

Abstract
Zolpidem tartrate is a non-benzodiazepine analogue of imidazopyridine of sedative and hypnotic category. It has a short half-life with usual dosage regimen being 5 mg, two times a day, or 10 mg, once daily. The duration of action is considered too short in certain circumstances. Thus, it is desirable to lengthen the duration of action. The formulation design was implemented by preparing extended-release tablets of zolpidem tartrate using the biphasic delivery system technology, where sodium starch glycolate acts as a superdisintegrant in immediate-release part and hydroxypropyl methyl cellulose as a release retarding agent in extended-release core. Tablets were prepared by direct compression. Both the core and the coat contained the drug. The pre-compression blends were evaluated for angle of repose, bulk density, and compressibility index. The tablets were evaluated for thickness, hardness, weight variation test, friability, and in vitro release studies. No interaction was observed between zolpidem tartrate and excipients from the Fourier transform infrared spectroscopy and differential scanning calorimetry analysis. The results of all the formulations prepared were compared with reference product Stilnoct®. Optimized formulations showed release patterns that match the United States Pharmacopeia (USP) guidelines for zolpidem tartrate extended-release tablets. The mechanism of drug release was studied using different mathematical models, and the optimized formulation has shown Fickian diffusion. Accelerated stability studies were performed on the optimized formulation.
KEY WORDS: biphasic delivery system technology, hydroxypropyl methyl cellulose, modified release, sodium starch glycolate, zolpidem tartrate
INTRODUCTION
Insomnia is a disorder that includes difficulty in initiating sleep, difficulty in maintaining sleep, or waking too early without being able to return to sleep, causing clinically significant daytime distress or functional impairment in social, occupational, and/or other important areas of functioning. It is important to consider the efficacy for sleep onset as well as sleep maintenance, when selecting a sleep medication (1).
Benzodiazepines (BZDs) were the popular drugs for insomnia, but in view of next-day "hangover" effects, dependency, and rebound insomnia, there was a need for drugs without such side effects. Several non-BZD hypnotics like the sedating antihistamines, the melatonin receptor agonists, certain antidepressants, and z-drugs like zopiclone and eszopiclone, zaleplon, and zolpidem as a result came to therapy (2).
Zolpidem tartrate (structure shown in Fig. 1) is an imidazopyridine derivative, non-benzodiazepine, non-barbiturate hypnosedative that is best prescribed as sleep aid, having largely replaced the benzodiazepine class as pharmacologic therapy for insomnia and related disorders. Zolpidem tartrate is reported to have similar sedative properties to the BZDs but minimal anxiolytic, muscle relaxant, and anticonvulsant properties. The low incidence of side effects is, in part, a consequence of a relatively short half-life in the circulation. Immediate-release dosage forms of zolpidem tartrate which were developed till recently have a rapid onset of action and short half-life, which shows limited efficacy for maintaining sleep throughout the night.
Fig. 1.
Structure of zolpidem tartrate
This contributed to the development of zolpidem extended-release dosage forms that enable to sustain release over a period compatible with the desired time of sleep and the time needed for elimination of the drug from the human body to a sufficiently low level (3–5).
Hydroxypropyl methyl cellulose (HPMC) is the dominant hydrophilic polymer carrier used for the preparation of oral controlled drug delivery systems (6). It has been used widely for the development of sustained-release dosage forms such as matrix tablets of high water-soluble drugs (7). A comparative study of propranolol hydrochloride release from matrix tablets with Kollidon® sustained release (SR) or hydroxypropyl methyl cellulose has been reported (8). The possible mechanisms of drug dispersion using different grades of HPMC have been discussed for directly compressed indomethacin tablets (9).
