

Abstract
Mitochondrial dysfunction and activation of the inflammatory system are two of the most consistently reported findings in bipolar disorder (BD). More specifically, altered levels of inflammatory cytokines and decreased levels of mitochondrial complex I subunits have been found in the brain and periphery of patients with BD, which could lead to increased production of mitochondrial reactive oxygen species (ROS). Recent studies have shown that mitochondrial production of ROS and inflammation may be closely linked through a redox sensor known as nod-like receptor pyrin domain-containing 3 (NLRP3). Upon sensing mitochondrial release of ROS, NLRP3 assembles the NLRP3 inflammasome, which releases caspase 1 to begin the inflammatory cascade. In this review, we discuss the potential role of the NLRP3 inflammasome as a link between complex I dysfunction and inflammation in BD and its therapeutic implications.
1. Introduction
Bipolar disorder (BD) is the sixth leading cause of disability worldwide (WHO) with a chronic course, where 25–50% of patients with BD attempt suicide and 50–67% of the patients experience at least one relapse [1]. Despite the urgent need to develop more effective treatments for this disorder, progress has been limited due to a lack of understanding of its pathology.
A growing number of studies are demonstrating mitochondrial dysfunction, especially that of complex I and inflammation in patients with BD [2–7]. For example, a recent review examining microarray findings in BD reported a decrease in complex I subunits that are responsible for transportation of electrons in patients with BD, which could result in increased leakage of electrons and production of reactive oxygen species (ROS) [6]. Altered levels of inflammatory cytokines were also shown in the brain and periphery of patients with BD, including IL-6, TNFα, IFN-γ, and IL-1β, suggesting that activation of the inflammatory system may also play a role in the pathophysiology of BD. Recent studies suggest that mitochondrial production of ROS may be linked to inflammatory activation [8, 9]. In fact, inhibition of complex I and subsequent increase in ROS production lead to increased levels of inflammatory factors such as IL-1β, caspase 1, and NF-κB [9, 10].
A potential link between mitochondrial dysfunction and inflammation may be the nod-like receptor pyrin domain-containing 3 (NLRP3) inflammasome, which is a redox sensor that can potentiate the activation of the inflammatory cascade by releasing caspase 1. Indeed, complex I inhibition resulted in the activation of the NLRP3 inflammasome, and decreasing mitochondrial ROS production was able to eliminate inflammasome activation [9, 11]. Therefore, the aim of this review is to explore the link between mitochondrial generation of oxidative stress and inflammation in BD, with a focus on the NLRP3 inflammasome.
2. BD and Mitochondrial Dysfunction: A Brief Overview
Mitochondria are energy producing organelles in the cell that generate ATP by transporting electrons through electron transport chain (ETC) complexes I-V. Moreover, they regulate calcium levels and apoptotic processes. Mitochondria are also the main producers of ROS [12]. Complex I, which is responsible for oxidizing NADH and transferring electrons to ubiquinone [13], contains four main subcomplexes: γ, β, α-λ, and λ that regulate its activity and ROS generation [14]. The α-λ and λ subcomplexes are located on the hydrophilic arm of complex I, which is responsible for electron transfer, and γ and β subcomplexes are located at the hydrophobic arm, which is responsible for proton pumping [13]. A recent review of microarray studies revealed that patients with BD have decreased mRNA levels of iron-sulfur cluster containing subunits in the hydrophilic portions that are specifically involved in electron transfer, including NDUFV1, NDUFS1, NDUFS8, and NDUFS7 [6, 15]. On the other hand, the same review revealed that while patients with SCZ have some alterations in mRNA levels of complex I subunits, they do not have alterations in the subunits that are directly involved in the electron transfer process [6, 16, 17]. In support of the microarray findings, decreased protein levels of NDUFS7 and complex I activity were also reported in patients with BD [7, 18]. These findings suggest that patients with BD may be more vulnerable to having increased levels of electron leakage compared to the normal population or patients with SCZ [6]. Leaked electrons from complex I can react with molecular oxygen to produce the superoxide anion, which can escape the mitochondria to undergo a series of reactions to form powerful ROS such as the hydroxyl radical [12]. Oxidative damage to lipids, DNA, and proteins in patients with BD is some of the most consistently reported alterations in BD, which is in agreement with these findings [2, 3, 19].
Superoxide anion and other ROS also play important roles as signaling molecules in the cell through redox sensors that undergo conformational changes, oligomerization, and/or translocation upon detecting ROS or downstream products of ROS release [20]. Nrf2, for example, migrates to the nucleus upon sensing ROS production, and thioredoxin undergoes a structural change upon being modified by ROS [20]. Recent studies have demonstrated that mitochondria may be a potent activator of the immune system through its ability to generate ROS and its interaction with redox sensors in the inflammatory system, such as NLRP3 [8, 9]. These findings suggest that mitochondrial dysfunction in BD may at least be partly responsible for cytokine activation in the central nervous system (CNS) and periphery of patients with BD.
3. BD and Inflammation
Alterations in the inflammatory pathway in patients with BD have been reported since 1995, when Maes et al. [21] found increased sIL-6R and sIL-2R levels in patients with mania. Indeed, medical complications related to activation of the inflammatory system such as cardiovascular diseases, diabetes, and obesity are frequently diagnosed in patients with BD [22–24]. Furthermore, patients with BD generally have an earlier onset of cardiovascular diseases [22]. Such findings have inspired the microglial theory, which states that proinflammatory cytokines produced as a result of microglial activation result in disruption of neuroprotective functions, leading to increased vulnerability in BD [23].
Majority of the studies examining inflammation in BD have focused on peripheral samples such as plasma and serum [25–47]. A summary of the findings discussed here can be found in Tables 1, 2, 3, and 4. Multiple studies have reported increased levels of sIL-2R, sIL-6R, TNF-α, sTNFR1, IL-1, ILl-12, and TGF-b in BD, while mixed results have been reported for other inflammatory factors, including IL-4, IL-2, IL-8, and IFN-γ [25, 27, 30, 32, 38, 40, 42, 44–47]. In this review, we will focus on TNF-α and IL-6 for the periphery and the IL-1 pathway for the CNS, as these factors have been consistently reported to be altered in patients with BD.
Table 1.
