amplitude. In the standard, physiologically based 1.5 mm Ca2+, skipping one stimulus led to an increase in the subsequent EPSCs (Fig. 5A, bottom magnified EPSCs), as we saw in the experiments of Figure 4. However, in 1 mm Ca2+, the skipped stimulus had little effect, whereas in 2 mm Ca2+, the skipped stimulus had an even greater effect (Fig. 5A, bottom magnified EPSCs, B).
We found similar behavior in a number of cells. EPSC amplitudes were averaged together based on either their latency (Fig. 5C) or pulse number (Fig. 5D) following the skipped stimulus. The first two pulses following the skipped stimulus were both significantly larger in 1.5 and 2 mm Ca2+, indicating that STP plays a large role during ongoing activity. In 1 mm Ca2+, STP probably lies between facilitation and depression, and skipping a stimulus shows little effect. These data indicate that the initial Pr continues to influence EPSC amplitude during ongoing activity.
The results so far indicate that depression plays an important role in setting EPSC amplitude at endbulbs. This is highly relevant to the situation in vivo because auditory nerve fibers are highly active, with spontaneous firing rates as high as 100 Hz, and peak firing rates of upwards of 300 Hz (Sachs and Abbas, 1974; Joris et al., 1994; Taberner and Liberman, 2005), so maintaining synaptic efficacy in the face of depression seems of great functional importance. In in vitro experiments, recovery from depression is accelerated by activity, to a τ of 40 ms (Yang and Xu-Friedman, 2008), through a calcium-dependent process that has been found at a number of synapses (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998; Sakaba and Neher, 2001). Surprisingly, however, recordings during normal activity in vivo or mimicked in vitro give the impression that rapid recovery is weak or absent (Fig. 1) (Hermann et al., 2007; Lorteije et al., 2009; Kuenzel et al., 2011), raising the question of whether ongoing activity is a fundamentally different situation.
To test this question directly, we used the skipped stimulus approach. We characterized EPSC amplitude with and without skipped stimuli (Fig. 6A, left). Then we treated synapses with 100 μm EGTA-AM for 5 min. This treatment reduces activity-dependent recovery without interfering with neurotransmitter release much (Yang and Xu-Friedman, 2008). In these experiments, the initial EPSC decreased to 82 ± 6% of control (N = 17 cells, p < 0.01, paired t test), whereas the steady-state EPSCs preceding the skip decreased considerably more, to 60 ± 5% of control after treatment with EGTA-AM (N = 12 cells, p ≪ 0.001, paired t test). This indicates that calcium-dependent recovery is important in controlling steady-state EPSC amplitude, but its role in shaping moment-by-moment changes in EPSC amplitude is unclear.
Figure 6.

Activity-dependent recovery and desensitization both influence EPSC amplitude during ongoing activity. A, C, Representative experiments, showing the effect of the skipped stimulus in control conditions (left) and after 5 min treatment with either 100 μm EGTA-AM (right in A) or during application of 50 μm CTZ (right in C). Long trains of stimuli were applied (top traces, 50 Hz Poisson-distributed), but only EPSCs near the skipped stimulus are shown. Changes in EPSC amplitude following the skipped stimulus are smaller after EGTA-AM or CTZ treatment. B, D, Effects of EGTA-AM treatment (12 cells, B) and CTZ (6 cells, D). Closed symbols represent individual experiments. Open symbols represent overall averages. D, Blue symbols represent experiments where the preceding pause was relatively long (up to 80 ms). Red symbols had short pauses. Points falling below the dashed unity line indicate smaller effects of the skipped stimulus after treatment in individual experiments. EPSC changes in control are significantly greater than those after EGTA-AM or CTZ treatment (p < 0.05 for both). E, Desensitization is greatest after a period of recovery. Plotted is the desensitization index desens = (EPSCctrlskip/EPSCctrl)/(EPSCCTZskip/EPSCCTZ) as a function of the preceding stimulus interval. The line shows the fit to the data, which are significantly correlated (r = 0.65, slope = 13.4, p < 0.05).
