

Abstract
Integrins are adhesion and survival molecules involved in axon growth during CNS development, as well as axon regeneration after injury in the peripheral nervous system (PNS). Adult CNS axons do not regenerate after injury, partly due to a low intrinsic growth capacity. We have previously studied the role of integrins in axon growth in PNS axons; in the present study, we investigate whether integrin mechanisms involved in PNS regeneration may be altered or lacking from mature CNS axons by studying maturing CNS neurons in vitro. In rat cortical neurons, we find that integrins are present in axons during initial growth but later become restricted to the somato-dendritic domain. We investigated how this occurs and whether it can be altered to enhance axonal growth potential. We find a developmental change in integrin trafficking; transport becomes predominantly retrograde throughout axons, but not dendrites, as neurons mature. The directionality of transport is controlled through the activation state of ARF6, with developmental upregulation of the ARF6 GEF ARNO enhancing retrograde transport. Lowering ARF6 activity in mature neurons restores anterograde integrin flow, allows transport into axons, and increases axon growth. In addition, we found that the axon initial segment is partly responsible for exclusion of integrins and removal of this structure allows integrins into axons. Changing posttranslational modifications of tubulin with taxol also allows integrins into the proximal axon. The experiments suggest that the developmental loss of regenerative ability in CNS axons is due to exclusion of growth-related molecules due to changes in trafficking.
Keywords: Arf6, axon initial segment, axonal transport, integrin, trafficking
Introduction
During development, the role of axons is to extend toward their targets. As they mature, the function of axons changes to neurotransmission and there is a sharp decline in their intrinsic growth capacity (Fawcett, 1992; Blackmore et al., 2012; Chisholm, 2013; Zou et al., 2013). As this happens, polarized distribution of many molecules develops. Proteins for neurotransmission reach the presynaptic axon terminal, whereas those intended for postsynaptic functions are restricted to dendrites (Conde and Caceres, 2009; Hirokawa et al., 2009; Rasband, 2010; Lasiecka and Winckler, 2011; Farías et al., 2012). Integrins are growth and survival molecules that are important for axon growth during development and for regeneration in the peripheral nervous system (PNS) in adulthood (Werner et al., 2000; Vogelezang et al., 2001, 2007; Gardiner et al., 2005). During CNS development, they are involved in axon growth and guidance (Huang et al., 2006; Myers et al., 2011) and, in adulthood, they regulate aspects of synaptic plasticity (McGeachie et al., 2011). In adulthood, there is an absence of integrins from mature CNS axons (Bi et al., 2001; Mortillo et al., 2012). We hypothesize that exclusion of integrins and other growth- and survival-related molecules is the reason for the lack of regenerative ability of mature axons.
Axon growth involves interactions between the cell surface, signaling pathways, and the cytoskeleton. Integrins are an example of membrane proteins involved in these processes, so their study can reveal mechanisms that are important for axon growth or regeneration and might indicate why the intrinsic growth capacity of CNS axons declines with maturity. Integrins are also potential therapeutics: expression of an appropriate integrin or its activator kindlin can enhance regeneration of adult PNS axons into the CNS environment (Andrews et al., 2009; Tan et al., 2012); however, this strategy will not work for CNS neurons because, like endogenous integrins, exogenous integrins are not transported into axons in mature CNS neurons in vivo (M.R. Andrews and J.W.F., unpublished findings). We have therefore investigated the mechanisms preventing axonal integrin traffic with the aim of discovering ways to enable integrin transport into mature CNS axons.
In previous work, we found that integrin trafficking in sensory axons (which transport integrins) is mediated by two GTPases, Rab11 and Arf6 (Eva et al., 2010, 2012), but these pathways and integrin trafficking in general have not been studied in CNS axons.
We first examined integrin trafficking in rat brain cortical neurons in vitro and found that integrins are restricted to the somatodendritic compartment after the initial stage of axon growth. A number of mechanisms contribute to the polarized localization of membrane proteins in neurons (Conde and Caceres, 2009; Hirokawa et al., 2009; Lewis et al., 2009; Rasband, 2010; Farías et al., 2012; Petersen et al., 2014). We have investigated which of these regulates integrin localization and whether manipulating them can restore integrin traffic. We find that the main factors excluding integrins from axons are a developmental shift to retrograde transport regulated by ARF6 that occurs throughout the axon, cytoskeletal modifications, and additional mechanisms associated with the axon initial segment (AIS). Manipulation of these mechanisms allows transport of integrin into CNS axons and increases axon growth. We suggest that selective exclusion of integrins and other growth-related molecules from mature CNS axons is the main reason for the loss of regenerative ability with maturity.
Materials and Methods
DNA constructs.
Integrin α9 enhanced green fluorescent protein (EGFP)-N3 was obtained from Dean Sheppard (University of California, San Francisco, CA). HA-p50 was a gift from Casper Hoogenraad (Utrecht University, Utrecht, The Netherlands; Hoogenraad et al., 2001). pcDNA3.1-ACAP1-myc was a gift from Victor Hsu (Department of Medicine, Harvard Medical School, Boston, MA) and was described previously (Eva et al., 2012). Myc-ARNO-E156K and GFP-ARNO-E156K were described previously (Venkateswarlu and Cullen, 2000; Bouschet et al., 2007).
Immunostaining.
Cell cultures on coverslips were fixed with either 4% paraformaldehyde (PFA) for 10 min or ice-cold methanol for 5 min at −20°C and blocked for 30 min in PBS containing 0.1% Triton X-100 and 2% goat serum. Cells were incubated with primary antibodies at 4°C overnight, washed, and incubated with Alexa Fluor-conjugated secondary antibodies (Invitrogen) diluted 1:500 for 1 h at room temperature before mounting in Fluorsave (Millipore). For live labeling of α9β1, cells were incubated with primary antibody for 45 min at 37°C, fixed with PFA, and incubated with secondary antibody. The following primary antibodies were used: anti-integrin β1 clone EP1041Y (04-1109, 1:100; Millipore), anti-integrin α5 (AB1928, 1:1000; Millipore), anti-integrin αV (AB1930, 1:1000; Millipore), anti-Ankyrin G (AnkG) clone N106/36 (1:100; NeuroMab), anti-neurofascin (clone A12/18, 1:200; NeuroMab), anti-α9β1 clone Y9A2 (ab27947, 1:200; Abcam), anti-tenascin-C (10337, 1:20; IBL), anti-MAP2 (AB5622, 1:300; Millipore), anti-Arf6 (ab77581, 1:500; Abcam), anti-ARNO (ab56510, 1:250; Abcam), anti-Rab11 (71-5300, 1:50; Invitrogen), and anti-HA (clone 16B12, 1:400; Covance). Polyclonal βIV spectrin antibody (1:100) was kindly provided by Matthew Rasband (Baylor College of Medicine, Houston, TX). Alexa Fluor 568-conjugated phalloidin (1:40; Invitrogen) was used to detect F-actin. Fluorescent images were acquired using a Leica DM6000 microscope and Leica LAS AF software.
Neuron cultures and transfection.
Primary cortical neuron cultures were prepared from embryonic day 18 (E18) rats. Neurons were dissociated with 0.1% trypsin (Invitrogen) for 15 min at 37°C, washed with HBSS, and cultured in neurobasal medium (NB; Invitrogen) supplemented with B27 (Invitrogen), Glutamax (Invitrogen), glutamic acid (Sigma), and PSF. Cells were plated on coverslips coated with poly-d-lysine and 2 mg/ml laminin. 2 d in vitro (DIV) and 9 DIV neurons were transfected by oscillating nanomagnetic transfection (magnefect nano system; nanoTherics). DNA (1 μg/well) was mixed with 1.8 μl of magnetic nanoparticles (NeuroMag), incubated for 20 min, and added to the neurons in 500 μl of NB. For cotransfection with α9-GFP and other plasmids (ACAP1, ARNO, or P50), DNA used was 0.7 μg of the α9-GFP and 0.7 μg of the other plasmid mixed with the same amount of nanomegnetic beads. All other procedures were kept the same. Plates were incubated for 30 min at 37°C over magnetic arrays that were moving laterally at 2 Hz and at 0.2 mm amplitude of displacement. After transfection, 500 μl of the original culture medium was added to each well and neurons were used for live imaging the next day.
Live-cell image acquisition and analysis.
Live-cell imaging was performed using spinning disk confocal microscopy, using an Olympus IX70 microscope with a Hamamatsu EM-CCD Image-EM camera and a PerkinElmer Ultra-VIEW scanner. Images were analyzed using MetaMorph software. To identify axons, neurons were live labeled with an Alexa Fluor 594-labeled neurofascin antibody (clone A12/18, 1:200; NeuroMab) for 45 min at 37°C. For antibody labeling, the Tris-containing buffer of anti-neurofascin was first exchanged into 0.05 m borate buffer by dialysis using a D-tube dialyzer (Novagen) and the antibody was then fluorescently labeled using a DyLight 594 Antibody Labeling Kit (Thermo Scientific). Axons were imaged at the AIS and just beyond the AIS initially and then further analyzed at regions ∼100 μm into the axon and toward the end of axons (300–600 μm beyond the cell body). Dendrites were analyzed within the first 100 μm of a primary dendrite. Images were acquired every 3 s for a time period of 3 min and kymographs were created to analyze direction of vesicle transport.
Vesicles were tracked for their visible lifetime and analyzed to determine the proportion of mobile and immobile vesicles and the proportion of mobile vesicles moving anterogradely, retrogradely, or bidirectionally. Vesicles with a total movement <2 μm during their visible lifetimes were classed as immobile. Vesicles moving in both directions but with net movement of <2 μm (during their visible lifetimes) were classed as bidirectional even though total movement may have been larger. Vesicles with net movements >2 μm in either direction by the end of their visible lifetimes were classed as anterograde or retrograde accordingly.
Direction of transport of at least 110 vesicles per condition was analyzed from at least 20 neurons obtained from three or more individual experiments. Statistically significant differences were calculated using Fisher's exact tests (pairwise comparisons for low numbers of vesicles) and Mann–Whitney tests (comparison of ranks for high number of vesicles).
Drug treatments.