Characteristics of studies examining cytokine alterations in peripheral samples of patients with bipolar disorder.
| Author name | Year | Sample size | Sample | Technique used | Cytokines examined |
|---|---|---|---|---|---|
| Tsai et al. [25] | 2001 | 31 manic, 31 remission, 31 control | Plasma | ELISA | sIL-2R, sIL-6R |
|
| |||||
| Su et al. [27] | 2002 | 20 BD-I manic, 15 control | Plasma | Stimulated sIL-2R, IL-10, IFN-g | |
|
| |||||
| Wadee et al. [28] | 2002 | 45 BD-I manic, 45 control | Serum | CRP | |
|
| |||||
| Kim et al. [92] | 2002 | 25 mania, 85 control | Plasma | ELISA | IL-12 |
|
| |||||
| Breunis et al. [30] | 2003 | 172 BD I and II, 66 matched control | Serum | sIL-2R | |
|
| |||||
| Boufidou et al. [31] | 2004 | BD I and II, 40 Li treated euthymic, 10 medication naïve, and 20 controls | Plasma | ELISA | Stimulated IL-2, IL-6, IL-10, IFN-g |
|
| |||||
| Kim et al. [93] | 2004 | 70 mania, 96 control | Plasma | ELISA | IFN-g, IL-4, TGF-b1 |
|
| |||||
| Liu et al. [94] | 2004 | 29 BD-I manic, 20 controls | Plasma | Stimulated IL-1RA, IL-2, IL-4, IL-10, IFN-g | |
|
| |||||
| Knijff et al. [95] | 2006 | 54 BD-I and II, 10 controls | Peripheral blood | FACS | Stimulated IL-2R w/dexamethasone suppression |
|
| |||||
| O'brien et al. [32] | 2006 | 21 control, 12 manic, 9 depressed | Plasma | ELISA | IL-6, IL-8, IL-10, TNF-alpha, sIL-6R |
|
| |||||
| Dickerson et al. [96] | 2007 | 122 BD-I and II, 165 controls | Serum | CRP | |
|
| |||||
| Huang and Lin [34] | 2007 | 13 BD-I manic, 23 MDD, 31 controls | Serum | hsCRP | |
|
| |||||
| Kim et al. [29] | 2007 | 37 BD-I manic, 74 controls | Plasma | ELISA | Stimulated IL-2, IL-4, IL-10, TNF-a, and IFN-g |
|
| |||||
| Knijff et al. [97] | 2007 | 80 BD-I and II, 59 controls | Serum | ELISA | IL-1b, I-6 |
|
| |||||
| Ortiz-Domínguez et al. [42] | 2007 | 33 controls, 20 patients, 10 in manic phase, 10 in depressed phase, | Serum | ELISA | TNF-a, IL-6, IL-1b, IL-2, and IL-4. |
|
| |||||
| Cunha et al. [98] | 2008 | 30 mania, 30 depressed, 20 euthymic, 32 controls | Serum | hsCRP | |
|
| |||||
| Kauer-Sant'Anna et al. [35] | 2009 | 60 matched controls, 30 early stage, 30 late stage | Serum | ELISA | BDNF, TNF-a, IL-6 and IL-1 |
|
| |||||
| Guloksuz [99] | 2010 | 31 euthymic, 16 control | Serum | Flow cytometry | IL-2, IL-4, IL-5, IL-10, IFN-g, TNF-a |
|
| |||||
| Brietzke and Teixeira [100] | 2010 | 30 euthymic, 30 control | Serum | ELISA | sTNFR1, sTNFR2 |
|
| |||||
| Kapczinski et al. [36] | 2011 | 20 manic, 20 depressed, 250 euthymic, 80 control | Serum | ELISA | TNF-a, IL-6, IL-10 |
|
| |||||
| Drexhage et al. [47] | 2010 | 38 Euthymic, 22 control | Serum | Flow cytometry, ELISA for sIL-2R | IFN-g, IL-17A, IL-10, IL-6, IL-4, IL-5, IL-8, TNF-a, IL-1b, sIL-2R |
|
| |||||
| Hope et al. [44] | 2011 | 17 “Elevated”, 58 Depressed, 26 Euthymic, 239 control | Plasma | EIA | sTNF-R1, IL1-Ra, IL-6 |
|
| |||||
| Barbosa et al. [101] | 2011 | 34 manic, 19 Euthymic, 38 control | Plasma | ELISA | sTNF-R1, IL1-Ra, IL-6 |
|
| |||||
| Guloksuz et al. [45] | 2012 | 45 euthymic with subsyndromal symptoms (BD+), 23 without subsyndromal symptoms (BD−), 23 control | Plasma | ELISA | Soluble tumor necrosis factor receptor-1 (sTNF-R1), soluble interleukin-6 receptor (sIL-6R), soluble interleukin-2 receptor (sIL-2R) |
|
| |||||
| Barbosa et al. [37] | 2012 | 25 euthymic, 25 control | Plasma | ELISA | TNF-a, sTNFR1, sTNFR2 |
|
| |||||
| Barbosa et al. [37] | 2012 | 30 euthymic, 30 control | Plasma | ELISA | TNF-a, sTNFR1, sTNFR2 |
|
| |||||
| Remlinger-Molenda et al. [40] | 2012 | 121 euthymic, 78 control | Serum | cytometry | IL-6, TNF-a, IL-10, IFN-g, IL-2, IL-1b |
|
| |||||
| Cetin et al. [41] | 2012 | 45 euthymic, 23 control | Plasma | ELISA | sTNF-R1, sIL-6R |
|
| |||||
| Tsai et al. [26] | 2012 | 33 manic, 33 remission, 33 control | Plasma | ELISA | IL1-Ra, sTNF-R1 |
Table 2.
Characteristics of studies examining inflammatory cytokines in the central nervous system of patients with BD.
| Author name | Article year | Sample size | Sample | Technique used | Cytokines examined |
|---|---|---|---|---|---|
| Rao et al. [52] | 2010 | 10 BD, 10 control | Postmortem frontal cortex BA 24 and BA 46 | Western plot, RT PCR, immunohistochemistry | NMDA receptors, NR-1 and NR-3A, IL-1β, IL-1R, MyD88, NF-kB (p50, p65), GFAP, iNOS, c-fos and CD11b, TNFα, neuronal nNOS, |
|
| |||||
| Söderlund et al. [53] | 2011 | BD euthymic patients, r type I (n = 15) or type II (n = 15) | CSF | An immunoassay-based protein array multiplex system | IL-1b, IL-6, |
|
| |||||
| Dean et al. [50] | 2013 | 10 MDD, 10 BD, 19 SZ, 30 control | Postmortem CNS tissue, BA24 and BA46 | Western plot, RT PCR | tmTNF-a, sTNF-a, TNF mRNA, TNFR1, TNFR2, IL-1beta, synaptophysin, PSD95, GFAP43, GFAP41, CD11b and pro-IL1B |
Table 3.