To gain better insight into how calcium-dependent recovery affects EPSC amplitude, we examined the effect of EGTA-AM after the skipped stimulus. Unlike control conditions, the effect of the skipped stimulus was greatly reduced after treatment with EGTA-AM (Fig. 6A, right). In 12 similar experiments, the effects of the skipped stimulus in control were quite variable (Fig. 6B), most likely because the train was different for each cell (similar to Fig. 4). In all cases, EGTA-AM treatment reduced the effect of the skipped stimulus, and overall the reduction was highly significant (ΔEPSC = 26.8 ± 5.9% in control, 6.6 ± 3.4% after EGTA-AM, p < 0.01). This means that activity-dependent recovery is normally highly important in regulating EPSC amplitude. Without it, depression is not as pronounced.
Endbulb EPSCs are also subject to significant postsynaptic receptor desensitization (Isaacson and Walmsley, 1996; Yang and Xu-Friedman, 2008; Chanda and Xu-Friedman, 2010a). AMPA receptors desensitize rapidly in the presence of glutamate, and it has been a long-standing question whether this actually contributes to STP under realistic conditions. This is an important question because the fast recovery from desensitization could play a role in sharpening the temporal responses of BCs. At high rates of activation, glutamate may become too depleted to cause significant desensitization (Chanda and Xu-Friedman, 2010a). However, brief cessations in spiking could permit enough recovery that desensitization could be induced.
We tested the role of desensitization by using a stimulus train in which there was a brief pause in ongoing activity before a period of high activity. An example is shown in Figure 6C, with a pause of 80 ms before two closely spaced EPSCs. The second EPSC in the pair showed a lot of depression, and skipping the previous stimulus caused a large increase in amplitude. Most of the depression appeared to result from desensitization under these conditions, as application of 50 μm cyclothiazide (CTZ) greatly reduced depression after the skipped stimulus (Fig. 6C). We quantified the increase in the first EPSC following the skipped stimulus and found that the amplitude increased much less in CTZ (Fig. 6D, blue symbols). For 6 cells with pauses in the Poisson train of up to 80 ms, the increase after the skip was much larger in control (173 ± 50%) than in CTZ (42 ± 9%, p < 0.01), indicating that desensitization played an important role in setting the amplitude of the EPSC. By contrast, for 4 cells studied using trains that had only brief pauses, or with a longer interval after the skipped EPSC, the effect of CTZ was smaller (Fig. 6D, red symbols). We computed a desensitization index by taking the ratio of the EPSC enhancement in control to the EPSC enhancement in CTZ: desens = (EPSCctrlskip/EPSCctrl)/(EPSCCTZskip/EPSCCTZ). When desensitization does not contribute to depression, then the effects of the skipped stimulus should be the same in control and CTZ, and desens should be near 1. When desensitization is important, the EPSC amplitude would increase in control after the skipped stimulus, but less so in CTZ, and desens should be >1. We found that desens varied considerably between experiments (Fig. 6E). There was a strong positive correlation between the desensitization index and the duration of the interval immediately preceding the skipped stimulus (Fig. 6E; r = 0.65, slope = 13.4, p < 0.05). Averaging all these cells together, CTZ significantly reduced the effect of the skipped stimulus (110 ± 37% in control vs 28 ± 9% in CTZ, p < 0.05). These results indicate that desensitization can be a major contributor to depression at steady state, with even brief pauses in presynaptic activity allowing sufficient recovery so that subsequent activity can cause desensitization.
The changes in EPSC amplitude following skipped stimuli appeared to be considerable, reflecting the role of STP in regulating EPSC amplitude. However, it remained a question whether these effects were functionally significant. To test this, we performed recordings of BCs in current clamp and stimulated single auditory nerve fibers in regular trains at rates where the probability of spiking was intermediate between 0% and 100%, which typically occurred for stimulation rates between 50 and 333 Hz (Fig. 7A). These experiments were conducted in the presence of 5 μm CPP, to block NMDA receptors. The probability of spiking dropped throughout the train, but following a skipped stimulus, spike probability increased (Fig. 7B). We wanted to quantify EPSP amplitude in these experiments. In BCs, it is often difficult to separate EPSP from spike because EPSPs are unusually large, whereas spikes are unusually small (Oertel, 1983). However, in some cases, it was possible to distinguish the EPSP from the spike in the derivative of the membrane potential over part of the train (Fig. 7C, inset). This showed that the skipped stimulus led to an increase in EPSP amplitude (Fig. 7C). We found similar effects in 8 experiments. After a skipped stimulus, the spike probability increased over control (Fig. 7D; p < 0.05 for the first 3 pulses after skip), which paralleled an increase in the EPSP (Fig. 7E; p < 0.01 for the 4 pulses after the skip). In addition, the EPSP immediately after the skip was significantly greater than the EPSP immediately before, by 15 ± 6% (p < 0.05, N = 8 experiments). Furthermore, the change in EPSP amplitude over the four stimuli following the skipped stimulus was highly correlated with the change in spike probability (Fig. 7F; slope = 0.59, r = 0.42, p < 0.05). The elevation in EPSP amplitude lasted for a duration that is consistent with the duration of the effect seen in Figures 4 and 5.