Neurons were treated after 14 DIV with Latrunculin (LatA; Merck), tubulin deacetylase inhibitor trichostatin A (TSA; Sigma-Aldrich) or Taxol (semisynthetic; Sigma-Aldrich). LatA and TSA were added to the cells at a final concentration of, respectively, 2.5 μm and 250 nm. Cells were incubated with LatA for 2 h at 37°C and with TSA overnight at 37°C. After treatment, cells were fixed and used for immunostaining. Neurons were treated with low-dose taxol (1 nm) for 3 h and either fixed and used for immunostaining or used directly for live-cell imaging. DMSO was used as a control for all drug treatments.
AnkG knock-down using adenovirus-mediated shRNA.
The adenoviruses were gifts from Dr. Matthew Rasband (Baylor College of Medicine, Houston, TX). E18 cortical neurons were infected at 3 DIV with adenovirus containing either AnkG shRNA or GFP as a control. Adenovirus sense sequences for AnkG shRNA was 5-GCCGTCAGTACCATCTTCT-3. Efficacy and specificity of these shRNA sequences were demonstrated previously (Hedstrom et al., 2007) and production of the adenovirus was as reported previously (Hedstrom et al., 2008).
Neurons were infected at 3 DIV for 3 h and then washed with warm HBSS before changing back to the conditioned medium from the same cells. After 72 h, cells were fixed with 4% PFA for 8 min at room temperature (RT) followed by 100% methanol at RT for 3 min. The cells were then immunostained for GFP, AnkG, or integrin β1.
Quantification of axonal and dendritic β1 integrin expression.
β1 Integrins were detected by immunofluorescence and images were acquired by fluorescence microscopy using a Leica DM6000B microscope with a 63× objective. To quantify β1 expression in axons, identical conditions for immunostainings were used within each experiment and images were acquired with identical microscope settings. Axons were identified as long and thin processes with positive AnkG staining. Cells with more than one AnkG positive process were excluded. Experiments were repeated three times and, each time, at least 30 axons per group were measured in each experiment. Dendritic fluorescence was quantified similarly, measuring the average intensity of staining of all primary dendrites of each cell analyzed.
Images were processed using ImageJ. Axons were traced with the freehand selection tool (line width = 3). Fluorescence intensity of each pixel along an axon was then obtained using the RGB Profiler plugin (Laummonerie and Mutterer, 2003) and data were analyzed in R (R Development Core Team, 2008). After background subtraction (radius = 50 pixel), fluorescent intensity was averaged for each 20 μm segment along the axon. Data from the three experiments representing the fluorescence intensity at each location (distance from the beginning of axon initial segment) were averaged and represented as means ± SEM at each 20 μm segment. Statistical analysis was performed using two-way ANOVA with β1 integrin fluorescent intensity as the dependent variable and treatment condition and axon position as independent factors. ANOVA was followed by Tukey's HSD post hoc test chosen for its conservativeness in comparing between groups.
Quantification of axonal length and branch number, and total dendritic length.
Axonal length was measured on the picture acquired using the above-mentioned method. Total axon length was calculated by adding together the lengths of each axon branch to the length of the primary axon. This figure was used to represent the axon length for each neuron. The axon was defined by the presence of AnkG or neurofascin staining. Branching number was defined as the total branching points on an axon. Data from each group were averaged and represented as means ± SEM. One-way ANOVA and Tukey's HSD post hoc test was used to compare between groups. Total dendrite length was measured by adding together the lengths of each of the primary dendrites per cell.
Results
The series of experiments that follow use integrins as a marker to demonstrate a mechanism for the loss of axon growth ability with maturation. We show in the first two sections that integrins become excluded from the axons of cortical neurons as the neurons mature and that this is associated with a change in the directionality of integrin transport, with integrins in maturing axons being transported retrogradely back to the cell body. We then test the various mechanisms that have been proposed for selective transport of molecules to the different parts of the neuron to find which of these is responsible for excluding integrins from axons and for switching transport to retrograde. Of the seven mechanisms that we tested, we found two that are relevant. Readers who wish to go directly to the positive results can skip to the sections entitled “Is the axon initial segment a barrier to integrin entry into axons?,” “Which component of the AIS restricts integrin mobility?,” and “Does the activation state of Arf6, controlled by GAPs and GEFs, change the directionality of transport in axons?” The main conclusion is that integrins become excluded from axons due to the properties of the axon initial segment and to activation of the recycling endosome GTPase ARF6 by the GEFs ARNO and EFA6, which switches integrin transport to retrograde. Forced inactivation of ARF6 restores anterograde transport and enables increased axon growth.
Integrins are progressively excluded from mature CNS axons
To examine the presence and distribution of integrins in CNS neurons, cortical neurons were immunostained for integrin β1 after being cultured for 1 d, 1 week, or 2 weeks (Fig. 1). Axons were identified through staining for the AIS marker AnkG except for day 1, when the AnkG was not yet expressed (Nakata et al., 2003; Song et al., 2009) and the axon was identified by morphology as the longest process. Early on (day 1, Fig. 1A–C), all processes contained vesicles that stained for β1 integrin, these molecules being uniformly distributed in the soma and all the processes of the neurons. In more mature neurons cultured for 1 week, β1 integrin was present at a reduced level in axons, being concentrated in the proximal 100 μm, but the soma and dendrites continued to contain many β1-integrin-positive vesicles (Fig. 1D). After 2 weeks of culture, β1 integrin was still present in the somatodendritic domain, but was excluded from axons beyond the AIS [see Figs. 1G, 4A (DMSO), 7A]. We also stained maturing neurons for the α integrins α5 and αV, finding the same distribution. The two α integrins were present ubiquitiously at 1 d in culture (Fig. 1B,C). By 7 DIV, the α integrins were much reduced in the axons beyond the AIS (Fig. 1E,F). These findings suggest that integrins are excluded from CNS axons as they mature and are consistent with previous in vivo observations (Bi et al., 2001; Mortillo et al., 2012).
Figure 1.
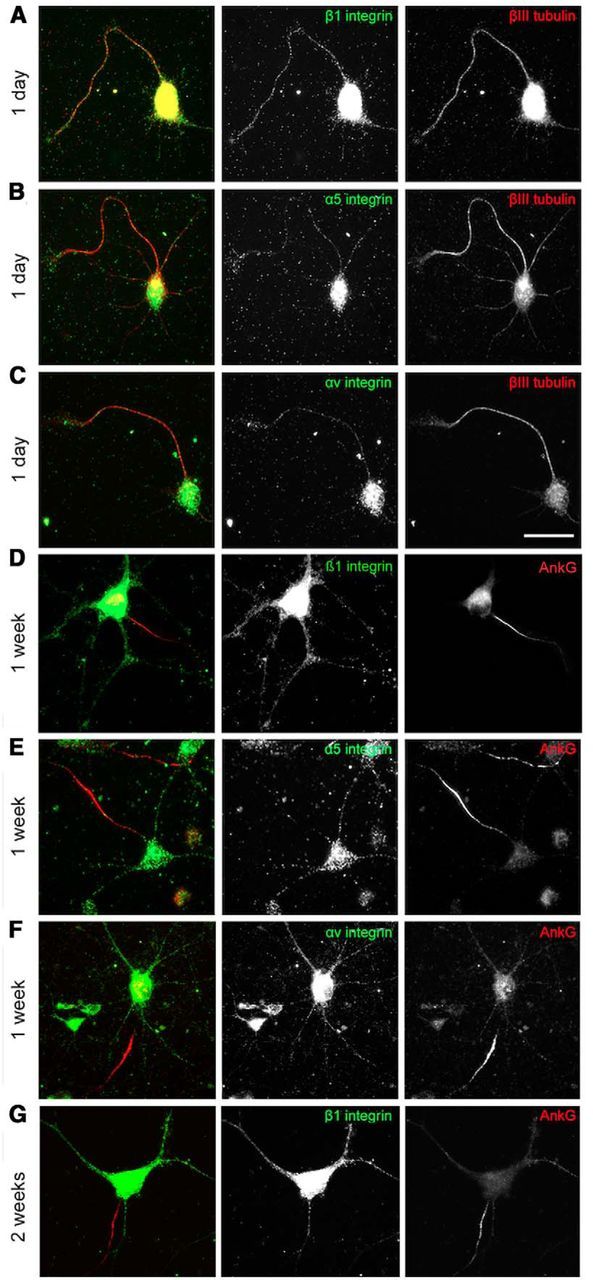
Integrins are not transported into the axons of mature CNS neurons. A–C, At 1 DIV, various types of integrins (β1, α5, and αv) are distributed ubiquitously in cell bodies and neurites. D, At 7 DIV, the β1 integrin level is gradually reduced in axons and dendrites. At 1 week (E, F), α integrins are restricted to the somatodendritic domain and absent in axons. At 2 weeks (G), β1 integrins disappear from axons too. Scale bars: A–C, 25 μm; D–G, 50 μm.
Figure 4.

Tubulin PTM modification results in increased axonal β1 integrin. A, B, Low-dose taxol treatment increased β1 integrin level in the axons with group difference of 3.149 ± 0.697 arbitrary units (p < 0.001). C, D, TSA-induced increased acetylation did not increase β1 integrin level in the axons. E, F, Taxol treatment increased α9-GFP vesicle anterograde movement. **p < 0.01; *p < 0.05. Scale bars in A–C, 25 μm.
Figure 7.

Direction of axonal integrin transport in mature CNS axons is controlled by ARF6. A, ARF6 was present in axons and dendrites of neurons at both 2 and 14 DIV. B, ARNO is increased in axons and dendrites of 14DIV neurons compared with 2 DIV and is expressed throughout the axon (MAP2 negative). C, Rab11 is present in dendrites but not in axons of 14 DIV neurons. D, For live imaging, neurons were cotransfected with α9-GFP and either ACAP1-myc or ARNO-E156K-myc and axons were identified with neurofascin. E, F, G, Kymographs and analysis of α9-GFP vesicle traffic in control axons or after transfection with ACAP1, ARNO-E156K, or ARNO-E156K combined with taxol treatment. ***p < 0.001; **p < 0.01; *p < 0.05.