Summary of findings from studies examining alterations in cytokines in peripheral samples from patients with bipolar disorder.
| Outcome | N studies | Manic vs. controls | Mania vs. euthymia | Depression vs. controls | Euthymia vs. control | Remission vs. control | BD vs. controls | Mania vs. depression | Early stage vs. control | Late stage vs. control | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| sIL-2R | 4 | + | + | + | + | + | + | + | [25, 27, 30, 45] | ||
| sIL-6R | 2 | + | [25, 41] | ||||||||
| IFN-g | 6 | +− | − | − | − | [29, 31, 40, 93, 94, 99] | |||||
| TNF-a | 11 | + | + | + | + | + | [29, 32, 35–37, 40, 42, 47, 99, 101] | ||||
| sTNFR1 | 7 | + | + | [26, 37, 41, 44, 100, 101] | |||||||
| sTNFR2 | 6 | NS | [37, 100, 101] | ||||||||
| TGF-b1 | 1 | + | [93] | ||||||||
| IL-1b | 4 | − | − | [40, 42, 47, 97] | |||||||
| IL-2 | 6 | − | − | − | − | [29, 31, 40, 42, 94, 99] | |||||
| IL-4 | 6 | +− | + | [29, 42, 47, 93, 94, 99] | |||||||
| IL-5 | 2 | [47, 99] | |||||||||
| IL-6 | 8 | + | + | − | − | − | [29, 31, 36, 40, 42, 44, 97] | ||||
| IL-8 | 2 | + | + | [32, 47] | |||||||
| IL-10 | 9 | − | − | [27, 29, 31, 32, 36, 40, 47, 94, 99] | |||||||
| IL-12 | 1 | + | [92] | ||||||||
| IL-1 | 2 | + | [35, 40] | ||||||||
| IL-1RA | 3 | + | + | [26, 44, 94] | |||||||
| CRP | 2 | + | + | + | + | [28, 96] | |||||
| hsCRP | 2 | + | [34, 98] |
Table 4.
Summary of findings from studies examining cytokine alterations in the central nerve system of patients with bipolar disorder.
| Outcome | N studies | BD vs. controls | References |
|---|---|---|---|
| NR-1 (mRNA and Protein) | 1 | + | [52] |
| NR-2A (mRNA and Protein) | 1 | + | [52] |
| IL-1β (mRNA and Protein) | 3 | + | [50, 52, 53] |
| IL-1R (mRNA and Protein) | 1 | + | [52] |
| MyD88 (mRNA and Protein) | 1 | + | [52] |
| GFAP (mRNA and Protein) | 2 | + | [50, 52] |
| iNOS (mRNA and Protein) | 1 | + | [52] |
| TNFα | 2 | + | [50, 52] |
| IL-6 | 1 | − | [53] |
| tmTNFa | 1 | + (at BA 24) | [50] |
| Astrocyte | 1 | + | [52] |
| Microglia markers | 1 | + | [52] |
| TNFR2 | 1 | − | [50] |
Despite the large number of studies examining inflammation in BD, there is a lack of agreement regarding the direction of alteration and the cytokines which are altered [23]. However, TNF-α and related factors such as sTNFR1 (soluble tumor necrosis factor receptor-1) have been consistently found to be elevated in the periphery of patients with BD [26, 29, 32, 35, 37, 40–42, 44–47]. TNF-α is proinflammatory cytokine, which is produced mainly by activated macrophages, CD4+ lymphocytes, and natural killer cells [48, 49]. Upon binding to its receptors, TNFR1 and TNFR2, TNF-α can trigger the activation of NF-κB and MAPK pathways [45, 50].
IL-6, which is a proinflammatory cytokine secreted by T cells and macrophages, was also found to be increased in peripheral samples from patients with BD in the majority of studies that were examined in this review. Indeed, IL-6 is one of the cytokines most commonly reported to be altered in BD [29, 31, 32, 40, 42]. IL-6 can mediate fever and acute phase responses. It can also cross the blood brain barrier and trigger the activation of prostaglandin synthesis, which has been implicated in BD [51].
To our knowledge, only three studies have examined inflammation in the CNS in BD [50, 52, 53]. Dean et al. [50] focused on TNF-α related factors and pathways in different brain areas (BA24 and BA46) using postmortem brain. Increased concentration of tmTNF-α was observed in BA24 for BD, but not in BA46. TNFR2 was found to be decreased in BD subjects [50]. Rao and colleagues [52] focused on the IL-1 pathway and markers of microglial activation using postmortem prefrontal cortex from patients with BD. Higher protein and mRNA levels of IL-1β, IL-1R, and MyD88 and microglial/astrocyte markers GFAP and iNOS were found in patients with BD [52]. This was in contrast to Dean et al. [50] who could not detect IL-1β in their samples [50]. Findings from Söderlund and others [53] were consistent with Rao et al.'s study [52], showing elevated IL-1β levels in the cerebral spinal fluid (CSF) of patients with BD compared to healthy controls. Also, patients who recently had manic or hypomanic episodes showed elevated IL-1β levels compared to those who did not [53]. Alterations in cytokine balance in the brain can lead to changes in neurotransmitter levels including dopamine [54, 55], cause microglial activation [56], and activate apoptotic processes [3, 57], all of which have been reported in patients with BD [3, 52, 58].
Interestingly, there has been a lack of agreement between the results found in peripheral samples and the CNS. For example, while TNF-α levels are not reported to differ between patients with BD and nonpsychiatric controls in the CNS, its levels are consistently reported to be altered in patients with BD using peripheral samples [29, 32, 35, 37, 40, 42, 47, 50, 52]. Moreover, while increased levels of cytokines in the IL-1 pathway have been reported in the central nervous system, studies examining peripheral samples have not reported alterations in this pathway [23, 40, 42, 47, 52, 53]. The difference between cytokine pathways activated in the periphery and the CNS in BD may be due to the presence of diseases that affect the periphery to a greater extent than the CNS, such as atherosclerosis [24]. The different immune cells that reside in the brain and outside of the blood brain barrier may also be underlying the differences in cytokine profile. On the other hand, the fact that inflammatory activation is found both in the CNS and periphery of patients with BD suggests that the same underlying factor may be causing their activation. Decreased expression of complex I subunits and subsequent generation of mitochondrial ROS may underlie activation of central and peripheral immune cells through the NLRP3 inflammasome [9] (Figure 1).
Figure 1.