A biphasic delivery system is formulated when a single constant rate of drug release cannot satisfy the therapeutic objective of maintaining sleep. Such a system can be achieved by the application of an immediate-release layer to the conventional layered matrix tablet. These systems can release the drug at two different rates or in two different periods of time: either quick/slow or slow/quick. A quick/slow-release system gives an initial burst of drug release followed by a constant rate of release over a defined period of time. It is useful when maximum relief needs to be achieved quickly and then followed by a sustained-release phase to avoid repeated administration (10). Drug release from a quick/slow biphasic delivery system for metoclopramide hydrochloride using the superdisintegrant Ac-di-sol for the fast-release layer and hydroxypropyl methyl cellulose K100M and Ucarflock 302 to modulate the release of the drug has been reported (11).
The pharmacokinetics of zolpidem, with around 2.5 h half-life, can lead to sub-therapeutic effects on sleep maintenance in the later portion of the night for some patients. In an effort to expand the coverage of sleep complaints and overcome the lack of efficacy in sleep maintenance, biphasic zolpidem tartrate tablet was prepared by using sodium starch glycolate as a superdisintegrant in the portion of immediate-release coat and cellulose polymers like different grades of hydroxypropyl methyl cellulose (HPMC K4M CR, HPMC K15M CR, and HPMC K100M CR) as release retardants in the core. The tablet was designed to mimic initial dosing while the controlled release of drug maintains a plasma concentration for a longer duration of time. This could reduce the number of nocturnal awakenings and provide clinical benefits beyond that of the original immediate-release formulation of zolpidem (12–14).
The major objectives of this study were to develop and evaluate a biphasic delivery system, to achieve a quick/slow release of the drug, to study the influence of the type of matrix core on the in vitro performance, to obtain a slow drug release period at a constant rate (zero-order kinetics), and to evaluate the combined effect of a fast-release coat together with a controlled-release core.
The tablet-in-tablet approach has some potential advantages against bilayer tablets. Hence, in the present work, formulations of tablet-in-tablet for zolpidem were prepared and compared with bilayer reference product Stilnoct®. It was used for the development of a quick/slow formulation with core containing a sustained-release tablet which is coated by compression over the whole surface with a fast-disintegrating formulation. From the viewpoint of manufacturing, this technology is an attractive alternative to bilayer or multilayer tablets. In the manufacturing of multilayer tablets, there is a chance of additional layers getting adhered to the pre-compressed layers during the double-layer or multilayer tableting process, thus making the process difficult. Furthermore, because this system uses conventional manufacturing methods, it is more acceptable to the industry.
The marketed formulation used as reference is available as bilayer tablets, but we have prepared a tablet-in-tablet biphasic delivery system. A bilayer tablet can exhibit certain disadvantages. In particular, both layers are adhered to each other by compression. Thus, they may be separated relatively easily by improper handling. Further, film coating of both layers is essentially necessary, which makes the process tedious. When wet granulation technique is used or necessary for the preparation of bilayer tablets, it makes the process costlier or less economical, to form a granulate which is then compressed into the tablet layer (15).
Pharmaceutical aspects of biphasic delivery compression-coated tablets in dosage form development are as follows: (a) to protect hygroscopic, light-sensitive, oxygen labile, or acid-labile drugs; (b) to separate incompatible drugs from each other and achieve sustained release; (c) and to modify drug release pattern (delayed, pulsatile, and programmable release for different drugs in one tablet).
Moreover, the tablet-in-tablet biphasic delivery system to be used as an oral dosage form provides the following advantages: (a) the immediate-release coat and SR core in their fixed positions, and therefore, no problem of separation (b) film coating is not required; (c) elimination of the bitter taste and unpleasant smell of the active pharmaceutical ingredient; (d) elimination of water or other solvent in the coating procedure and thereby decreasing the possible degradation of the active pharmaceutical ingredient; (e) easier and more economical manufacturing processes; and (f) additionally, the compression coatings may include flavoring agents and pharmaceutically acceptable colorants or opacifiers which could improve the patient’s compliance and acceptance with the drug regimen (16).
The author(s) worked for the development of biphasic delivery system of zolpidem tartrate as there is no comparative study report on different grades of HPMC used for development of such a biphasic delivery system and its industrial or commercial benefit over the marketed bilayer tablets when compared.