We found previously that NMDA receptor activation supported increased firing probability during long trains of activation in vitro (Pliss et al., 2009). It is possible that NMDA-receptor activation in vivo could compensate for depression of AMPA-receptor-mediated EPSPs and prevent a decrease in spiking. To test this, we conducted skipped stimulus experiments with the NMDA-receptor mediated currents intact (no CPP). We still found a strong enhancement of spiking following a skipped stimulus (Fig. 8A, left). The degree of enhancement was similar to that seen after addition of 5 μm CPP (Fig. 8A, right, B). We obtained similar results in 5 experiments (Fig. 8C). Pspike increased significantly following the skipped stimulus with and without the NMDAR component present (p = 0.03 in control, p = 0.02 in CPP), but the increase did not differ between the two conditions (p = 0.93). Thus, NMDAR activation did not compensate for the skipped stimulus' effect on the AMPA component.
Figure 8.

The NMDA receptor component of the EPSC does not compensate for skipped stimuli. A, Representative current-clamp experiment similar to Figure 7, but without (left) and with (right) 5 μm CPP to block NMDAR. Top traces and rasters, Regular trains (20 pulses, 200 Hz). Bottom traces and rasters, With one stimulus skipped, showing elevated spike probability for the subsequent stimulus. B, Quantitative effects of the skipped stimulus for the experiment in A. C, Average effects from five similar experiments. Asterisk indicates significantly elevated Pspike (p < 0.05) for both control and CPP.
The increase in spiking in Figures 7 and 8 could have resulted from the increase in EPSP amplitude, but it may have also been influenced by spike initiation. The skipped stimulus may provide time for the BC to emerge from the refractoriness associated with Na-channel inactivation and K-channel activation. Indeed, spike refractoriness has been suggested to play a larger role than STP during ongoing activity (Lorteije et al., 2009; Kuenzel et al., 2011). We wanted to distinguish these two effects of skipping a stimulus, so we turned to the dynamic-clamp technique. Dynamic clamp allows the experimenter full control over amplitude and timing of synaptic inputs, which allows us to dissociate the effects of the pause from the increased conductance. We developed 4 conductance protocols using a model of neurotransmitter release based on the endbulb (Yang and Xu-Friedman, 2013). Briefly, the model incorporates vesicle depletion, receptor desensitization, activity-dependent recovery, and the stochastic properties of neurotransmitter release. Conductances in the first protocol were based on a regular train of stimuli (Fig. 9A, top); and in the second, the identical conductances were used with the addition of a delay (Δt) at the same point in the train that we normally skipped one stimulus (after pulse #15) (Fig. 9B, top). The conductances of the third protocol were based on a regular train with one skipped stimulus. In the model, this skip led to a modest increase of 34% in the subsequent conductance (Fig. 9C, top). The fourth protocol used conductances identical to the third, but removing the delay associated with the skipped stimulus (Fig. 9D, top). Conductance amplitudes from these four protocols were convolved with a unitary EPSC, and applied to BCs in dynamic clamp (Fig. 9A–D, middle traces). BC spiking was quite similar over the initial parts of the train that shared identical conductances (Fig. 9, black portions of traces), reflecting the fidelity of BCs. However, spike probability diverged when the delay was added or removed (Fig. 9, red portions of traces).
Figure 9.