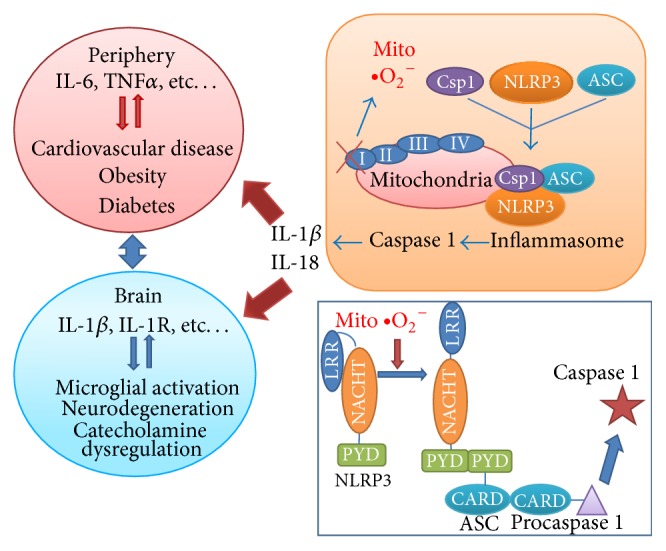
Mitochondrial complex I dysfunction in patients with BD could lead to increased release of superoxide anions, resulting in greater reactive oxygen species (ROS) production. This release of ROS causes a conformational change in NLRP3 such that the pyrin domain (PYD) becomes available recruit ASC. The combining of NLRP3 and ASC that allows for the recruitment of caspase 1 (csp1) through ASC's CARD domain, causing the formation of the NLRP3 inflammasome. The inflammasome then migrates to the mitochondria, allowing it to be close to the site of damage. Activated NLRP3 inflammasome releases caspase 1 into the cytosol, which then cleaves and activates two downstream cytokines, Il-1beta and Il-18, causing them to be released into the extracellular space. These two cytokines cause the activation of downstream pathways, which may differ depending on the type of immune cell. Indeed, NLRP3 inflammasome activation may underlie the different patterns of cytokine activation observed in the brain and peripheral samples of patients with BD, where alterations in cytokines pertaining to the IL-1 pathway have been reported for the brain, while a more general pattern of cytokine activation involving IL-6 and TNF-alpha has been reported in the periphery. Cytokine activation in the periphery can lead to various immune disorders, including cardiovascular disease and diabetes, while, in the brain, it could lead to alterations in neurotransmitters and neurodegeneration.
4. The NLRP3 Inflammasome
Recently, studies have shown that oxidative stress and mitochondrial dysfunction have important roles in regulating immune cells of the CNS and the periphery [59]. NLRP3 is a pattern recognition receptor in the inflammatory system that was shown to act as a redox sensor [9]. Cytosolic and membrane-associated pattern recognition receptors can detect danger signals induced by physical and psychological stress [60]. Membrane associated pattern recognition receptors include toll-like receptors that recognize pathogen associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), which then leads to the release of proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 [61]. Cytosolic pattern recognition receptors include NOD-like receptors, RIG-like receptors, and DNA sensors [62]. NLRP3 is the most widely studied receptor in the NOD-like receptor family [61]. NLRP3 contains a pyrin domain, a C-terminal leucine-rich domain, and a central nucleotide binding domain [61, 63]. NLRP3 is implicated in a wide variety of inflammatory conditions as it is activated by many different triggers including microbial infection, lipopolysaccharide, tissue damage, ATP, nigericin, and monosodium urate [64, 65]. When NLRP3 is inactive, it resides in the cytoplasm with its leucine-rich domain bound to the central nucleotide binding domain, preventing oligomerization [64]. Upon activation, NLRP3 migrates to the mitochondria associated endoplasmic reticulum membranes and the mitochondria [9]. This was demonstrated by increased colocalization between NLRP3 and mitotracker, which is a fluorescent marker for the mitochondria, and increased levels of NLRP3 in the mitochondrial fraction as well as mitochondria-associated membranes following NLRP3-inflammasome activation [9]. Since ROS are highly reactive and can only travel short distances, it would be ideal for NLRP3 to be localized the mitochondria where ROS is released [9, 66]. However, more studies are needed to test the generalizability of these findings in different systems and in humans. Activation of NLRP3 also causes NLRP3 oligomerization and recruitment of apoptosis-associated speck-like protein containing a CARD (ASC) through pyrin-pyrin domain interaction [9]. Procaspase 1 is also recruited through a CARD-CARD interaction between ASC and procaspase 1, completing the process of NLRP3 inflammasome assembly and activation [63]. NLRP3 inflammasome then releases caspase 1, also known as IL-1β converting enzyme. Caspase 1 cleaves pro-IL-1β and pro-IL-18 to their mature biologically active forms [67]. IL-1β is then released from the cells and binds to the IL-1 type-I receptor, a plasma membrane receptor, and IL-1 receptor-accessory protein to trigger the inflammatory cascade involving downstream signaling molecules such as MYD88 and NF-κB [68]. This leads to increased expression and activation of other inflammatory mediators such as IL-6, TNF-α, and prostaglandin E2 [69, 70].
5. Mitochondrial Dysfunction and the NLRP3 Inflammasome
Since many different PAMPs and DAMPs can activate NLRP3, it is unlikely that its ligand binding site recognizes all the molecules known to trigger the assembly of the NLRP3 inflammasome [71]. Mitochondrial dysfunction and subsequent production of ROS have received much attention as the common pathway by which different PAMPs and DAMPs trigger inflammasome activation [8, 9, 11, 72]. For example, addition of rotenone, a complex I inhibitor, induces a dose-dependent increase in IL-1β secretion [73], while in Nlrp3 KO mice, the addition of a mitochondrial ETC inhibitor fails to increase IL-1β and caspase 1 release [9]. Furthermore, inhibiting liposome-induced mitochondrial ROS release was followed by a decrease in the level of NLRP3-inflammasome activation [11]. While the exact pathway by which mitochondrial ROS leads to NLRP3 inflammasome activation and assembly remains elusive, two possible mechanisms have been proposed: thioredoxin-interacting protein- (TXNIP-) NLRP3 interaction and mitochondrial DNA (mtDNA) release [9, 65].
TXNIP is a tumor suppressor gene and its primary role is to inhibit the redox protein thioredoxin to suppress cell proliferation [9, 74]. Mitochondrial ROS production causes the dissociation between TXNIP and thioredoxin in the mitochondria, causing migration of TXNIP to the cytoplasm, which allows it to directly bind and activate cytoplasmic NLRP3 [74]. Zhou et al. (2011) showed that inflammation stimulating substances such as monosodium urate (MSU), silica, and ATP produce significantly less caspase 1 and IL-1β in TXNIP deficient mice, indicating decreased level of NLRP3-inflammasome activation [9]. In addition, in a high glucose concentration environment, islet cells from Txnip −/− and Nlrp3 −/− mice showed reduced level of IL-1β secretion compared to wild-type mice [9]. TXNIP was also observed to be increased in patients with type II diabetes by a number of different studies [75–77]. Patients with BD are three times more likely to be diagnosed with type II diabetes compared to the general population [78], suggesting that NLRP3 inflammasome activation mediated by TXNIP could be underlying increased peripheral and CNS inflammation in patients with BD.
Another potential mediator between mitochondrial ROS and NLRP3 inflammasome assembly is mtDNA. The role of mtDNA release from the mitochondria to the cytoplasm in NLRP3-inflammasome activation has been suggested following the observation that mtDNA directly binds and activates the NLRP3-inflammasome [10, 79]. Opening of mitochondrial membrane permeability transition pores (MPTs), which allows for mtDNA to escape the mitochondria, is often preceded by mitochondrial ROS production [80]. Also, adding ATP and lipopolysaccharide, which are two well-known stimulators of the NLRP3-inflammasome, increases mitochondrial ROS production and oxidized mtDNA levels in NLRP3 immunoprecipitates [79]. Importantly, it was also found that adding NLRP3 stimuli into cells lacking mtDNA (p0 cells) does not result in IL-1β secretion [79], and that the addition of mito-TEMPO, a mitochondrial-ROS scavenger, to bone marrow derived macrophages inhibits IL-1β and IL-18 secretion in a dose-dependent manner [81, 82]. Furthermore, preventing the opening of MPTs through the addition of cyclosporine A and thereby preventing mtDNA release inhibit LPS- and ATP-induced IL-1β secretion [10].