Overall direction of integrin transport changes with development
To gain some insight into how integrins traffic in CNS neurons and how they might be excluded from axons, we first visualized axonal integrin transport in real time. We transfected 9 and 2 DIV cortical neurons with GFP-tagged α9 integrin (α9-GFP) and used these neurons for live-cell imaging using spinning disc confocal microscopy at 10 and 3 DIV. We have focused on α9 integrin because our previous work has shown that expression of α9 integrin in sensory neurons can enhance regeneration into the spinal cord (Andrews et al., 2009), that α9-GFP targets correctly in stable cell lines, and that it is functional upon overexpression (Eva et al., 2010, 2012). To distinguish axons from dendrites during live-cell imaging, we live-labeled the AIS with Alexa Fluor 594-conjugated anti-neurofascin (Fig. 2A). We initially analyzed transport in the proximal part of the axon due to reports that the initial segment can regulate the polarized distribution of certain membrane proteins (Kobayashi et al., 1992; Bradke and Dotti, 2000; Nakata and Hirokawa, 2003; Song et al., 2009). Overexpressed α9-GFP was present in vesicles in axons at both 3 DIV and at a lower level at 10 DIV. Vesicles were traced in the initial segment and just beyond and their movements analyzed (Eva et al., 2010, 2012). Each vesicle was classified as anterograde, retrograde, bidirectional, or immobile as described in the Materials and Methods section. We observed a striking difference in the overall direction of transport at the two time points (Movies 1, 2). There was a considerable increase in the proportion of vesicles transported retrogradely at 10 DIV (57%) compared with 3 DIV (35%; Fig. 2B,C). However, anterograde transport changed little. We also calculated the percentage of axons in which 0, 1, 2, 3, or ≥4 vesicles were moving anterogradely, retrogradely, or bidirectionally or that were immobile. At 3 DIV, most axons had either 0 or 1 retrograde vesicles, whereas, at 10 DIV, the majority of axons contained 2–4 retrograde vesicles (p < 0.001). This contrasts with vesicle movement in mature dendrites, in which anterograde and bidirectional transport was maintained (Fig. 2B, Movie 3). At 10 DIV, ∼70% of dendrites exhibited both anterograde and retrograde transport and many vesicles showed bidirectional movement (Fig. 2C). We investigated axon transport more distally in axons. We analyzed transport at 10 DIV at two regions, one region ∼100 μm into the axon and one region 300–600 μm beyond the cell body. At both of these regions, we found the same prevailing retrograde transport that we observed in the initial part of the axon (Fig. 2D, Movies 4, 5). These results demonstrate that axonal integrins are mostly being transported back toward the cell body at the time at which integrins are lost from axons, but that transport into dendrites is maintained. This retrograde transport occurs both within the initial segment and throughout the axon.
Figure 2.
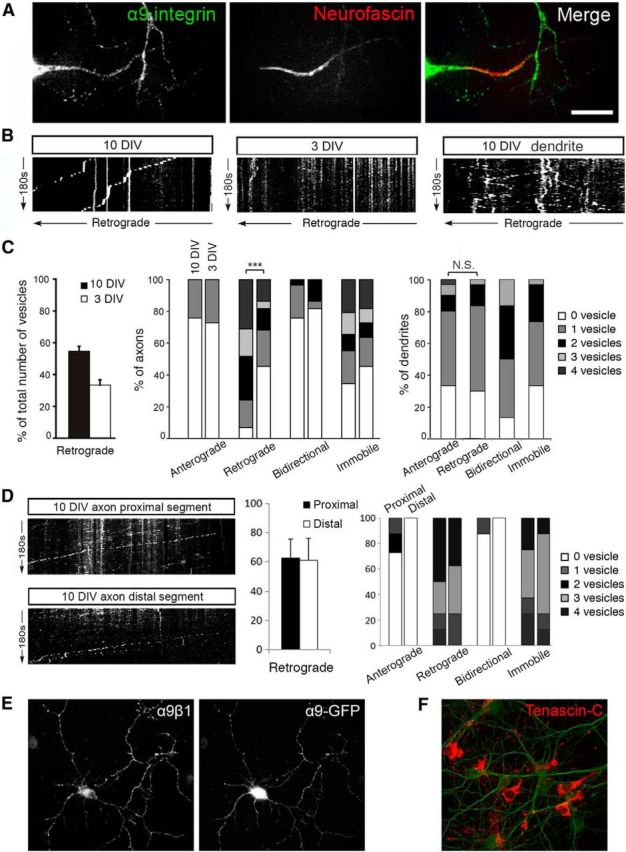
In mature CNS axons, α9-GFP vesicles are retrogradely transported to the cell body. A, Day 10 cortical neuron transfected with α9-GFP was stained with Neufascin to identify the axon, where the vesicle movement was imaged. Transfected α9-GFPs are slightly missorted, with more integrin molecules found in the axons. Scale bar, 25 μm. B, Kymographs of α9-GFP vesicles traffic in axons at 10 DIV (left) and 3 DIV (middle) and in a dendrite at 10 DIV (right). In axons, less retrograde transportation was observed in younger cultures compared with 10 DIV cultures; in dendrites, vesicles were transported in both directions at 10 DIV. C, From 3 to 10 DIV (left), the overall percentage of retrogradely moving vesicles increases; a greater percentage of axons were observed to contain retrogradely moving vesicles and the number of vesicles increased as axons mature (middle; ***p < 0.001, Mann–Whitney test); in dendrites at 10 DIV, no significant difference was observed between number of anterogradely and retrogradely moving α9 vesicles (right). D, Kymographs and analysis of α9-GFP vesicle traffic in axons at 10 DIV at a region 100 μm into the axon (proximal) and toward the end of the axon (distal, 300–600 μm). E, α9β1 dimer was formed in neurons transfected with α9-GFP. F, Neuron cultures produce tenascin-C on the substrate. Scale bars, 50 μm.
In mature cortical neurons, α9 integrin vesicles are retrogradely transported from axon to cell body. Cortical neurons were transfected with α9-GFP (green) and live-imaged at 10 DIV for 3 min. Axons were identified by neurofascin live staining in red at the AIS (Fig. 6).
In young cortical neurons, α9 integrin vesicles were not transported back from axon to cell body. Cortical neurons were transfected with α9-GFP (green) and live-imaged at 3 DIV for 3 min.
In mature cortical dendrites, α9 integrin vesicles traffic in both directions. Cortical neurons were cotransfected with α9-GFP, and dendrites were live-imaged at 10 DIV.
Live imaging of α9-GFP vesicle traffic in axons at 10 DIV at a region proximal to the cell body, typically 100 μm into the axon.
Live imaging of α9-GFP vesicle traffic in axons at 10 DIV at a region distal from the cell body. This was performed at a region as far into the axon as could be traced by α9-GFP signal, typically 300–600 μm into the axon.
What mechanisms might exclude integrins from maturing axons?
In the following sections, we have tested various hypotheses that might explain the exclusion of integrins from axons, and the developmental change in their directionality of transport. We have asked the following: (1) Is exclusion of integrins due to lack of integrin subunit partners, preventing heterodimer formation? (2) Do integrins leave axons because there is no ligand in the environment? (3) Is the axon intial segment a barrier to integrin entry into axons? (4) If so, is this due to its dense actin network or due to modifications of tubulin? (5) Is the change in directionality of transport due to competition between anterograde and retrograde flow? (6) Does recognition of binding sites on integrins by the mu1A subunit of the clathrin adapter protein-1 complex (AP-1) direct integrins away from axons? (7) Does the activation state of Arf6, controlled by GAPs and GEFs, change the directionality of transport in axons?
Is exclusion of integrins due to lack of integrin subunit partners, preventing heterodimer formation?
Inability to form heterodimers due to lack of partners was excluded for two reasons. First, both α and the matching β1 integrin subunits are present in the somatodendritic domain at the time when they are being excluded from axons (Fig. 1). Second, we used an antibody that recognizes only the α9β1 dimer but neither subunit individually. In neurons transfected with α9 integrin, α9β1 heterodimers were present (Fig. 2E).
Do integrins leave axons because there is no ligand in the environment?
This argument is unlikely because all neuronal processes share the same environment in vitro, yet integrin is only excluded from axons. To comfirm the presence of the ligand in our cultures, we immunolabeled tenascin-C (ligand for α9β1 integrin) and demonstrated that it is distributed extensively throughout the cultures, most likely as a result of astrocyte secretion (Fig. 2F).
Is the axon initial segment a barrier to integrin entry into axons?
Because of the sharp cutoff of endogenous integrins at the AIS, we investigated whether the AIS might form a barrier to integrin transport into axons, as was previously suggested as a mechanism of polarized distribution (Kobayashi et al., 1992; Bradke and Dotti, 2000; Nakata and Hirokawa, 2003; Song et al., 2009). We also wondered whether interfering with the AIS might enhance axon growth potential. We first demolished the AIS by reducing expression of AnkG, which is essential for AIS integrity. We used a well characterized adenovirus expressing a GFP-shRNA construct to silence AnkG, which has been previously validated in neurons (Hedstrom et al., 2007, 2008). We transduced cortical neurons at 3 DIV and, by 72 h, AnkG was no longer detectable by immunostaining in >90% of the GFP-positive cells (Fig. 3A). After AnkG knock-down, we could no longer identify axons using AIS neurofascin labeling, so for measurements of integrin distribution, we selected neurons with an obvious single long process with axonal morphology and shorter tapered dendrites. β1 integrin levels were measured using immunostaining as a ratio of GFP level. Neurons lacking AnkG showed a significant increase of integrin β1 in axons (AnkG silenced vs GFP group is 3.917 ± 0.369 by Tukey post hoc analysis, p < 0.001) and it was detectable much farther along the axons compared with GFP controls transduced by the same adenovirus (Fig. 3B–D). However, admission of integrins to the proximal part of the axons was not sufficient to increase axon growth in our cultures. At 7 DIV, the length of control GFP axons was 393.7 ± 77.14 μm, whereas the AnkG knock-down axons averaged 430.2 ± 94.33 μm (p = 0.205 by t test). We found some endogenous integrins on the surface of axons after silencing of AnkG, as well as within the axons, suggesting that they have been inserted into the membrane, but also raising the possibility that some of this entry may be due to lateral diffusion within the surface membrane. Our results suggest that the AIS exerts some control over the entry of integrins into the axon, but that its removal does not allow for increased axon growth.