Skipped stimuli increase subsequent spiking, partly through enhanced spike initiation, but primarily through increases in synaptic conductance. A–D, Four representative conductance protocols used to drive dynamic-clamp experiments. Synaptic conductances were drawn from a stochastic model of neurotransmitter release (Yang and Xu-Friedman, 2013) for a 200 Hz train of stimuli. Conductances were modeled for a regular train (“Ctrl” protocol, A) as well as one with a skipped stimulus (“Skip” protocol, C). A delay equivalent to the skip was inserted into the control protocol (“Ctrl + Δt” protocol, B) or removed from the skipped protocol (“Skip − Δt” protocol, D), but preserving the identical conductance amplitudes. The experimentally relevant conductances are highlighted in red. Middle traces show representative dynamic-clamp trials for the conductances at top. Responses to the experimentally relevant conductances are highlighted in red. Bottom raster plots show spikes over many similar trials. Red markers here and in E, F represent the important conductances to compare. E, F, Spike probability for the conductances near the “skipped” stimulus for the representative experiment in A–D (E), and 6 similar experiments (F). G, Changes in spike probability resulting from the delay (Δt) or change in conductance (ΔG) associated with skipped stimuli. Black bar represents the overall increase in spike probability, comparing the control and skip protocols. The contribution of the delay (Δt, blue bars) is isolated by comparing the spike probability for control protocols with and without the added delay (open minus closed circles), and for skip protocols with and without the delay removed (closed minus open triangles). The delay alone increased spike probability by approximately one-third of the overall increase. The contribution of the conductance (ΔG, red bars) is isolated by comparing the spike probability for skip protocols against the parallel control protocols with the same timing (open triangle minus closed circle, and closed triangle minus open circle). The conductance alone increased spike probability by approximately two-thirds of the overall increase. H, Measuring refractory period using conductance threshold. The conductance required to trigger a spike after a 14-pulse, 200 Hz conditioning train was normalized to threshold without conditioning. Threshold increased considerably for intervals <3 ms. Each point is the average of 5 cells, except the first two points, which are 1 cell and 4 cells. The line is a fit to the data of the form y = 1 + A exp (−t/τ), with A = 2.16 ± 0.83 and τ = 1.97 ± 0.61 ms.
These four protocols were applied multiple times, modeling new conductances after each round, and the resulting spikes were detected (Fig. 9A–D, bottom rasters) to calculate spike probability (Fig. 9E). We hypothesized that, if the refractory period were important, then adding a delay (from “Ctrl” to “Ctrl + Δt”) should increase spike probability, and removing the delay (from “Skip” to “Skip − Δt”) should decrease spike probability while all the conductance values were held constant. Indeed, the spike probability did increase when a delay was added in control (compare red open and closed circles in Fig. 9E), and the spike probability decreased when the delay was removed from skipped conductances (compare red open and closed triangles in Fig. 9E).
We also hypothesized that, if synaptic conductance were important, then increasing one synaptic conductance should increase spike probability while the presence or absence of the delay was held constant. Indeed, the probability of spiking was greater in the skipped protocol with the delay removed (“Skip − Δt,” Fig. 9E, open red triangle) than in the control protocol that had no delay (“Ctrl”; Fig. 9E, closed red circle). Similarly the probability of spiking was greater in the skipped protocol (“Skip”; Fig. 9E, closed red triangle) than in the control protocol when a delay was added (“Ctrl + Δt”; Fig. 9E, open red circle).