While the exact mechanism for how mitochondrial dysfunction triggers the assembly of the NLRP3 inflammasome remains to be elucidated, recent studies suggest that release of mitochondrial ROS plays a significant role in this pathway, either through activation of an intermediate redox sensor, such as TXNIP, or by activating apoptotic pathways causing the opening of MPTs [9, 10]. These findings suggest that amelioration of mitochondrial ROS production may aid in decreasing NLRP3-inflammasome activation, which could contribute to decreasing cytokine release in patients with BD.
6. Perspectives
With the discovery of immunological alterations in BD, much attention has been given to the possibility of implementing anti-inflammatory agents to treat symptom severity and cognitive decline [24]. An anti-inflammatory drug that was examined in patients with BD is celecoxib, which is a cyclooxygenase-2 inhibitor. Studies performed on rats showed that celecoxib can decrease IL-1β concentration in the hypothalamus, prefrontal cortex, and the hippocampus [83, 84]. Celecoxib was also shown to have a significant antidepressant effect in patients with BD, suggesting that anti-inflammatory medications targeting IL-1β may be helpful for patients with BD [85]. Aspirin (acetylsalicylic acid), which also inhibits the activity of cyclooxygenase 2 as well as cyclooxygenase 1, is also receiving much attention as a potential treatment option for bipolar depression [86, 87]. Cyclooxygenase enzymes are involved in the arachidonic acid cascade, which can lead to the activation of neuroinflammation pathways [88, 89]. Indeed, low-dose aspirin was found to decrease medication events (change in type of drug, increase in dose, or the number of prescribed drugs) in patients with BD, suggesting that aspirin may aid in stabilizing the symptoms [87].
Since NLRP3 inflammasome is strongly linked to mitochondrial dysfunction and subsequent production of ROS, improving mitochondrial function may contribute to decreasing inflammation in BD. A potential treatment is melatonin, which is a well-established antioxidant and an anti-inflammatory agent that was also demonstrated to target and accumulate in the mitochondria, improve mitochondrial respiration, and inhibit lipopolysaccharide-induced cytokine release [90, 91]. Interestingly, melatonin was found to be decreased in patients with BD, which may underlie disruptions in sleep patterns frequently observed in these patients. These findings suggest that melatonin may aid in decreasing mitochondrial ROS production and subsequent NLRP3 inflammasome activation in BD.
7. Conclusion
Mitochondrial complex I dysfunction and chronic inflammation are two of the most consistent findings in BD [2, 3]. Mitochondria are potent activators of the immune system, and this may occur in part through the NLRP3 inflammasome, which is assembled and activated following mitochondrial release of ROS [9]. Since complex I dysfunction in BD could lead to increased production of mitochondrial ROS, NLRP3 inflammasome mediated activation of the inflammatory system may underlie increased cytokine release in the CNS and periphery of patients with BD. Future studies examining the role of the NLRP3 inflammasome in BD will contribute to elucidating the link between two prominent pathophysiological alterations in this disorder, which may reveal pathways that can be used for the development of novel therapeutic interventions that can target both systems to improve symptomatology and cognitive functioning.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Fountoulakis K. N., Gonda X., Siamouli M., Rihmer Z. Psychotherapeutic intervention and suicide risk reduction in bipolar disorder: a review of the evidence. Journal of Affective Disorders. 2009;113(1-2):21–29. doi: 10.1016/j.jad.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 2.Clay H. B., Sillivan S., Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. International Journal of Developmental Neuroscience. 2011;29(3):311–324. doi: 10.1016/j.ijdevneu.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berk M., Kapczinski F., Andreazza A. C., et al. Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neuroscience & Biobehavioral Reviews. 2011;35(3):804–817. doi: 10.1016/j.neubiorev.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Kato T., Kato N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disorders. 2000;2(3):180–190. doi: 10.1034/j.1399-5618.2000.020305.x. [DOI] [PubMed] [Google Scholar]
- 5.Konradi C., Sillivan S. E., Clay H. B. Mitochondria, oligodendrocytes and inflammation in bipolar disorder: evidence from transcriptome studies points to intriguing parallels with multiple sclerosis. Neurobiology of Disease. 2012;45(1):37–47. doi: 10.1016/j.nbd.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scola G., Kim H. K., Young L. T., Andreazza A. C. A fresh look at complex i in microarray data: clues to understanding disease-specific mitochondrial alterations in bipolar disorder. Biological Psychiatry. 2013;73(2):e4–e5. doi: 10.1016/j.biopsych.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Andreazza A. C., Shoo L., Wang J. F., Trevor Young L. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Archives of General Psychiatry. 2010;67(4):360–368. doi: 10.1001/archgenpsychiatry.2010.22. [DOI] [PubMed] [Google Scholar]
- 8.López-Armada M. J., Riveiro-Naveira R. R., Vaamonde-García C., Valcárcel-Ares M. N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13(2):106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Zhou R., Yazdi A. S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 10.Nakahira K., Haspel J. A., Rathinam V. A. K., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Z., Zhai Y., Liang S., et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nature Communications. 2013;4, article 1611 doi: 10.1038/ncomms2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halliwell B. Reactive oxygen species and the central nervous system. Journal of Neurochemistry. 1992;59(5):1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 13.Green D. R., Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 14.Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2002;52(3–5):159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 15.Janssen R. J. R. J., Nijtmans L. G., van den Heuvel L. P., Smeitink J. A. M. Mitochondrial complex I: structure, function and pathology. Journal of Inherited Metabolic Disease. 2006;29(4):499–515. doi: 10.1007/s10545-006-0362-4. [DOI] [PubMed] [Google Scholar]
- 16.Vawter M. P., Tomita H., Meng F., et al. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Molecular Psychiatry. 2006;11(7):663–679. doi: 10.1038/sj.mp.4001830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prabakaran S., Swatton J. E., Ryan M. M., et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Molecular Psychiatry. 2004;9(7):684–697. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- 18.Andreazza A. C., Wang J. F., Salmasi F., Shao L., Young L. T. Specific subcellular changes in oxidative stress in prefrontal cortex from patients with bipolar disorder. Journal of Neurochemistry. 2013;127(4):552–561. doi: 10.1111/jnc.12316. [DOI] [PubMed] [Google Scholar]