MATERIALS AND METHODS
Materials
Zolpidem tartrate was provided by M/s Aarti Drugs Ltd. (Mumbai, India). HPMC (Methocel) K4M CR, K15M CR, and K100M CR were obtained from Colorcon Asia Pvt. Ltd. (Mumbai, India). Lactose monohydrate (Pharmattose) was obtained from Loba Chemie Pvt. Ltd. (Mumbai, India). Tartaric acid was obtained from Triveni Chemicals (Vapi, India). Magnesium stearate was obtained from Yashica Pharmaceuticals Pvt. Ltd. (Thane, India). Sodium starch glycolate was obtained from Prachin Chemicals (Ahmedabad, India). Microcrystalline cellulose (Avicel PH 102) was supplied by Juku Chemicals Pvt. Ltd. (Chennai, India). LubriTose AN was obtained from Kerry Ingredients India Pvt. Ltd. (Bangalore, India). All other materials and chemicals procured for the studies were of analytical grade and were used as such as obtained.
Preparation of Biphasic Delivery System Tablets
The biphasic delivery system tablets were prepared by compressing the core components to a smaller tablet, forming a central core, followed with a compression of coat component powder mixture to produce a final tablet.
Slow-Release Component (Core Tablet)
The modified-release tablets were prepared by direct compression method. Active ingredient along with the excipients except magnesium stearate were accurately weighed and passed through mesh 40 and blended for 10 min. Magnesium stearate was weighed, passed through mesh 60, and mixed for 1–3 min. The formula for core tablet formulations contained 10%, 15%, and 20% w/w of HPMC of core tablet weight. Lactose monohydrate is used as a tablet diluent. Tartaric acid is used to get drug release independent of external pH by maintaining the pH of the core in natural pH conditions. So, the dissolution is independent of pH in the second release pulse. Magnesium stearate is used as a lubricant. The resulting blend was subjected to compression using compression machine (KMP–8, Kambert mini rotary compression machine) employing 6-mm round standard concave punch. The total weight of tablets was kept constant at 70 mg. In addition to being used for biphasic release system preparations, they were used as single units to evaluate the effect of compression on the structure and in vitro dissolution behavior.
Fast-Release Component (External Layer)
The powder used to coat the core was formulated to obtain a quick release of the drug. The composition contained zolpidem tartrate, sodium starch glycolate, microcrystalline cellulose (Avicel PH 102), and LubriTose AN. Sodium starch glycolate was used as a superdisintegrant for the immediate release of the drug. Avicel PH 102 was used due to its good compaction and disintegration properties. LubriTose AN contains anhydrous lactose and glyceryl monostearate which act as tablet diluent and lubricant, respectively. The active ingredient and excipients were accurately weighed and passed through mesh 40 and then blended for 10 min before compression.
Biphasic Delivery System
For the preparation of the biphasic delivery system, the die of the tableting machine was first filled manually with the half the amount of the fast-release component, and then, core tablet was placed carefully at the center. The remaining half of the fast-releasing powder was added to coat the core tablet. The formulations differed in the grade and concentration of HPMC used in the preparation of the core tablet and in the concentration of sodium starch glycolate used in the immediate-release part. Even the amount of zolpidem tartrate used in core and coat part was varied as per requirement. Composition of formulations F1 to F15 is given in Table I. Compressed core tablet systems were prepared by direct compression, using 9-mm standard concave punch. The total weight of tablet obtained was 280 mg.
Table I.