We found similar results in 6 experiments (Fig. 9F). We used these data to quantify the relative contribution of the refractory period versus synaptic conductance to the total increase in spiking following a skipped stimulus. First, we calculated the full increase in spike probability resulting from one skipped EPSP by comparing the control versus skip protocols (Fig. 9F, closed red triangle minus the closed red circle, G, black bar), which was 0.34 ± 0.04. This reflected the effects of both the time delay (Δt) and the change in synaptic conductance (ΔG), and was similar in principle to the changes in spiking seen in the current-clamp experiments of Figures 7 and 8. Next, we isolated the contribution of the refractory period to this overall increase in spike probability in two ways (Fig. 9G, blue bars). (1) We compared the spike probability of the control protocol to the control protocol immediately after the added delay (Fig. 9F, open vs closed red circles), which was a difference of 0.10 ± 0.03 (Fig. 9G). (2) We compared the spike probability of the skipped protocol to the skipped protocol with the delay removed (Fig. 9F, open vs closed triangles), which was a difference of 0.12 ± 0.04 (Fig. 9G). Next, we isolated the contribution of the synaptic conductance to the overall increase in spike probability in two ways (Fig. 9G, red bars). (1) We compared the spike probability of the control versus skipped protocols with no delay (Fig. 9F, open red triangle vs closed red circle), which was a difference of 0.22 ± 0.02 (Fig. 9G). (2) We compared the control versus skipped protocols with the delay (Fig. 9F, closed red triangle vs open red circle), which was a difference of 0.24 ± 0.02 (Fig. 9G). Thus, for these experimental conditions, approximately one-third of the increase in spiking after a skipped stimulus was due to the delay and its contribution to recovery from the refractory period. A greater contribution, approximately two-thirds, was due to the increase in synaptic conductance as influenced by the processes of STP. Based on these dynamic-clamp data, it would appear that in the current-clamp experiment in Figures 7 and 8, most of the increased spiking following a skipped stimulus resulted from the increase in EPSP amplitude, with a smaller contribution from recovery from inactivation.
The relative contributions of conductance changes versus recovery from inactivation are likely context-dependent. The specific pattern of activity determines the effects of the skipped stimulus on synaptic conductance (Figs. 4, 6), and the effect of the refractory period diminishes with recovery time. We measured the time course of the refractory period by looking at changes in the minimum conductance needed to fire a spike. For a rested cell, the threshold was 17.5 ± 4.3 nS (mean ± SD, N = 5 cells). Then we applied a train of conditioning, suprathreshold conductances at 200 Hz for 14 pulses in dynamic clamp, followed by test conductances at different intervals and amplitudes. The threshold conductance after the conditioning train was normalized to the threshold without conditioning. Average threshold conductance is shown for 5 cells in Figure 9H. In these experiments, spikes were not reliably detectable for very large conductances, so changes in threshold were based on fewer cells for the shortest intervals (1 ms, N = 1 cell; 2 ms, N = 4 cells). These data indicate that the refractory period does influence spiking at very short intervals, and decays away by 10 ms. This analysis suggests that there would still be a small refractoriness for the conditions in Figures 7, 8, and 9 (i.e., 5 ms), consistent with the quantification in Figure 9H.
Discussion
Our results indicate that STP is present and plays an important role in regulating EPSC amplitude during ongoing activity, which strongly suggests that STP is a major player in controlling synaptic strength in vivo. Standard analyses are insufficient to reveal the presence of STP. However, the skipped stimulus paradigm we developed shows that an EPSC influences subsequent neurotransmitter release for >50 ms. Factors that have a large influence on EPSC amplitude in simplified experiments in vitro continue to operate even for synapses at steady state. These changes in EPSC amplitude influence spiking in the postsynaptic BCs; thus, STP is functionally important in how information is transmitted across the endbulb synapse.
These conclusions are in line with many studies implicating regulation of STP at different synapses. Different synapses show characteristic STP ranging from depressing to facilitating, which are reliable enough to use for identifying synapse types. Even individual axons can show distinct plasticity depending on the cell type on which they terminate, a phenomenon referred to as target-cell-specific synaptic plasticity (Reyes et al., 1998). In the cochlear nucleus, auditory nerve fiber synapses show greater depression onto BCs than onto nearby multipolar cells (Chanda and Xu-Friedman, 2010a). In addition, STP of auditory nerve fibers that converge on the same BC are more similar than auditory nerve fibers that terminate on different BCs, which we called “coordinated plasticity” (Yang and Xu-Friedman, 2009, 2012). This reflects an extremely tight regulation of STP and Pr in the cochlear nucleus. This tight regulation would suggest that STP must have important functional consequences during normal activity.