Figure 3.
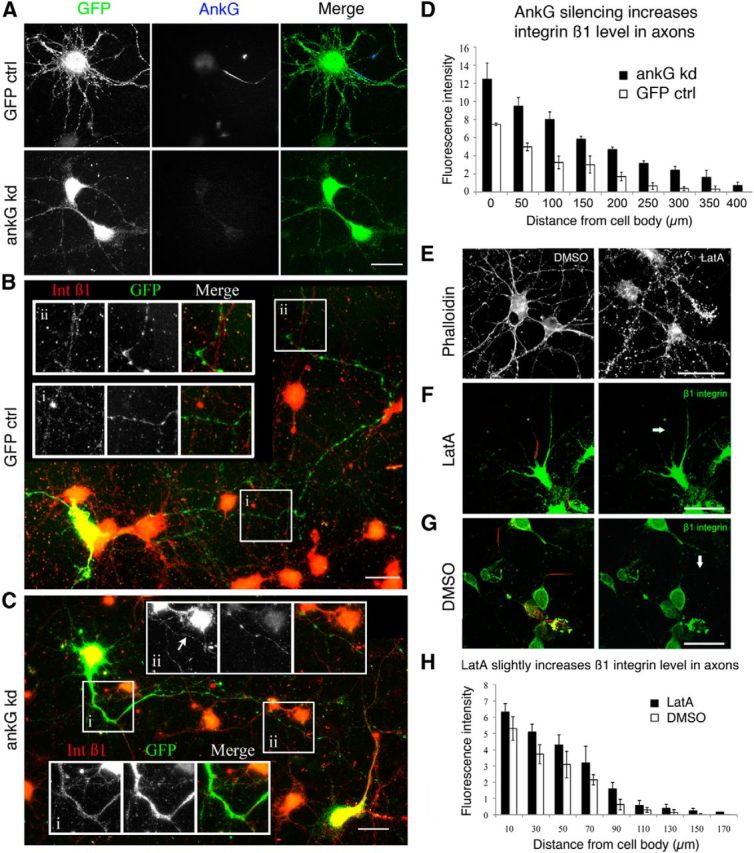
Disruption of the AIS increased β1 integrin expression in axons. A, Adenovirus-mediated delivery of AnkG shRNA resulted in silencing of AnkG in cortical neurons 72 h after infection. B–D, At 72 h after transduction, integrin β1 level is increased in axon (C) compared with GFP control (B) by 3.917 ± 0.369 units (p < 0.001, group difference by Tukey post hoc; D). E, LatA results in disruption of actin filaments (phalloidin staining) and results in a small but significant increase in β1 integrin level in the axons (F–H; p = 0.002). Note that the experimental proceddures of LatA treatment and AnkG knock down were very different, the measurements are not to be compared between experiments. Scale bars, A-C, 25 μm. E–G, 50 μm.
Which component of the AIS restricts integrin mobility?
Having established that the AIS plays a role in the polarized distribution of integrins in neurons, we asked how it might do so. The barrier is proposed to act in two main ways: by the dense actin network or through posttranslational modifications to tubulin in the AIS. To determine whether an intact actin cytoskeleton plays a role in impeding integrin transport into axons, neurons were treated with the actin-filament disrupter LatA or DMSO. After treatment with LatA, staining with phalloidin showed substantial disruption of actin filaments (Fig. 3E). We found a small but significant increase in the β1 integrin level by immunostaining in the region of the AIS (p = 0.002; Fig. 3F–H). LatA treatment also led to a small reduction in integrin fluorescence in dendrites, although this was not statistically significant (fluorescence of 11.58 arbitrary units ± 0.54 SEM for taxol-treated neurons, compared with 12.42 ± 0.28 for DMSO-treated controls, p = 0.176, Student's t test). We can conclude that disrupting the actin network partially releases the blockade of integrin transport, but only into the initial part of CNS axons.
Next, we investigated whether posttranslational modifications (PTMs) of microtubules (MTs) are involved in polarized integrin distribution. PTMs have been shown to provide information for directional transport. For example, kinesin-1 preferentially accumulates in axons, particularly at the tip (Nakata and Hirokawa, 2003), and the kinesin kif3C accumulates in growth cones influenced by tubulin tyrosination and other factors (Gumy et al., 2013). Low-dose taxol changes the location of several PTMs and has been shown to disrupt the selective location of kinesin-1 and kif3C (Hammond et al., 2010; Gumy et al., 2013). Neurons were treated with low-dose taxol (1 nm) or DMSO for 3 h and endogenous β1 integrin was measured in axons by immunostaining. After taxol treatment, there was a significant increase in β1 integrin in the first 150 μm of the axon (3.149 ± 0.697 arbitrary units, p < 0.001, Tukey post hoc analysis; Fig. 4A,B). Taxol treatment led to a small reduction in integrin fluorescence in dendrites compared with DMSO-treated controls, although this was not statistically significant (fluorescence of 13.33 arbitrary units ± 0.47 SEM for taxol-treated neurons compared with 14.43 ± 0.45 for DMSO-treated controls, p = 0.0947, Student's t test). PTMs within the AIS therefore exert some control over the entry of integrins into axons but, again, this is only apparent within the initial part of the axon. Altering PTMs with taxol does not allow for integrin transport into the distal part of the axon.
We then investigated whether taxol changes integrin vesicle traffic direction. α9-GFP-transfected neurons were treated with taxol and subjected to live-cell imaging of the proximal part of the axon. In 10 DIV neurons, which normally show predominantly retrograde integrin flow, we found that taxol treatment increased anterograde transport—72.5% of the taxol-treated neurons have 3 or more vesicles transported anterogradely in axons during the 3 min observation period, compared with none in the control neurons (p = 0.01)—and decreased retrograde transport—42.5% contain retrogradely transported vesicles in the taxol-treated samples compared with 100% in the control group (p = 0.002; Fig. 4E,F). Axon growth measured at 10 DIV showed no effect of taxol treatment (639.1 ± 91.8 for GFP vs 672.2 ± 81.4 for GFP + taxol).
Due to its multiple effects on PTMs, it is rarely possible to identify which of the many effects of taxol is responsible for a particular change (Janke and Bulinski, 2011). However, taxol increases acetylation and increased acetylation by tubulin deacetylase inhibitor TSA influences kinesin localization (Hammond et al., 2010). We tested whether TSA would alter integrin transport in a similar fashion to taxol, however we did not observe a significant effect (p = 0.564; Fig. 4C,D). Together, these results suggest the AIS plays a part in the exclusion of integrins from axons and that local tubulin PTM of axonal MTs are involved in this exclusion through influencing the distribution and directionality of transport of β1 integrin. It is probable that a combination of tubulin PTMs is involved.
Is the change in directionality of transport due to an imbalance between anterograde and retrograde flow?
To answer this question, we first had to find the molecule responsible for retrograde transport of integrins; we hypothesized that it is mediated by dyneins. To test this, we inhibited dynein function by overexpressing HA-tagged dynamitin/p50, which results in dissasembly of dynactin, a complex that mediates the binding of dynein to its cargo (Burkhardt et al., 1997; Melkonian et al., 2007; Kapitein et al., 2010). Neurons were cotransfected with α9-GFP and p50-HA; the cotransfection was confirmed by fixing and labeling with anti-HA (Fig. 5A). We found that inhibiting dynein function almost completely abolished retrograde transport of α9-GFP vesicles in axons (p < 0.001) and the number of immobile vesicles increased compared with cells expressing α9-GFP only (p = 0.02; Fig. 5C,D, Movie 6). This suggests that the retrograde transport of α9-GFP vesicles in axons is mediated by dyneins. However, there was no change in anterograde transport after retrograde transport suppression (Fig. 5D), suggesting that the change in the directionality of integrin transport with neuronal maturity is not due to a change in the balance of a competition between anterograde and retrograde transport machinery.
Figure 5.

Retrograde transport of α9-GFP in axons is mediated by dyneins. A, Neurons were cotransfected with α9-GFP and p50-HA. Scale bar, 30 μm. B, Axons were identified with Neurofascin live staining. C, D, Inhibiting dynein function resulted in an abolishment of retrograde transport but increased immobile vesicles ***p < 0.001; **p < 0.01.
Interfering with dynein stops α9 integrin retrograde transportation in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and HA-p50 and live-imaged at 10 DIV.
Does recognition of binding sites on integrins by AP-1-mediated sorting mechanisms direct integrins away from axons?
Recent work has demonstrated that the distribution of some transmembrane receptors between the axonal and somatodendritic compartment is determined by the recognition of signals within the cytosolic domains of the proteins interacting with the mu1A subunit of the AP-1 complex (Margeta et al., 2009; Farías et al., 2012). Examination of the intracellular domains of α integrins shows integrin subunits α 3, 4, 5, 7, and 9 contain a potential YXXΦ motif that could potentially recognize AP-1. The intracellular domain of integrin β1 has two NPXY motifs independently involved in endocytic sorting and traffic (Margadant et al., 2012; Steinberg et al., 2012).
We used a previously validated mutated dominant negative form of the AP-1 mu1 subunit (Farías et al., 2012) to investigate whether this mechanism is involved in the exclusion of integrins from axonal transport. Day 9 primary cortical neurons were transfected with mutated AP-1 mu1 (mu1-W408S) subunit (Fig. 6A,C) or the wild-type (WT) form (Fig. 6B,D). High levels of expression were seen on the following day, neurons were then fixed and stained to measure the distribution of endogenous integrin β1 (Fig. 6A,B) or α5 (Fig. 6C,D) in axons identified by neurofascin staining. We found no difference in the integrin level in axons of neurons expressing mu1-WT or mu1-W408S (Fig. 6E), indicating that direction by the NPXY motifs on the β1 tail are probably dominant over AP-1-dependent sorting for the restriction of integrin transport into axons. This finding and highlights the probability that there are several mechanisms regulating the polarized distribution of membrane proteins in neurons.