Composition of Formulations F1 to F15
| Formulation no. | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 | F13 | F14 | F15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ingredients | Quantity in mg per tablet | ||||||||||||||
| Fast-release coat | |||||||||||||||
| Zolpidem tartrate | 6. 25 | 6.25 | 6.25 | 6.50 | 6.50 | 6. 25 | 6.25 | 6.25 | 6.50 | 6.25 | 6. 25 | 6.25 | 6.25 | 6.50 | 6.50 |
| SSG | 8.40 | 8.40 | 8.40 | 8.40 | 10.5 | 8.40 | 8.40 | 8.40 | 8.40 | 10.5 | 8.40 | 8.40 | 8.40 | 8.40 | 10.5 |
| PH 102 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| LubriTose AN | 105.35 | 105.35 | 105.35 | 105.10 | 103 | 105.35 | 105.35 | 105.35 | 105.10 | 103 | 105.35 | 105.35 | 105.35 | 105.10 | 103 |
| Total weight | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 | 210 |
| Slow-release core | |||||||||||||||
| Zolpidem tartrate | 6.25 | 6.25 | 6.25 | 6.00 | 6.00 | 6.25 | 6.25 | 6.25 | 6.00 | 6.00 | 6.25 | 6.25 | 6.25 | 6.00 | 6.00 |
| HPMC K100M | 7 | 14 | 10.5 | 10.5 | 10.5 | – | – | – | – | – | – | – | – | – | – |
| HPMC K15M | – | – | – | – | – | 7 | 14 | 10.5 | 10.5 | 10.5 | – | – | – | – | – |
| HPMC K4M | – | – | – | – | – | – | – | – | – | – | 7 | 14 | 10.5 | 10.5 | 10.5 |
| Lactose monohydrate | 50 | 43 | 46.5 | 46.75 | 46.75 | 50 | 43 | 46.5 | 46.75 | 46.75 | 50 | 43 | 46.5 | 46.75 | 46.75 |
| Tartaric acid | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Magnesium stearate | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 | 0.75 |
| Total weight | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 |
| Total weight of press-coated tablet | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 | 280 |
Evaluation of Tablets
Physical Characterization of Core Tablets and Biphasic Delivery System Tablets
Core tablets and the biphasic delivery system tablets were characterized for weight variation (analytical balance AUX220, Shimadzu), thickness (digital vernier caliper ABSOLUTE 500-196-20, Mitutoyo, Mumbai, India), hardness test (Monsanto hardness tester MHT-20, Campbell Electronics, Mumbai, India), and friability (friabilator, Electrolab EF-1W, Mumbai, India).
In Vitro Release Testing
The in vitro release tests were performed using United States Pharmacopeia (USP) dissolution apparatus type I (Electro lab USP XXII, TDT-08L, Mumbai, India) equipped with basket which was operated at 100 rpm speed. The dissolution media used were 500 mL of 0.01 N hydrochloric acid (pH 2). The temperature was maintained at 37 ± 0.5°C. At predetermined time intervals, 10-mL sample was withdrawn and was replaced with an equal volume of fresh dissolution medium. The samples were analyzed spectrophotometrically using UV–vis spectrophotometer (Shimadzu, model UV-1650 PC, Japan) at 295 nm. The cumulative fraction of the drug release was calculated from the total amount of zolpidem tartrate and plotted as a function of time. Dissolution studies (n = 3) were performed on both press-coated tablets and core tablets to investigate the effect of compression on the dissolution behavior (17).
The dissolution profiles from press-coated tablets were compared with marketed product Stilnoct® using a similarity factor (f2) (18):
| 1 |
where Rt and Tt are the percentage of drug dissolved at each time point for the test and reference product, respectively. The US Food and Drug Administration and the European Agency for the Evaluation of Medicinal Products have suggested that two dissolution profiles can be considered similar if f2 is between 50 and 100 (19,20).