However, a number of in vivo studies have indicated that STP in vivo can differ considerably from what is characterized in vitro. For example, the retinogeniculate synapse depresses in vitro (Turner and Salt, 1998; Chen et al., 2002), but in vivo, pairs of presynaptic action potentials are more effective at driving postsynaptic responses, suggesting the synapses facilitate (Usrey et al., 1998). In addition, the calyx of Held synapse in the MNTB can show facilitation or depression in vitro (von Gersdorff and Borst, 2002; Wong et al., 2003; Müller et al., 2010; Grande and Wang, 2011) but in vivo shows little evidence of any deterministic changes in EPSP amplitude (Lorteije et al., 2009). The reasons for these differences are not known. One possibility is that STP is usually studied in rested synapses in vitro, which is clearly unnatural (Hermann et al., 2007), and perhaps also misleading about ongoing activity. Our data suggest that this is unlikely to be the case because we found that the same basic processes of STP are at work both in rested synapses and during ongoing activity.
Another possibility, suggested by Lorteije et al. (2009), is that STP differs in vitro and in vivo because Pr is different. This could occur as a result of changes to the modulatory environment following slice preparation, which could sever the axons carrying modulatory activity. STP could differ because these systems are recruited in vivo, but not in vitro. Indeed, GABAB receptors mediate strong modulation in much of the auditory brainstem, and these receptors appear to be recruited by activity in the medial and lateral superior olive (Magnusson et al., 2008; Stange et al., 2013), although their roles in the MNTB and AVCN remain unresolved (Chanda and Xu-Friedman, 2010b; Wang et al., 2013). A second difference could be the external free calcium concentration, which is thought to be 1.5–1.7 mm (Manthei et al., 1973; Frosini et al., 1993). Small errors in this measurement would be magnified by the cooperativity of neurotransmitter release (Dodge and Rahamimoff, 1967) and could lead to distorting Pr and STP in vitro.
Our results highlight the important role of specific mechanisms underlying STP during ongoing activity, particularly activity-dependent recovery. When activity-dependent recovery was blocked by EGTA-AM, both the steady-state EPSC and the effect of the skipped stimulus decreased, indicating that this rapid form of recovery dominates EPSC amplitude. There are also slower forms of recovery at the endbulb that have a τ of recovery >1 s (Yang and Xu-Friedman, 2008), but we did not detect an influence of release on this timescale. This may be because our individual trials were too short or because its importance during ongoing activity diminishes.
Our results also reveal that desensitization influences EPSC amplitude during ongoing activity. There has been conflicting information about desensitization, in part because its prominence can change over development (Brenowitz and Trussell, 2001; Wang and Manis, 2008). Desensitization persists in mature endbulbs but is significant primarily when there is little depletion of neurotransmitter (Yang and Xu-Friedman, 2008; Chanda and Xu-Friedman, 2010a). Using the skipped stimulus paradigm, we found that even brief 50–100 ms pauses in activity allow significant recovery so that EPSCs can trigger desensitization. Thus, desensitization is likely to commonly influence EPSPs in vivo. Because recovery from desensitization is so rapid (Trussell et al., 1993), this would tend to make BCs less responsive to closely spaced auditory nerve spikes and therefore to rapid temporal features of sounds.
These components of STP have been identified and characterized in synapses in vitro with long periods of recovery between trials, which does not mimic physiological conditions. Our results suggest that this does not invalidate the conclusions from those experiments. However, the relative role of each factor contributing to STP will depend on the specific conditions. We found that EPSCs were more depressed following periods of high activity, whereas desensitization was more powerful after a pause in activity.
Our skipped stimulus paradigm offers insight into whether those processes actually amount to any changes in spiking as well. Using this approach, we observed an increase in spiking following a skipped stimulus. Our dynamic-clamp experiments suggest that most of this increase was a result of the change in EPSP amplitude, with a smaller contribution from recovery from inactivation. This contrasts with in vivo experiments that showed decreased spiking for a 1 ms period following a spike. Our skipped stimulus approach allowed us to detect and separate the effects of spike refractoriness and depression, and depression had the larger effect.
Footnotes
This work was supported by National Institutes of Health Grant R03 DC012433 to H.Y. and National Science Foundation Grant 1208131 to M.A.X.-F. We thank J. Goetz, R. La Rosa, T. Ngodup, Y. Yang, and X. Zhuang for helpful comments on the paper.
The authors declare no competing financial interests.