- 19.Andreazza A. C., Kauer-Sant'Anna M., Frey B. N., et al. Oxidative stress markers in bipolar disorder: a meta-analysis. Journal of Affective Disorders. 2008;111(2-3):135–144. doi: 10.1016/j.jad.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Jones D. P. Redox sensing: orthogonal control in cell cycle and apoptosis signalling. Journal of Internal Medicine. 2010;268(5):432–448. doi: 10.1111/j.1365-2796.2010.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maes M., Bosmans E., Calabrese J., Smith R., Meltzer H. Y. Interleukin-2 and interleukin-6 in schizophrenia and mania: effects of neuroleptics and mood stabilizers. Journal of Psychiatric Research. 1995;29(2):141–152. doi: 10.1016/0022-3956(94)00049-W. [DOI] [PubMed] [Google Scholar]
- 22.Kilbourne A. M., Cornelius J. R., Han X., et al. Burden of general medical conditions among individuals with bipolar disorder. Bipolar Disorders. 2004;6(5):368–373. doi: 10.1111/j.1399-5618.2004.00138.x. [DOI] [PubMed] [Google Scholar]
- 23.Munkholm K., Braüner J. V., Kessing L. V., Vinberg M. Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. Journal of Psychiatric Research. 2013;47(9):1119–1133. doi: 10.1016/j.jpsychires.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein B. I., Fagiolini A., Houck P., Kupfer D. J. Cardiovascular disease and hypertension among adults with bipolar I disorder in the United States. Bipolar Disorders. 2009;11(6):657–662. doi: 10.1111/j.1399-5618.2009.00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai S.-Y. M., Yang Y.-Y., Kuo C.-J., Chen C.-C., Leu S.-J. C. Effects of symptomatic severity on elevation of plasma soluble interleukin-2 receptor in bipolar mania. Journal of Affective Disorders. 2001;64(2-3):185–193. doi: 10.1016/S0165-0327(00)00252-4. [DOI] [PubMed] [Google Scholar]
- 26.Tsai H.-C., Lu M.-K., Yang Y.-K., et al. Empirically derived subgroups of bipolar I patients with different comorbidity patterns of anxiety and substance use disorders in Han Chinese population. Journal of Affective Disorders. 2012;136(1-2):81–89. doi: 10.1016/j.jad.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 27.Su K.-P., Leu S.-J. C., Yang Y.-Y., Shen W. W., Chou Y.-M., Tsai S.-Y. M. Reduced production of interferon-gamma but not interleukin-10 in bipolar mania and subsequent remission. Journal of Affective Disorders. 2002;71(1–3):205–209. doi: 10.1016/S0165-0327(01)00369-X. [DOI] [PubMed] [Google Scholar]
- 28.Wadee A. A., Kuschke R. H., Wood L. A., Berk M., Ichim L., Maes M. Serological observations in patients suffering from acute manic episodes. Human Psychopharmacology. 2002;17(4):175–179. doi: 10.1002/hup.390. [DOI] [PubMed] [Google Scholar]
- 29.Kim Y.-K., Jung H.-G., Myint A.-M., Kim H., Park S.-H. Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. Journal of Affective Disorders. 2007;104(1–3):91–95. doi: 10.1016/j.jad.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Breunis M. N., Kupka R. W., Nolen W. A., et al. High numbers of circulating activated T cells and raised levels of serum IL-2 receptor in bipolar disorder. Biological Psychiatry. 2003;53(2):157–165. doi: 10.1016/S0006-3223(02)01452-X. [DOI] [PubMed] [Google Scholar]
- 31.Boufidou F., Nikolaou C., Alevizos B., Liappas I. A., Christodoulou G. N. Cytokine production in bipolar affective disorder patients under lithium treatment. Journal of Affective Disorders. 2004;82(2):309–313. doi: 10.1016/j.jad.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 32.O'Brien S. M., Scully P., Scott L. V., Dinan T. G. Cytokine profiles in bipolar affective disorder: focus on acutely ill patients. Journal of Affective Disorders. 2006;90(2-3):263–267. doi: 10.1016/j.jad.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 33.Dickerson F., Stallings C., Origoni A., et al. A combined marker of inflammation in individuals with mania. PLoS ONE. 2013;8(9) doi: 10.1371/journal.pone.0073520.e73520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang T. L., Lin F. C. High-sensitivity C-reactive protein levels in patients with major depressive disorder and bipolar mania. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2007;31(2):370–372. doi: 10.1016/j.pnpbp.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 35.Kauer-Sant'Anna M., Kapczinski F., Andreazza A. C., et al. Brain-derived neurotrophic factor and inflammatory markers in patients with early- vs. late-stage bipolar disorder. The International Journal of Neuropsychopharmacology. 2009;12(4):447–458. doi: 10.1017/S1461145708009310. [DOI] [PubMed] [Google Scholar]
- 36.Kapczinski F., Dal-Pizzol F., Teixeira A. L., et al. Peripheral biomarkers and illness activity in bipolar disorder. Journal of Psychiatric Research. 2011;45(2):156–161. doi: 10.1016/j.jpsychires.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 37.Barbosa I. G., Rocha N. P., Miranda A. S. D., et al. Increased levels of adipokines in bipolar disorder. Journal of Psychiatric Research. 2012;46(3):389–393. doi: 10.1016/j.jpsychires.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 38.Barbosa I. G., Rocha N. P., Bauer M. E., et al. Chemokines in bipolar disorder: trait or state? European Archives of Psychiatry and Clinical Neuroscience. 2013;263(2):159–165. doi: 10.1007/s00406-012-0327-6. [DOI] [PubMed] [Google Scholar]
- 39.Barbosa I. G., Morato I. B., de Miranda A. S., Bauer M. E., Soares J. C., Teixeira A. L. A preliminary report of increased plasma levels of IL-33 in bipolar disorder: further evidence of pro-inflammatory status. Journal of Affective Disorders. 2014;157:41–44. doi: 10.1016/j.jad.2013.12.042. [DOI] [PubMed] [Google Scholar]
- 40.Remlinger-Molenda A., Wojciak P., Michalak M., Karczewski J., Rybakowski J. K. Selected cytokine profiles during remission in bipolar patients. Neuropsychobiology. 2012;66(3):193–198. doi: 10.1159/000339949. [DOI] [PubMed] [Google Scholar]
- 41.Cetin T., Guloksuz S., Cetin E. A., et al. Plasma concentrations of soluble cytokine receptors in euthymic bipolar patients with and without subsyndromal symptoms. BMC Psychiatry. 2012;12, article 158 doi: 10.1186/1471-244X-12-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ortiz-Domínguez A., Hernández M. E., Berlanga C., et al. Immune variations in bipolar disorder: phasic differences. Bipolar Disorders. 2007;9(6):596–602. doi: 10.1111/j.1399-5618.2007.00493.x. [DOI] [PubMed] [Google Scholar]