Release Drug Data Modeling
The suitability of several equations that are reported in the literature to identify the mechanisms for the release of zolpidem tartrate was tested with respect to the release data. Some diffusion models (Korsmeyer-Peppas) are expected to be valid only up to ~60% cumulative drug released, so the data for analysis were restricted to that range, excluding also the lag time (21,22). The data were evaluated according to the following equations:
where Mt is the amount of drug dissolved in time t, M0 is the initial amount of drug, K0 is the zero-order release constant, C is the concentration of drug at time t, C0 is the initial concentration of drug, K1 is the first-order release constant, KH is the Higuchi rate constant, Mt/M∞ is a fraction of drug released at time t, KK is Korsmeyer-Peppas release constant, n is the release exponent that characterizes the mechanism of drug release, W0 is the initial amount of drug in the pharmaceutical dosage form, Wt is the remaining amount of drug in the pharmaceutical dosage form at time t, and Ks is the Hixson-Crowell release constant. The value of n indicates whether the release mechanism is Fickian diffusion, case II transport, or anomalous transport. As the tablets in the present study are convex in shape, the limits considered were n = 0.45 (indicates a Fickian diffusion-controlled drug release) and n = 0.89 (indicates a case II relaxational release transport) and values between 0.45 and 0.89 (indicate both phenomena of coupling of drug diffusion in the hydrated matrix with polymer relaxation, commonly called anomalous non-Fickian transport). Values of n greater than 0.89 indicate super case II transport, in which a pronounced acceleration in solute release by a film occurs toward the latter stages of release experiments, resulting in a more rapid relaxation-controlled transport (30–33).
Compatibility Studies
Drug-excipient compatibility study was conducted by preparing homogenous mixture of excipients with drug and filled in transparent glass vials. Glass vials were stored at 40 ±2°C/75 ± 5% relative humidity (RH) for 3 months. Samples were observed periodically for any physical change at 1, 2, and 3 months, and Fourier transform infrared spectroscopy (FTIR) and differential scanning calorimetry (DSC) studies for the samples were conducted after 3 months.
DSC Study
Differential scanning calorimetry studies of pure drug and polymers as well as drug–polymer mixtures were performed using a Toledo DSC (Mettler Star SW 9.20) to determine compatibility. The analysis was performed at a rate of 40°C min-1 from 20°C to 300°C temperature ranges under nitrogen flow of 25 mL min−1.
Fourier Transform Infrared Spectroscopy
Compatibility studies of pure drug, polymers, and the physical mixture of drug and polymers were carried out using FTIR spectrophotometer (Shimadzu FTIR 8400-S) in the scanning range of 400–4,000 cm−1 by KBr disc method.
Stability Studies
Tablets were strip packed and stored at stability chamber (Tabai Espec Corp., Osaka, Japan) at 40°C/75% RH for 12 weeks. After 3 months, tablet strips were taken out from the chamber and tested for appearance, hardness, thickness, assay, and in vitro drug release.
RESULTS
Evaluation of Tablets
Physical Characterization of Core Tablets and Biphasic Delivery System Tablets
The physical properties (weight, thickness, hardness, and friability) of the core tablets and biphasic delivery system tablets for all the formulations were noted. The average weight of all core tablets was in the range 70 ± 2.5 mg. The biphasic delivery system tablets were in the weight range of 280 ± 2 mg. The thickness of the core tablets was in the range of 2.31 ± 0.29 mm, while that of biphasic delivery system tablets was in the range of 4.09 ± 0.09 mm. The prepared core tablets as well as biphasic delivery system tablets in all the trials possessed good mechanical strength with sufficient hardness of 5.5 ± 0.5 and 6.5 ± 0.5 kp, respectively. The friability of all the tablets was found to be within the limit (<1%). The percentage of drug content among different formulations of the tablets ranged 99.36 ± 1.54% which was within acceptable limits.
Dissolution Testing of Biphasic Delivery System Tablets
The release profile of all the prepared biphasic delivery system tablets is shown in Table II, while Table III shows the release profiles of core tablets of all the prepared formulations. Figure 2 shows the release profiles of zolpidem tartrate from the biphasic delivery system of formulations F5, F10, F14, and F15 comparing with the marketed product Stilnoct ER 12.5 mg. Figure 3 shows the contributions of each component (quick/slow) to the release profile of zolpidem tartrate from biphasic delivery system tablets of F14 containing HPMC K4M as a prolonged release component. In the release of the drug from the core tablets, different dissolution profiles were observed. From the plots of Fig. 3, it can be seen that the release rates are affected by the composition of the core present in the biphasic delivery system.
Table II.