References
- Brenowitz S, Trussell LO. Maturation of synaptic transmission at end-bulb synapses of the cochlear nucleus. J Neurosci. 2001;21:9487–9498. doi: 10.1523/JNEUROSCI.21-23-09487.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao XJ, Oertel D. Auditory nerve fibers excite targets through synapses that vary in convergence, strength, and short-term plasticity. J Neurophysiol. 2010;104:2308–2320. doi: 10.1152/jn.00451.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Xu-Friedman MA. A low-affinity antagonist reveals saturation and desensitization in mature synapses in the auditory brainstem. J Neurophysiol. 2010a;103:1915–1926. doi: 10.1152/jn.00751.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Xu-Friedman MA. Neuromodulation by GABA converts a relay into a coincidence detector. J Neurophysiol. 2010b;104:11466–11475. doi: 10.1152/jn.00474.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron. 2002;33:779–788. doi: 10.1016/S0896-6273(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Response of hippocampal synapses to natural stimulation patterns. Neuron. 1999;22:157–166. doi: 10.1016/S0896-6273(00)80687-X. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Katz B, Kuffler SW. Nature of the “endplate potential” in curarized muscle. J Neurophysiol. 1941;4:362–387. [Google Scholar]
- Feng TP. Studies on the neuromuscular junction: XXVI. The changes in the endplate potential during and after prolonged stimulation. Chin J Physiol. 1941;16:341–372. [Google Scholar]
- Fortune ES, Rose GJ. Short-term synaptic plasticity as a temporal filter. Trends Neurosci. 2001;24:381–385. doi: 10.1016/S0166-2236(00)01835-X. [DOI] [PubMed] [Google Scholar]
- Frosini M, Gorelli B, Matteini M, Palmi M, Valoti M, Sgaragli GP. HPLC determination of inorganic cation levels in CSF and plasma of conscious rabbits. J Pharmacol Toxicol Methods. 1993;29:99–104. doi: 10.1016/1056-8719(93)90057-L. [DOI] [PubMed] [Google Scholar]
- Glantz SA. Primer of biostatistics. New York: McGraw-Hill; 1997. [Google Scholar]
- Grande G, Wang LY. Morphological and functional continuum underlying heterogeneity in the spiking fidelity at the calyx of Held synapse in vitro. J Neurosci. 2011;31:13386–13399. doi: 10.1523/JNEUROSCI.0400-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann J, Pecka M, von Gersdorff H, Grothe B, Klug A. Synaptic transmission at the calyx of Held under in vivo like activity levels. J Neurophysiol. 2007;98:807–820. doi: 10.1152/jn.00355.2007. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Amplitude and time course of spontaneous and evoked excitatory postsynaptic currents in bushy cells of the anteroventral cochlear nucleus. J Neurophysiol. 1996;76:1566–1571. doi: 10.1152/jn.1996.76.3.1566. [DOI] [PubMed] [Google Scholar]
- Johnson DH. The relationship between spike rate and synchrony in responses of auditory-nerve fibers to single tones. J Acoust Soc Am. 1980;68:1115–1122. doi: 10.1121/1.384982. [DOI] [PubMed] [Google Scholar]
- Joris PX, Carney LH, Smith PH, Yin TC. Enhancement of neural synchronization in the anteroventral cochlear nucleus: I. Responses to tones at the characteristic frequency. J Neurophysiol. 1994;71:1022–1036. doi: 10.1152/jn.1994.71.3.1022. [DOI] [PubMed] [Google Scholar]
- Kiang NY. Discharge Patterns of single fibers in the cat's auditory nerve. Cambridge, MA: Massachusetts Institute of Technology; 1965. [Google Scholar]
- Kuenzel T, Borst JG, van der Heijden M. Factors controlling the input–output relationship of spherical bushy cells in the gerbil cochlear nucleus. J Neurosci. 2011;31:4260–4273. doi: 10.1523/JNEUROSCI.5433-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorteije JA, Rusu SI, Kushmerick C, Borst JG. Reliability and precision of the mouse calyx of Held synapse. J Neurosci. 2009;29:13770–13784. doi: 10.1523/JNEUROSCI.3285-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL. Short-term changes in synaptic efficacy. In: Edelman GM, Gall WE, Cowan WM, editors. Synaptic function. New York: Wiley; 1987. pp. 21–56. [Google Scholar]