- 43.Cunha A. B. M., Frey B. N., Andreazza A. C., et al. Serum brain-derived neurotrophic factor is decreased in bipolar disorder during depressive and manic episodes. Neuroscience Letters. 2006;398(3):215–219. doi: 10.1016/j.neulet.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 44.Hope S., Dieset I., Agartz I., et al. Affective symptoms are associated with markers of inflammation and immune activation in bipolar disorders but not in schizophrenia. Journal of Psychiatric Research. 2011;45(12):1608–1616. doi: 10.1016/j.jpsychires.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 45.Guloksuz S., Altinbas K., Aktas Cetin E., et al. Evidence for an association between tumor necrosis factor-alpha levels and lithium response. Journal of Affective Disorders. 2012;143(1–3):148–152. doi: 10.1016/j.jad.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 46.Brietzke E., Stabellini R., Grassi-Oliveira R., Lafer B. Cytokines in bipolar disorder: recent findings, deleterious effects but promise for future therapeutics. CNS Spectrums. 2011;16(7):157–168. doi: 10.1017/S1092852912000338. [DOI] [PubMed] [Google Scholar]
- 47.Drexhage R. C., Knijff E. M., Padmos R. C., et al. The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Review of Neurotherapeutics. 2010;10(1):59–76. doi: 10.1586/ern.09.144. [DOI] [PubMed] [Google Scholar]
- 48.Carswell E. A., Old L. J., Kassel R. L., Green S., Fiore N., Williamson B. An endotoxin induced serum factor that cuases necrosis of tumors. Proceedings of the National Academy of Sciences of the United States of America. 1975;72(9):3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koj A., Magielska-Zero D., Bereta J., Kurdowska A., Rokita H., Gauldie J. The cascade of inflammatory cytokines regulating synthesis of acute phase proteins. The Tokai Journal of Experimental and Clinical Medicine. 1988;13(6):255–264. [PubMed] [Google Scholar]
- 50.Dean B., Gibbons A. S., Tawadros N., Brooks L., Everall I. P., Scarr E. Different changes in cortical tumor necrosis factor-α-related pathways in schizophrenia and mood disorders. Molecular Psychiatry. 2013;18(7):767–773. doi: 10.1038/mp.2012.95. [DOI] [PubMed] [Google Scholar]
- 51.Banks W. A., Kastin A. J., Broadwell R. D. Passage of cytokines across the blood-brain barrier. NeuroImmunomodulation. 1995;2(4):241–248. doi: 10.1159/000097202. [DOI] [PubMed] [Google Scholar]
- 52.Rao J. S., Harry G. J., Rapoport S. I., Kim H. W. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Molecular Psychiatry. 2010;15(4):384–392. doi: 10.1038/mp.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Söderlund J., Olsson S. K., Samuelsson M., et al. Elevation of cerebrospinal fluid interleukin-1β in bipolar disorder. Journal of Psychiatry & Neuroscience. 2011;36(2):114–118. doi: 10.1503/jpn.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Felger J. C., Li L., Marvar P. J., et al. Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain, Behavior, and Immunity. 2013;31:153–160. doi: 10.1016/j.bbi.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Felger J. C., Miller A. H. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Frontiers in Neuroendocrinology. 2012;33(3):315–327. doi: 10.1016/j.yfrne.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dheen S. T., Kaur C., Ling E.-A. Microglial activation and its implications in the brain diseases. Current Medicinal Chemistry. 2007;14(11):1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- 57.Berk M., Berk L., Dodd S., et al. Stage managing bipolar disorder. Bipolar Disorders. 2014;16(5):471–477. doi: 10.1111/bdi.12099. [DOI] [PubMed] [Google Scholar]
- 58.Berk M., Dodd S., Kauer-Sant'anna M., et al. Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatrica Scandinavica. 2007;(434):41–49. doi: 10.1111/j.1600-0447.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- 59.Sorbara M. T., Girardin S. E. Mitochondrial ROS fuel the inflammasome. Cell Research. 2011;21(4):558–560. doi: 10.1038/cr.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boller T., Felix G. A renaissance of elicitors: perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annual Review of Plant Biology. 2009;60:379–407. doi: 10.1146/annurev.arplant.57.032905.105346. [DOI] [PubMed] [Google Scholar]
- 61.Bryant C., Fitzgerald K. A. Molecular mechanisms involved in inflammasome activation. Trends in Cell Biology. 2009;19(9):455–464. doi: 10.1016/j.tcb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 62.Beutler B., Jiang Z., Georgel P., et al. Genetic analysis of host resistance: toll-like receptor signaling and immunity at large. Annual Review of Immunology. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 63.Agostini L., Martinon F., Burns K., McDermott M. F., Hawkins P. N., Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–325. doi: 10.1016/S1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 64.Tschopp J., Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nature Reviews Immunology. 2010;10(3):210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 65.Schroder K., Zhou R., Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327(5963):296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 66.Harwell B. Biochemistry of oxidative stress. Biochemical Society Transactions. 2007;35(5):1147–1150. doi: 10.1042/BST0351147. [DOI] [PubMed] [Google Scholar]
- 67.Perregaux D., Gabel C. A. Interleukin-1β maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. The Journal of Biological Chemistry. 1994;269(21):15195–15203. [PubMed] [Google Scholar]
- 68.Eder C. Mechanisms of interleukin-1β release. Immunobiology. 2009;214(7):543–553. doi: 10.1016/j.imbio.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 69.Bryan N. B., Dorfleutner A., Kramer S. J., Yun C., Rojanasakul Y., Stehlik C. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. Journal of Inflammation. 2010;7, article 23 doi: 10.1186/1476-9255-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weber A., Wasiliew P., Kracht M. Interleukin-1β (IL-1β) processing pathway. Science Signaling. 2010;3(105):p. cm2. doi: 10.1126/scisignal.3105cm2. [DOI] [PubMed] [Google Scholar]
- 71.Lamkanfi M., Dixit V. M. Inflammasomes: guardians of cytosolic sanctity. Immunological Reviews. 2009;227(1):95–105. doi: 10.1111/j.1600-065X.2008.00730.x. [DOI] [PubMed] [Google Scholar]
- 72.Liu D., Rhebergen A. M., Eisenbarth S. C. Licensing adaptive immunity by NOD-like receptors. Frontiers in Immunology. 2013;4, article 486 doi: 10.3389/fimmu.2013.00486.Article 486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li N., Ragheb K., Lawler G., et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. The Journal of Biological Chemistry. 2003;278(10):8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 74.Lane T., Flam B., Lockey R., Kolliputi N. TXNIP shuttling: missing link between oxidative stress and inflammasome activation. Frontiers in Physiology. 2013;4, article 50 doi: 10.3389/fphys.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parikh H., Carlsson E., Chutkow W. A., et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Medicine. 2007;4(5, article e158) doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Minn A. H., Hafele C., Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces β-cell apoptosis. Endocrinology. 2005;146(5):2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- 77.Chen J., Saxena G., Mungrue I. N., Lusis A. J., Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and β-cell apoptosis. Diabetes. 2008;57(4):938–944. doi: 10.2337/db07-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calkin C. V., Gardner D. M., Ransom T., Alda M. The relationship between bipolar disorder and type 2 diabetes: more than just co-morbid disorders. Annals of Medicine. 2013;45(2):171–181. doi: 10.3109/07853890.2012.687835. [DOI] [PubMed] [Google Scholar]