Figure 6.
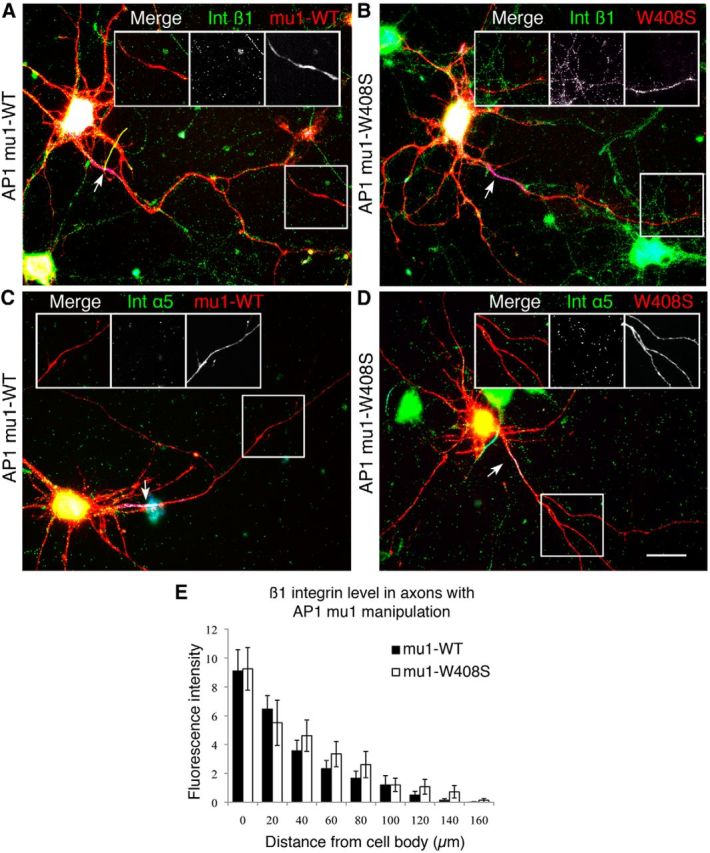
AP-1-dependent somatodendritic sorting is not the main mechanism for integrin axonal exclusion. Cortical neurons were transfected with WT (A, C) and the mutated (B, D) AP-1 mu1 subunit and the integrin level was measured in axons with β1 (A, B) and α5 (C, D). Arrows point to the AIS, which is stained with AnkG in blue and is used to identify axon branches. E, Expression of mutated AP-1 did not increase the concentration of β1 or a5 integrins in axons and did not allow integrin α5 to enter the axon far beyond axon initial segments. Scale bars, 25 μm.
Does the activation state of Arf6, controlled by GAPs and GEFs, change the directionality of transport in axons?
In adult DRG axons, there is no block of integrin transport into axons. Many of the vesicles that contain integrin are marked with the GTPase Arf6 and manipulation of the activation state of Arf6 alters the direction of integrin transport, with activation by GEFs promoting retrograde transport and reduced axon growth, whereas inactivation by GAPs had the opposite effect (Eva et al., 2012). We hypothesized that Arf6 activation/inactivation might have a similar effect on the direction of integrin transport in CNS axons.
We first visualized the presence and distribution of Arf6 in cortical neurons by immunostaining, finding it in axons and dendrites at both 2 and 14 DIV (Fig. 7A). Conversely, the GEF ARNO was barely detectable at 2 DIV, but was strongly labeled in dendrites and throughout the axon at 14 DIV, as indicated by the absence of the dendritic marker MAP2 (Fig. 7B). Expression of another Arf6 GEF, EFA6, also increases with development (Sakagami et al., 2006). Interestingly, we also found that Rab11 was largely excluded from mature axons, being present at low levels in axons compared with dendrites in 14 DIV neurons (Fig. 7C).
We then manipulated Arf6 activity in 9 DIV neurons by expressing constructs that alter Arf6 activation. ACAP1 is an Arf6 GAP that inactivates Arf6 and favors anterograde integrin transport when overexpressed in sensory neurons (Eva et al., 2012); ARNO-E156K (a catalytically inactive form of ARNO) also inactivates Arf6 with similar effects. However, these molecules do not simply affect ARF6 activation; ACAP1 also functions as a coat protein and ARNO associates with trafficking-related scaffolding molecules and paxillin (Li et al., 2007, Mo et al., 2012, Torii et al., 2010). These constructs were cotransfected with α9-GFP to allow live imaging and analysis of axonal traffic. Cotransfection was confirmed afterward by fixing and labeling for the epitope tag (Fig. 7D). Axons were identified with anti-neurofascin-594.
Quantification of the direction of transport of α9-GFP vesicles showed that ACAP1 transfection resulted in a large increase in the percentage of axons containing vesicles moving in an anterograde direction (44% compared with 9% in cells with α9-GFP alone, p = 0.04) and less with retrograde transport (52% vs 95% in α9-GFP alone, p = 0.002; Fig. 7E–G, Movie 7). Similar to overexpression of ACAP1, overexpression of ARNO-E156K resulted in increased anterograde (53% of neurons vs 9% in controls, p = 0.003) and decreased retrograde transport (87% of neurons vs 95% in controls, p = 0.001; Fig. 7E–G, Movie 8). ACAP1 and ARNO-E156K not only changed the overall percentage of vesicles moving anterogradely and retrogradely (Fig. 7F), but also the percentage of axons containing multiple vesicles moving in either direction (Fig. 7G). Interestingly, overexpression of ARNO-E156K also resulted in an increased percentage of axons showing no immobile vesicles (Fig. 7G). This might suggest that overexpression of ARNO-E156K forces immobile vesicles in the axons to move anterogradely. Combining ARNO-E156K with taxol further increased the percentage of anterograde-transporting vesicles compared with ARNO-E156K alone from 36% to 57% (p = 0.028, Mann–Whitney test; Fig. 7E–G, Movie 9). These results suggest that modulation of ARF6 by the two GEFS ARNO and EFA6 is probably responsible for the developmental change from bidirectional to retrograde flow of integrin. Transfection with ACAP1 and ARNO-E156K will reverse the activation of ARF6, but will also have other effects due to the coat protein actions of ACAP1 and the interactions of ARNO with trafficking-related scaffolding proteins and paxillin (Mo et al., 2012, White et al., 2010).
Overexpression of ARF6 GAP ACAP1 increases anterograde transportation and decreases retrograde transportation of α9 integrin in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and ARF6 GAP ACAP1-myc.
Interfering with ARF6 GTP by overexpressing ARNO-E156K increases anterograde transportation and decreases retrograde transportation of α9 integrin in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and a catalytically inactive form of ARF6 GEF ARNO, ARNO-E156K.
Combining ARNO-E156K and taxol further increases anterograde transportation of α9 integrin in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and an ARNO-E156K and treated with taxol.
We then investigated whether changing the direction of transport could result in increased localization of endogenous integrins to maturing axons and if the changes could result in an enhancement of axon growth. We chose to do this using ARNO-E156K either alone or in conjunction with taxol treatment. We measured endogenous integrin β1 in axons transfected with ARNO-E156K in the presence or absence of taxol. Nine DIV cortical neurons were transfected with GFP, ARNO-E156K, or ARNO-E156K combined with taxol (Fig. 8A–C). ARNO-E156K expression resulted in a significant increase in axonal integrin compared with GFP controls, with an overall difference of 3.168 ± 0.362 units (p < 0.001; Fig. 8D). Combining ARNO-E156K and taxol treatment increased the difference to GFP control to 6.223 ± 1.436 units (p < 0.001) and to the ARNO-E156K alone group in the initial 200 μm (t = 0.00269, paired t test; Fig. 8D). Importantly, this increase in integrin transport was also associated with an increase in axon growth (903 ± 146 vs 694 ± 134, ARNO-E156K vs GFP, t < 0.001, unpaired t test; Fig. 8E), but not the branching number (6.48 ± 1.13 vs 5.83 ± 1.06, t = 0.384). The combination with taxol did not further increase axon length. Overexpression of WT ARNO did not produce any significant change in integrin level or axon length compared with controls, with integrin excluded from axons (data not shown). Expressing ARNO E156K also had an effect on dendrites, leading to shorter dendrites compared with GFP-expressing controls (total ARNO E156K dendrite length 399.2 ± 20 μm compared with 498.2 ± 15.1 for GFP expressing controls, p < 0.001, Student's t test). They also had more primary dendrites per cell (average number of 8 ± 0.29 for ARNO E156K compared with 5.3 ± 0.2 for controls, p < 0.0001, t test). Neurons expressing ACAP1 also had shorter dendrites compared with GFP-expressing controls (total ACAP1 dendrite length 287.4 ± 12.3 compared with 510.2 ± 15.9 for GFP-expressing controls, p < 0.0001, Student's t test), although there was no difference in the number of primary dendrites per cell between the two groups.
Figure 8.