- Magnusson AK, Park TJ, Pecka M, Grothe B, Koch U. Retrograde GABA signaling adjusts sound localization by balancing excitation and inhibition in the brainstem. Neuron. 2008;59:125–137. doi: 10.1016/j.neuron.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Manthei RC, Wricht DC, Kenny AD. Altered CSF constituents and retrograde amnesia in rats: a biochemical approach. Physiol Behav. 1973;10:517–521. doi: 10.1016/0031-9384(73)90214-X. [DOI] [PubMed] [Google Scholar]
- Müller M, Goutman JD, Kochubey O, Schneggenburger R. Interaction between facilitation and depression at a large CNS synapse reveals mechanisms of short-term plasticity. J Neurosci. 2010;30:2007–2016. doi: 10.1523/JNEUROSCI.4378-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D. Synaptic responses and electrical properties of cells in brain slices of the mouse anteroventral cochlear nucleus. J Neurosci. 1983;3:2043–2053. doi: 10.1523/JNEUROSCI.03-10-02043.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. J Physiol. 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliss L, Yang H, Xu-Friedman MA. Context-dependent effects of NMDA receptors on precise timing information at the endbulb of held in the cochlear nucleus. J Neurophysiol. 2009;102:2627–2637. doi: 10.1152/jn.00111.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF. Physiology of synaptic transmission and short-term plasticity. In: Cowan WM, Sudhof TC, Stevens CF, editors. Synapses. Baltimore: Johns Hopkins UP; 2001. pp. 135–175. [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Sachs MB, Abbas PJ. Rate versus level functions for auditory-nerve fibers in cats: tone-burst stimuli. J Acoust Soc Am. 1974;56:1835–1847. doi: 10.1121/1.1903521. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/S0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Stange A, Myoga MH, Lingner A, Ford MC, Alexandrova O, Felmy F, Pecka M, Siveke I, Grothe B. Adaptation in sound localization: from GABA(B) receptor-mediated synaptic modulation to perception. Nat Neurosci. 2013;16:1840–1847. doi: 10.1038/nn.3548. [DOI] [PubMed] [Google Scholar]
- Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T. Complexin-I is required for high-fidelity transmission at the endbulb of Held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner AM, Liberman MC. Response properties of single auditory nerve fibers in the mouse. J Neurophysiol. 2005;93:557–569. doi: 10.1152/jn.00574.2004. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-Z. [DOI] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Characterization of sensory and corticothalamic excitatory inputs to rat thalamocortical neurones in vitro. J Physiol. 1998;510:829–843. doi: 10.1111/j.1469-7793.1998.829bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usrey WM, Reppas JB, Reid RC. Paired-spike interactions and synaptic efficacy of retinal inputs to the thalamus. Nature. 1998;395:384–387. doi: 10.1038/26487. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Borst JG. Short-term plasticity at the calyx of Held. Nat Rev Neurosci. 2002;3:53–64. doi: 10.1038/nrn705. [DOI] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wang T, Rusu SI, Hruskova B, Turecek R, Borst JG. Modulation of synaptic depression of the calyx of Held synapse by GABAB receptors and spontaneous activity. J Physiol. 2013;591:4877–4894. doi: 10.1113/jphysiol.2013.256875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Manis PB. Short-term synaptic depression and recovery at the mature mammalian endbulb of Held synapse in mice. J Neurophysiol. 2008;100:1255–1264. doi: 10.1152/jn.90715.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Relative roles of different mechanisms of depression at the mouse endbulb of Held. J Neurophysiol. 2008;99:2510–2521. doi: 10.1152/jn.01293.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Impact of synaptic depression on spike timing at the endbulb of Held. J Neurophysiol. 2009;102:1699–1710. doi: 10.1152/jn.00072.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Emergence of coordinated plasticity in the cochlear nucleus and cerebellum. J Neurosci. 2012;32:7862–7868. doi: 10.1523/JNEUROSCI.0167-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Xu-Friedman MA. Stochastic properties of neurotransmitter release expand the dynamic range of synapses. J Neurosci. 2013;33:14406–14416. doi: 10.1523/JNEUROSCI.2487-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]