- 79.Shimada K., Crother T. R., Karlin J., et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lemasters J. J., Theruvath T. P., Zhong Z., Nieminen A.-L. Mitochondrial calcium and the permeability transition in cell death. Biochimica et Biophysica Acta: Bioenergetics. 2009;1787(11):1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trnka J., Blaikie F. H., Logan A., Smith R. A., Murphy M. P. Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radical Research. 2009;43(1):4–12. doi: 10.1080/10715760802582183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang J., Stoyanovsky D. A., Belikova N. A., et al. A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiation Research. 2009;172(6):706–717. doi: 10.1667/RR1729.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Myint A. M., Steinbusch H. W. M., Goeghegan L., Luchtman D., Kim Y. K., Leonard B. E. Effect of the COX-2 inhibitor celecoxib on behavioural and immune changes in an olfactory bulbectomised rat model of depression. NeuroImmunoModulation. 2007;14(2):65–71. doi: 10.1159/000107420. [DOI] [PubMed] [Google Scholar]
- 84.Casolini P., Catalani A., Zuena A. R., Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. Journal of Neuroscience Research. 2002;68(3):337–343. doi: 10.1002/jnr.10192. [DOI] [PubMed] [Google Scholar]
- 85.Nery F. G., Monkul E. S., Hatch J. P., et al. Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Human Psychopharmacology. 2008;23(2):87–94. doi: 10.1002/hup.912. [DOI] [PubMed] [Google Scholar]
- 86.Savitz J., Preskorn S., Teague T. K., Drevets D., Yates W., Drevets W. Minocycline and aspirin in the treatment of bipolar depression: a protocol for a proof-of-concept, randomised, doubleblind, placebo-controlled, 2 × 2 clinical trial. BMJ Open. 2012;2(1) doi: 10.1136/bmjopen-2011-000643.000643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stolk P., Souverein P. C., Wilting I., et al. Is aspirin useful in patients on lithium? A pharmacoepidemiological study related to bipolar disorder. Prostaglandins Leukotrienes and Essential Fatty Acids. 2010;82(1):9–14. doi: 10.1016/j.plefa.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weerasinghe G. R., Rapoport S. I., Bosetti F. The effect of chronic lithium on arachidonic acid release and metabolism in rat brain does not involve secretory phospholipase A2 or lipoxygenase/cytochrome P450 pathways. Brain Research Bulletin. 2004;63(6):485–489. doi: 10.1016/j.brainresbull.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 89.Ramadan E., Basselin M., Rao J. S., et al. Lamotrigine blocks NMDA receptor-initiated arachidonic acid signalling in rat brain: implications for its efficacy in bipolar disorder. International Journal of Neuropsychopharmacology. 2012;15(7):931–943. doi: 10.1017/S1461145711001003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lowes D. A., Almawash A. M., Webster N. R., Reid V. L., Galley H. F. Melatonin and structurally similar compounds have differing effects on inflammation and mitochondrial function in endothelial cells under conditions mimicking sepsis. British Journal of Anaesthesia. 2011;107(2):193–201. doi: 10.1093/bja/aer149. [DOI] [PubMed] [Google Scholar]
- 91.Srinivasan V., Spence D. W., Pandi-Perumal S. R., Brown G. M., Cardinali D. P. Melatonin in mitochondrial dysfunction and related disorders. International Journal of Alzheimer's Disease. 2011;2011:16. doi: 10.4061/2011/326320.326320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim Y. K., Suh I. B., Kim H., et al. The plasma levels of interleukin-12 in schizophrenia, major depression, and bipolar mania: Effects of psychotropic drugs. Molecular Psychiatry. 2002;7(10):1107–1114. doi: 10.1038/sj.mp.4001084. [DOI] [PubMed] [Google Scholar]
- 93.Kim Y.-K., Myint A.-M., Lee B.-H., et al. T-helper types 1, 2, and 3 cytokine interactions in symptomatic manic patients. Psychiatry Research. 2004;129(3):267–272. doi: 10.1016/j.psychres.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 94.Liu H.-C., Yang Y.-Y., Chou Y.-M., Chen K.-P., Shen W. W., Leu S.-J. Immunologic variables in acute mania of bipolar disorder. Journal of Neuroimmunology. 2004;150(1-2):116–122. doi: 10.1016/j.jneuroim.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 95.Knijff E. M., Breunis M. N., van Geest M. C., et al. A relative resistance of T cells to dexamethasone in bipolar disorder. Bipolar Disorders. 2006;8(6):740–750. doi: 10.1111/j.1399-5618.2006.00359.x. [DOI] [PubMed] [Google Scholar]
- 96.Dickerson F., Stallings C., Origoni A., Boronow J., Yolken R. Elevated serum levels of C-reactive protein are associated with mania symptoms in outpatients with bipolar disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2007;31(4):952–955. doi: 10.1016/j.pnpbp.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 97.Knijff E. M., Breunis M. N., Kupka R. W., et al. An imbalance in the production of IL-1beta and IL-6 by monocytes of bipolar patients: restoration by lithium treatment. Bipolar Disorders. 2007;9(7):743–753. doi: 10.1111/j.1399-5618.2007.00444.x. [DOI] [PubMed] [Google Scholar]
- 98.Cunha A. B., Andreazza A. C., Gomes F. A., et al. Investigation of serum high-sensitive C-reactive protein levels across all mood states in bipolar disorder. European Archives of Psychiatry and Clinical Neuroscience. 2008;258(5):300–304. doi: 10.1007/s00406-007-0797-0. [DOI] [PubMed] [Google Scholar]
- 99.Guloksuz S., Cetin E. A., Cetin T., Deniz G., Oral E. T., Nutt D. J. Cytokine levels in euthymic bipolar patients. Journal of Affective Disorders. 2010;126(3):458–462. doi: 10.1016/j.jad.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 100.Brietzke E., Teixeira A. L. Similar immune profile in bipolar disorder and schizophrenia: Selective increase in soluble tumor necrosis factor receptor I and von Willebrand factor. Bipolar Disorders. 2010;12(4):453–454. doi: 10.1111/j.1399-5618.2010.00822.x. [DOI] [PubMed] [Google Scholar]
- 101.Barbosa I. G., Huguet R. B., Mendonça V. A., et al. Increased plasma levels of soluble TNF receptor i in patients with bipolar disorder. European Archives of Psychiatry and Clinical Neuroscience. 2011;261(2):139–143. doi: 10.1007/s00406-010-0116-z. [DOI] [PubMed] [Google Scholar]