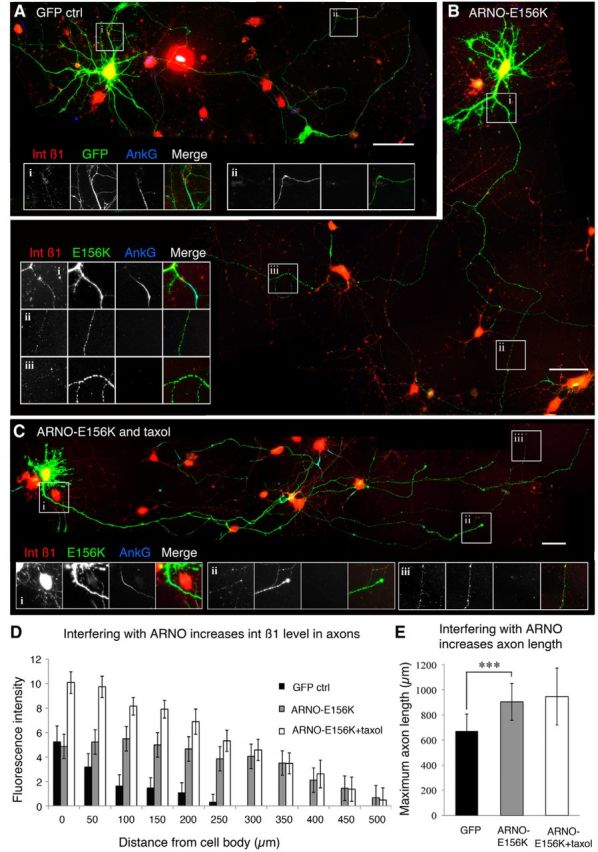
ARNO-E156K significantly increased integrin level in axons as well as axon length. A, In day 9 cortical neurons transfected with GFP, integrin β1 was absent in axons beyond the AIS (stained with AnkG). B, Overexpression of ARNO-E156K increased β1 integrin level in axons including in the distal part (ii,iii). C, Combination of ARNO-E156K with taxol treatment showed a similar effect. D, Integrin β1 fluorescent intensity in axons is increased with ARNO-E156K or the combination treatment (ARNO-E156K vs GFP: 3.168 ± 0.362; ARNO-E156K + taxol vs GFP: 6.223 ± 1.436; p < 0.001 in both cases). Compared with ARNO-E156K alone, the combination increased integrin β1 in the proximal 200 μm (t = 0.0027, paired t test), but not in the distal part. E, ARNO-E156K significantly increased axon length; combination with taxol had no additional effect. ***p < 0.001. Scale bars in A–C, 50 μm.
Putting these findings together, we found that, as with sensory axons, the overall direction of integrin transport in CNS axons is controlled by the activation state of Arf6. Forced inactivation of ARF6 causes an increase in anterograde transport of α9-GFP vesicles, higher integrin levels, and increased axon growth. For PC12 cells growing on tenascin-C, the effects of ARF6 manipulations were α9 integrin dependent (Eva et al., 2012), but in the present experiments with axons growing in a complex environment, it is likely that some of the other growth-related molecules transported in ARF6 vesicles may also be important.
In summary, integrins are excluded from axons by the properties of the AIS, with posttranslational modifications to tubulin playing a part, and by the developmental change in the overall direction of integrin transport driven by ARF6 activation. Inactivation of ARF6 restores integrin transport to distal cortical axons and increases axon growth.
Discussion
Our experiments demonstrate a developmental change in neuronal integrin trafficking, leading to the exclusion of integrins from cortical axons after the initial phase of axon growth. This change affects axon growth and suggests a mechanism that contributes to the developmental loss of intrinsic axon growth potential in mature CNS axons.
Mechanisms of integrin transport
Integrins traffic in vesicles marked by GTPases that regulate vesicle behavior. These mechanisms are well understood in cancer cells, the migration and invasion of which involve integrins (Arjonen et al., 2011; Margadant et al., 2011). We previously investigated integrin trafficking in adult sensory neurons (which transport integrins in both central and peripheral branches) and found that integrin trafficking shares similar mechanisms to cancer cells (Eva et al., 2010, 2012). Integrins associate with vesicles marked by Rab11 and ARF6, which regulate integrin trafficking in the growth cone and in axons. Rab11 vesicles undergo mainly short range movements, whereas for long-range transport, integrins are in vesicles regulated by ARF6. We now find both Rab11 and ARF6 are present in cortical neurons, although axonal Rab11 levels decline with maturation and it becomes principally localized to dendrites. We demonstrate that ARF6 regulates a mechanism that leads to predominantly retrograde transport of integrins away from axons as they become more mature. Retrograde transport is dynein dependent because blocking dynein with dynamitin/p50 stops retrograde flow of integrin vesicles.
Mechanisms of selective localization in neurons
Many molecules, including some glutamate receptors, various postsynaptic proteins, transferrin receptor, MT-associated protein MAP2, and others, are restricted to the somatodendritic domain in mature neurons (Dotti and Poo, 2003; Farías et al., 2012). Several mechanisms have been demonstrated for selective localization of molecules. The AIS of CNS neurons may act as a selective transport filter, preserving neuronal polarization by restricting diffusion of proteins between the somatodendritic and axonal domains (Kobayashi et al., 1992; Winckler et al., 1999; Dotti and Poo, 2003; Nakata and Hirokawa, 2003; Song et al., 2009; Farías et al., 2012). The AIS exhibits a dense network of actin, which can restrict access to axons by acting as size filter or by supporting retrograde myosin-driven transport (Arnold, 2009; Lewis et al., 2009; Song et al., 2009) and actin is also important for directing molecules to dendrite or axon-directed vesicles (Petersen et al., 2014). The AIS also has cross-linked and tyrosinated MTs (Rasband, 2010). PTMs of MTs at the AIS and elsewhere can alter the distribution of kinesins (Hammond et al., 2010; Nakata et al., 2011; Gumy et al., 2013). Different cargos are linked to different kinesins, several of which are developmentally regulated, and the distribution of cargo is therefore influenced by the type of kinesin and the cargo (Huang and Banker, 2012; Nakata et al., 2011; Heintz et al., 2014). Subcellular localization of some molecules in many cell types, including neurons, is influenced by adaptor proteins such as AP-1, with the mu1A subunit of AP-1 being implicated in localization in neurons (Farías et al., 2012). The balance between anterograde and retrograde transport can determine localization into neuronal dendrites (Kapitein et al., 2010). Our studies were designed to find out which of these possible mechanisms is responsible for the exclusion of integrins from mature cortical neurons.
Why do integrins become excluded from axons?
In the cortical neurons that we studied, integrins were initially evenly present in all the neuronal processes, but from 7 DIV onward, they became progressively excluded from axons, remaining present in the soma and dendrites. Because the distribution of integrins showed an anatomical cutoff at the AIS, an obvious hypothesis was that this structure is a barrier to integrin transport. We found direct evidence for this when we disrupted the AIS through knock-down of AnkG, which allowed integrin to enter the axons and to be transported for a considerable distance along their length. We then investigated whether the AIS filtering could be ascribed to the actin or tubulin components within it. We found evidence for a minor role of the actin cytoskeleton, disruption of which admitted an increased amount of integrin into the initial segment, as seen also for the transferrin receptor (Petersen et al., 2014). However, modification of the MTs had a greater effect, with low-dose taxol treatment allowing integrins into the initial segment and proximal axon. Taxol has many effects on MTs, including on acetylation, tyrosination, and polyglutamylation (Hammond et al., 2010; Nakata et al., 2011; Kee et al., 2012). Increasing acetylation with a deacetylase inhibitor did not have the same effect. Low-dose taxol treatment can enhance axon regeneration over inhibitory substrates both in vitro and in vivo (Mansfield and Gordon-Weeks, 1991; Hellal et al., 2011). This has been ascribed to its effects on MT dynamics, but our results suggest that an effect on transport may also be significant.
For some molecules that are restricted to the somatodendritic compartment, compartmentalization relies on the adaptor protein AP-1 and clathrin (Margeta et al., 2009; Farías et al., 2012). Adaptor proteins such as AP-1 recognize cargos via a YXXØ (present in some α integrins) or [DE]XXXL[LI] motif (Bonifacino and Traub, 2003; Caster et al., 2013; Ren et al., 2013); however, we did not find evidence that the mu1A subunit of AP-1 controls integrin distribution. Integrin trafficking is also directed by the two NPXY motifs on the intracellular tail of β1 and other β chains, which bind competitively to many ligands (Legate and Fassler, 2009, Morse et al., 2014). Our results suggest that trafficking direction by the β chain is dominant over the α chains.
However, we believe that the main mechanism for selective integrin distribution is through regulation of trafficking via the small GTPase ARF6. That this type of mechanism controls the selective distribution of integrin was suggested when we found that, as cortical axons mature, the overall direction of integrin transport changes to predominantly retrograde in axons, but not in dendrites. It seems probable that this predominantly retrograde movement empties the axons of integrin and excludes integrin entry. Our previous work in sensory axons has shown that rapid transport occurs in vesicles marked by ARF6. Moreover, the activity of ARF6, controlled by balanced activity of GAPs and GEFs, affects vesicle transport direction. Activation of ARF6 by GEFs favors retrograde axon transport and internalization at the growth cone, whereas inactivation by GAPs has the opposite effect (Eva et al., 2012). We found evidence that this switch also controls the direction of transport in cortical neurons. Developmental activation of ARF6 by GEFs is responsible for the switch to retrograde flow and anterograde flow and axon growth can be restored by overexpressing a GAP or the dominant-negative GEF ARNO-E156K. During cortical neuronal maturation, two ARF6 GEFs, ARNO and EFA6, are strongly upregulated (Sakagami et al., 2006) and our findings suggest that developmental upregulation of these two GEFs is involved in excluding integrins from axons. A number of GEFs and GAPs localize to dendrites (Scholz et al., 2010; Oku and Huganir, 2013). Interestingly, in DRG neurons, we have found that ARNO mRNA is preferentially expressed in embryonic rather than adult neurons (Gumy et al., 2011).
ARF6 and axon growth
Axon growth involves the coordination of many processes, including the transport, internalization, and recycling of integrins. In these processes, ARF6 is involved at several stages, including recruitment of coat proteins and effector molecules, generation of phosphoinositides and phosphatidic acid, clathrin-mediated events, and polarized delivery of other GTPases (Hernández-Deviez et al., 2004, 2005, 2007; Bouschet et al., 2007). ARF6 GAPs and GEFs, can also act as scaffolds for the assembly of signaling complexes (for review, see Donaldson and Jackson, 2011). ACAP-1 also associates directly with β1 integrin and, together with Rab11 and ARF6, participates in its recycling (Powelka et al., 2004; Li et al., 2005). ARNO associates with various scaffolds and with paxillin (Mo et al., 2012, Torii et al., 2010, White et al., 2010). In sensory axons, the balance of ARF6 GAPs and GEFs translates to effects on axon growth, with ARF6 inactivation by GAPs increasing growth (Eva et al., 2012). Our findings in cortical neurons now show that a similar mechanism operates in CNS axons. However, in the CNS, the developmental activation of ARF6 by GEFs leads to the exclusion of integrins from axons by favoring retrograde transport. This mechanism likely involves an interaction between the scaffolding molecules JIP3 and JIP4, ARF6, and kinesin and dynein motors (Isabet et al., 2009; Suzuki et al., 2010; Montagnac et al., 2011).
CNS axon regeneration
Axons will only regenerate if they are equipped with the appropriate machinery. Our results show that CNS axons lack at least one critical component, integrins. Sensory axons transport integrins and, if transduced to express α9 integrin and the activator kindlin-1, can regenerate profusely in the spinal cord (Andrews et al., 2009, Tan et al., 2012, Cheah et al., 2013). However, α9 integrin expressed in cortical neurons in vivo does not enter axons. We have identified mechanisms that exclude integrins from cortical axons. These may also exclude additional molecules that affect axonal regenerative ability, including ribosomal protein, which is excluded from CNS retinal axons, diminishing their regenerative ability through local translation (Verma et al., 2005), and trkB, which is excluded from corticospinal axons (Hollis et al., 2009). Overall, it seems probable that the poor intrinsic regenerative performance of CNS axons is partly due to the exclusion of key growth-related molecules.
Notes
Supplemental material for this article is available at www.brc.cam.ac.uk. This material has not been peer reviewed.
Footnotes
This work was supported by the Medical Research Council, the Christopher and Dana Reeve Foundation, European Union Framework 7 Project Plasticise, the European Research Council, the John and Lucille van Geest Foundation, and the National Institute for Health Research Cambridge Biomedical Research Centre. We thank Matthew N. Rasband for kindly providing the adenoviruses for AnkG silencing experiment, Juan Bonifacino for AP-1 constructs, and Menghon Cheah for assistance.
J.W.F. is a paid consultant for Acorda Therapeutics and Vertex Pharmaceuticals. The remaining authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Andrews MR, Czvitkovich S, Dassie E, Vogelaar CF, Faissner A, Blits B, Gage FH, ffrench-Constant C, Fawcett JW. Alpha9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. J Neurosci. 2009;29:5546–5557. doi: 10.1523/JNEUROSCI.0759-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjonen A, Kaukonen R, Ivaska J. Filopodia and adhesion in cancer cell motility. Cell Adh Migr. 2011;5:421–430. doi: 10.4161/cam.5.5.17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold DB. Actin and microtubule-based cytoskeletal cues direct polarized targeting of proteins in neurons. Sci Signal. 2009;2:pe49. doi: 10.1126/scisignal.283pe49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi X, Lynch G, Zhou J, Gall CM. Polarized distribution of integrin alpha5 in dendrites of hippocampal and cortical neurons. J Comp Neurol. 2001;435:184–193. doi: 10.1002/cne.1201. [DOI] [PubMed] [Google Scholar]
- Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, Lee JK, Goldberg JL, Lemmon VP, Bixby JL. Kruppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci U S A. 2012;109:7517–7522. doi: 10.1073/pnas.1120684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Bouschet T, Martin S, Kanamarlapudi V, Mundell S, Henley JM. The calcium-sensing receptor changes cell shape via a β-arrestin-1–ARNO–Arf6–ELMO protein network. J Cell Sci. 2007;120:2489–2497. doi: 10.1242/jcs.03469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Changes in membrane trafficking and actin dynamics during axon formation in cultured hippocampal neurons. Microsc Res Tech. 2000;48:3–11. doi: 10.1002/(SICI)1097-0029(20000101)48:1<3::AID-JEMT2>3.0.CO%3B2-O. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caster AH, Sztul E, Kahn RA. A role for cargo in the activation of ADP-ribosylation factors (Arf) and adaptor recruitment. J Biol Chem. 2013;288:14788–14804. doi: 10.1074/jbc.M113.453621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah M, Andrews MR, Verhaagen J, Fassler R, Faissner A, Fawcett JW. Alpha9 integrin activation in neurite outgrowth and axon regeneration. Soc Neurosci Abstr. 2013;39:341.17. [Google Scholar]
- Chisholm AD. Cytoskeletal dynamics in Caenorhabditis elegans axon regeneration. Annu Rev Cell Dev Biol. 2013;29:271–297. doi: 10.1146/annurev-cellbio-101512-122311. [DOI] [PubMed] [Google Scholar]
- Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. ARF family G proteins and their regulators, roles in membrane transport, development and disease. Nat Rev Mol Cell Biol. 2011;12:362–375. doi: 10.1038/nrm3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Poo MM. Neuronal polarization, building fences for molecular segregation. Nat Cell Biol. 2003;5:591–594. doi: 10.1038/ncb0703-591b. [DOI] [PubMed] [Google Scholar]
- Eva R, Dassie E, Caswell PT, Dick G, ffrench-Constant C, Norman JC, Fawcett JW. Rab11 and its effector Rab coupling protein contribute to the trafficking of β1 integrins during axon growth in adult dorsal root ganglion neurons and PC12 cells. J Neurosci. 2010;30:11654–11669. doi: 10.1523/JNEUROSCI.2425-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eva R, Crisp S, Marland JR, Norman JC, Kanamarlapudi V, ffrench-Constant C, Fawcett JW. Arf6 directs axon transport and traffic of integrins and regulates axon growth in adult DRG neurons. J Neurosci. 2012;32:10352–10364. doi: 10.1523/JNEUROSCI.1409-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farías GG, Cuitino L, Guo X, Ren X, Jarnik M, Mattera R, Bonifacino JS. Signal-mediated., AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron. 2012;75:810–823. doi: 10.1016/j.neuron.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW. Axon growth and regeneration: intrinsic neuronal effects. Trends Neurosci. 1992;15:5–8. doi: 10.1016/0166-2236(92)90338-9. [DOI] [PubMed] [Google Scholar]
- Gardiner NJ, Fernyhough P, Tomlinson DR, Mayer U, von der Mark H, Streuli CH. Alpha7 integrin mediates neurite outgrowth of distinct populations of adult sensory neurons. Mol Cell Neurosci. 2005;28:229–240. doi: 10.1016/j.mcn.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Gumy LF, Yeo GS, Tung YC, Zivraj KH, Willis D, Coppola G, Lam BY, Twiss JL, Holt CE, Fawcett JW. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA. 2011;17:85–98. doi: 10.1261/rna.2386111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Chew DJ, Tortosa E, Katrukha EA, Kapitein LC, Tolkovsky AM, Hoogenraad CC, Fawcett JW. The kinesin-2 family member KIF3C regulates microtubule dynamics and is required for axon growth and regeneration. J Neurosci. 2013;33:11329–11345. doi: 10.1523/JNEUROSCI.5221-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol Biol Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN. Neurofascin assembles a specialized extra-cellular matrix at the AIS. J Cell Biol. 2007;178:875–886. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the AIS and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz TG, Heller JP, Zhao R, Caceres A, Eva R, Fawcett JW. Kinesin KIF4A transports integrin beta1 in developing axons of cortical neurons. Mol Cell Neurosci. 2014;63:60–71. doi: 10.1016/j.mcn.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, Hoogenraad CC, Bradke F. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–931. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Deviez DJ, Wilson JM. Functional assay of ARNO and Arf6 in neurite elongation and branching. Methods Enzymol. 2005;404:242–252. doi: 10.1016/S0076-6879(05)04023-1. [DOI] [PubMed] [Google Scholar]
- Hernández-Deviez DJ, Roth MG, Casanova JE, Wilson JM. ARNO and Arf6 regulate axonal elongation and branching through downstream activation of phosphatidylinositol 4-phosphate 5-kinase alpha. Mol Biol Cell. 2004;15:111–120. doi: 10.1091/mbc.E03-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Deviez D, Mackay-Sim A, Wilson JM. A role for Arf6 and ARNO in the regulation of endosomal dynamics in neurons. Traffic. 2007;8:1750–1764. doi: 10.1111/j.1600-0854.2007.00649.x. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Hollis ER, 2nd, Jamshidi P, Löw K, Blesch A, Tuszynski MH. Induction of corticospinal regeneration by lentiviral trkB-induced Erk activation. Proc Natl Acad Sci U S A. 2009;106:7215–7220. doi: 10.1073/pnas.0810624106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Akhmanova A, Howell SA, Dortland BR, De Zeeuw CI, Willemsen R, Visser P, Grosveld F, Galjart N. Mammalian golgi-associated bicaudal-D2 functions in the dynein-dynactin pathway by interacting with these complexes. EMBO J. 2001;20:4041–4054. doi: 10.1093/emboj/20.15.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CF, Banker G. The translocation selectivity of the kinesins that mediate neuronal organelle transport. Traffic. 2012;13:549–564. doi: 10.1111/j.1600-0854.2011.01325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Shimazu K, Woo NH, Zang K, Müller U, Lu B, Reichardt LF. Distinct roles of the beta 1-class integrins at the developing and the mature hippocampal excitatory synapse. J Neurosci. 2006;26:11208–11219. doi: 10.1523/JNEUROSCI.3526-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabet T, Montagnac G, Regazzoni K, Raynal B, El Khadali F, England P, Franco M, Chavrier P, Houdusse A, Ménétrey J. The structural basis of Arf effector specificity, the crystal structure of Arf6 in a complex with JIP4. EMBO J. 2009;28:2835–2845. doi: 10.1038/emboj.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton, mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, van der Zwan WA, Wulf PS, Keijzer N, Hoogenraad CC. Probing intracellular motor protein activity using an inducible cargo trafficking assay. Biophys J. 2010;99:2143–2152. doi: 10.1016/j.bpj.2010.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee HL, Dishinger JF, Blasius TL, Liu CJ, Margolis B, Verhey KJ. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol. 2012;14:431–437. doi: 10.1038/ncb2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Storrie B, Simons K, Dotti CG. A functional barrier to movement of lipids in polarized neurons. Nature. 1992;359:647–650. doi: 10.1038/359647a0. [DOI] [PubMed] [Google Scholar]
- Lasiecka ZM, Winckler B. Mechanisms of polarized membrane trafficking in neurons–focusing in on endosomes. Mol Cell Neurosci. 2011;48:278–287. doi: 10.1016/j.mcn.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laummonerie C, Mutterer J. RGB Profiler ImageJ Plugin. 2003. [Accessed May 8, 2015]. Available from: http://rsbweb.nih.gov/ij/plugins/download/RGB_Profiler.java.
- Legate KR, Fässler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci. 2009;122:187–198. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- Lewis TL, Jr, Mao T, Svoboda K, Arnold DB. Myosin-dependent targeting of transmembrane proteins to neuronal dendrites. Nat Neurosci. 2009;12:568–576. doi: 10.1038/nn.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ballif BA, Powelka AM, Dai J, Gygi SP, Hsu VW. Phosphorylation of ACAP1 by Akt regulates the stimulation-dependent recycling of integrin beta1 to control cell migration. Dev Cell. 2005;9:663–673. doi: 10.1016/j.devcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Li J, Peters PJ, Bai M, Dai J, Bos E, Kirchhausen T, Kandror KV, Hsu VW. An ACAP1-containing clathrin coat complex for endocytic recycling. J Cell Biol. 2007;178:453–464. doi: 10.1083/jcb.200608033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield SG, Gordon-Weeks PR. Dynamic post-translational modification of tubulin in rat cerebral cortical neurons extending neurites in culture: effects of taxol. J Neurocytol. 1991;20:654–666. doi: 10.1007/BF01187067. [DOI] [PubMed] [Google Scholar]
- Margadant C, Monsuur HN, Norman JC, Sonnenberg A. Mechanisms of integrin activation and trafficking. Curr Opin Cell Biol. 2011;23:607–614. doi: 10.1016/j.ceb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Margadant C, Kreft M, de Groot DJ, Norman JC, Sonnenberg A. Distinct roles of talin and kindlin in regulating integrin α5β1 function and trafficking. Curr Biol. 2012;22:1554–1563. doi: 10.1016/j.cub.2012.06.060. [DOI] [PubMed] [Google Scholar]
- Margeta MA, Wang GJ, Shen K. Clathrin adaptor AP-1 complex excludes multiple postsynaptic receptors from axons in C. elegans. Proc Natl Acad Sci U S A. 2009;106:1632–1637. doi: 10.1073/pnas.0812078106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachie AB, Cingolani LA, Goda Y. A stabilising influence: Integrins in regulation of synaptic plasticity. Neurosci Res. 2011;70:24–29. doi: 10.1016/j.neures.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkonian KA, Maier KC, Godfrey JE, Rodgers M, Schroer TA. Mechanism of dynamitin-mediated disruption of dynactin. J Biol Chem. 2007;282:19355–19364. doi: 10.1074/jbc.M700003200. [DOI] [PubMed] [Google Scholar]
- Mo J, Choi S, Ahn PG, Sun W, Lee HW, Kim H. PDZ-scaffold protein, Tamalin promotes dendritic outgrowth and arborization in rat hippocampal neuron. Biochem Biophys Res Commun. 2012;422:250–255. doi: 10.1016/j.bbrc.2012.04.136. [DOI] [PubMed] [Google Scholar]
- Montagnac G, de Forges H, Smythe E, Gueudry C, Romao M, Salamero J, Chavrier P. Decoupling of activation and effector binding underlies Arf6 priming of fast endocytic recycling. Curr Biol. 2011;21:574–579. doi: 10.1016/j.cub.2011.02.034. [DOI] [PubMed] [Google Scholar]
- Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry. 2014;53:810–820. doi: 10.1021/bi401596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortillo S, Elste A, Ge Y, Patil SB, Hsiao K, Huntley GW, Davis RL, Benson DL. Compensatory redistribution of neuroligins and N-cadherin following deletion of synaptic β1-integrin. J Comp Neurol. 2012;520:2041–2052. doi: 10.1002/cne.23027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JP, Santiago-Medina M, Gomez TM. Regulation of axonal outgrowth and pathfinding by integrin-ECM interactions. Dev Neurobiol. 2011;71:901–923. doi: 10.1002/dneu.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J Cell Biol. 2003;162:1045–1055. doi: 10.1083/jcb.200302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Niwa S, Okada Y, Perez F, Hirokawa N. Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J Cell Biol. 2011;194:245–255. doi: 10.1083/jcb.201104034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku Y, Huganir RL. AGAP3 and Arf6 regulate trafficking of AMPA receptors and synaptic plasticity. J Neurosci. 2013;33:12586–12598. doi: 10.1523/JNEUROSCI.0341-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JD, Kaech S, Banker G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J Neurosci. 2014;34:4135–4147. doi: 10.1523/JNEUROSCI.3779-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powelka AM, Sun J, Li J, Gao M, Shaw LM, Sonnenberg A, Hsu VW. Stimulation-dependent recycling of integrin beta1 regulated by Arf6 and Rab11. Traffic. 2004;5:20–36. doi: 10.1111/j.1600-0854.2004.00150.x. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008. [Google Scholar]
- Rasband MN. The AIS and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- Ren X, Farías GG, Canagarajah BJ, Bonifacino JS, Hurley JH. Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell. 2013;152:755–767. doi: 10.1016/j.cell.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakagami H, Suzuki H, Kamata A, Owada Y, Fukunaga K, Mayanagi H, Kondo H. Distinct spatiotemporal expression of EFA6D, a guanine nucleotide exchange factor for Arf6, among the EFA6 family in mouse brain. Brain Res. 2006;1093:1–11. doi: 10.1016/j.brainres.2006.02.058. [DOI] [PubMed] [Google Scholar]
- Scholz R, Berberich S, Rathgeber L, Kolleker A, Köhr G, Kornau HC. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66:768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM. A selective filter for cytoplasmic transport at the AIS. Cell. 2009;136:1148–1160. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Steinberg F, Heesom KJ, Bass MD, Cullen PJ. SNX17 protects integrins from degradation by sorting between lysosomal and recycling pathways. J Cell Biol. 2012;197:219–230. doi: 10.1083/jcb.201111121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Arikawa C, Kuwahara Y, Itoh K, Watanabe M, Watanabe H, Suzuki T, Funakoshi Y, Hasegawa H, Kanaho Y. The scaffold protein JIP3 functions as a downstream effector of the small GTPase Arf6 to regulate neurite morphogenesis of cortical neurons. FEBS Lett. 2010;584:2801–2806. doi: 10.1016/j.febslet.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Tan CL, Andrews MR, Kwok JC, Heintz TG, Gumy LF, Fässler R, Fawcett JW. Kindlin-1 enhances axon growth on inhibitory chondroitin sulfate proteoglycans and promotes sensory axon regeneration. J Neurosci. 2012;32:7325–7335. doi: 10.1523/JNEUROSCI.5472-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii T, Miyamoto Y, Sanbe A, Nishimura K, Yamauchi J, Tanoue A. Cytohesin-2/ARNO, through its interaction with focal adhesion adaptor protein paxillin, regulates preadipocyte migration via the downstream activation of Arf6. J Biol Chem. 2010;285:24270–24281. doi: 10.1074/jbc.M110.125658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswarlu K, Cullen PJ. Signalling via ADP-ribosylation factor 6 lies downstream of phosphatidylinositide 3-kinase. Biochem J. 2000;345:719–724. doi: 10.1042/0264-6021:3450719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelezang MG, Liu Z, Relvas JB, Raivich G, Scherer SS, ffrench-Constant C. α4 integrin is expressed during peripheral nerve regeneration and enhances neurite outgrowth. J Neurosci. 2001;21:6732–6744. doi: 10.1523/JNEUROSCI.21-17-06732.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelezang M, Forster UB, Han J, Ginsberg MH, ffrench-Constant C. Neurite outgrowth on a fibronectin isoform expressed during peripheral nerve regeneration is mediated by the interaction of paxillin with alpha4beta1 integrins. BMC Neurosci. 2007;8:44. doi: 10.1186/1471-2202-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Willem M, Jones LL, Kreutzberg GW, Mayer U, Raivich G. Impaired axonal regeneration in 7 integrin-deficient mice. J Neurosci. 2000;20:1822–1830. doi: 10.1523/JNEUROSCI.20-05-01822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DT, McShea KM, Attar MA, Santy LC. GRASP and IPCEF promote ARF-to-Rac signaling and cell migration by coordinating the association of ARNO/cytohesin 2 with Dock180. Mol Biol Cell. 2010;21:562–571. doi: 10.1091/mbc.E09-03-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- Zou Y, Chiu H, Zinovyeva A, Ambros V, Chuang CF, Chang C. Developmental decline in neuronal regeneration by the progressive change of two intrinsic timers. Science. 2013;340:372–376. doi: 10.1126/science.1231321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In mature cortical neurons, α9 integrin vesicles are retrogradely transported from axon to cell body. Cortical neurons were transfected with α9-GFP (green) and live-imaged at 10 DIV for 3 min. Axons were identified by neurofascin live staining in red at the AIS (Fig. 6).
In young cortical neurons, α9 integrin vesicles were not transported back from axon to cell body. Cortical neurons were transfected with α9-GFP (green) and live-imaged at 3 DIV for 3 min.
In mature cortical dendrites, α9 integrin vesicles traffic in both directions. Cortical neurons were cotransfected with α9-GFP, and dendrites were live-imaged at 10 DIV.
Live imaging of α9-GFP vesicle traffic in axons at 10 DIV at a region proximal to the cell body, typically 100 μm into the axon.
Live imaging of α9-GFP vesicle traffic in axons at 10 DIV at a region distal from the cell body. This was performed at a region as far into the axon as could be traced by α9-GFP signal, typically 300–600 μm into the axon.
Interfering with dynein stops α9 integrin retrograde transportation in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and HA-p50 and live-imaged at 10 DIV.
Overexpression of ARF6 GAP ACAP1 increases anterograde transportation and decreases retrograde transportation of α9 integrin in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and ARF6 GAP ACAP1-myc.
Interfering with ARF6 GTP by overexpressing ARNO-E156K increases anterograde transportation and decreases retrograde transportation of α9 integrin in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and a catalytically inactive form of ARF6 GEF ARNO, ARNO-E156K.
Combining ARNO-E156K and taxol further increases anterograde transportation of α9 integrin in mature cortical axons. Cortical neurons were cotransfected with α9-GFP and an ARNO-E156K and treated with taxol.