In Vitro Dissolution Profile of Zolpidem Tartrate from Compressed Core Tablets of Formulations F1–F15 and Stilnoct® 12.5 mg
| Time (min) | 30 (mean cumulative percent ± SD drug released) | 60 (mean cumulative percent ± SD drug released) | 90 (mean cumulative percent ± SD drug released) | 120 (mean cumulative percent ± SD drug released) | 180 (mean cumulative percent ± SD drug released) | 240 (mean cumulative percent ± SD drug released) | 300 (mean cumulative percent ± SD drug released) | 360 (mean cumulative percent ± SD drug released) |
|---|---|---|---|---|---|---|---|---|
| F1 | 50.57 ± 1.32 | 54.23 ± 1.76 | 67.53 ± 2.17 | 75.97 ± 1.98 | 82.94 ± 2.15 | 90.91 ± 0.92 | 95.98 ± 0.74 | 99.39 ± 0.85 |
| F2 | 47.14 ± 1.98 | 49.03 ± 1.48 | 55.28 ± 2.11 | 58.91 ± 3.12 | 65.11 ± 1.78 | 71.17 ± 2.35 | 79.98 ± 3.14 | 89.47 ± 2.12 |
| F3 | 48.03 ± 2.15 | 52.79 ± 1.84 | 58.81 ± 1.43 | 69.48 ± 2.16 | 74.03 ± 1.74 | 81.54 ± 2.44 | 88.47 ± 1.99 | 94.39 ± 1.07 |
| F4 | 50.62 ± 1.68 | 55.97 ± 2.04 | 62.12 ± 2.12 | 72.65 ± 1.88 | 82.28 ± 2.19 | 89.72 ± 1.97 | 95.99 ± 1.36 | 98.55 ± 1.02 |
| F5 | 52.17 ± 1.79 | 57.52 ± 1.38 | 63.67 ± 1.45 | 74.20 ± 1.86 | 83.83 ± 1.23 | 91.27 ± 1.06 | 97.54 ± 0.98 | 100.10 ± 0.98 |
| F6 | 51.07 ± 1.87 | 55.80 ± 1.76 | 69.13 ± 1.54 | 78.11 ± 1.98 | 88.02 ± 1.99 | 97.71 ± 0.87 | 100.13 ± 0.99 | 100.70 ± 0.89 |
| F7 | 48.72 ± 2.34 | 52.84 ± 1.87 | 59.52 ± 2.11 | 67.15 ± 1.98 | 73.41 ± 1.76 | 78.01 ± 2.16 | 86.12 ± 1.98 | 93.36 ± 1.21 |
| F8 | 49.82 ± 3.12 | 54.24 ± 2.19 | 64.59 ± 1.77 | 73.53 ± 1.84 | 83.47 ± 1.07 | 89.84 ± 2.21 | 95.40 ± 1.65 | 97.65 ± 1.09 |
| F9 | 51.77 ± 2.67 | 56.18 ± 2.98 | 65.89 ± 1.74 | 74.88 ± 1.38 | 84.77 ± 1.89 | 90.93 ± 2.13 | 97.02 ± 0.98 | 100.15 ± 0.66 |
| F10 | 53.98 ± 3.56 | 58.39 ± 2.44 | 68.10 ± 2.16 | 77.09 ± 1.96 | 86.98 ± 1.73 | 93.14 ± 0.98 | 99.23 ± 0.99 | 102.36 ± 1.01 |
| F11 | 52.96 ± 1.31 | 57.55 ± 1.74 | 74.26 ± 2.08 | 82.63 ± 1.88 | 94.54 ± 2.09 | 99.23 ± 0.94 | 100.51 ± 0.78 | 101.02 ± 0.88 |
| F12 | 49.14 ± 2.01 | 53.4 ± 1.51 | 62.74 ± 1.99 | 74.28 ± 3.13 | 81.62 ± 1.78 | 85.97 ± 2.44 | 89.18 ± 3.09 | 91.60 ± 2.11 |
| F13 | 50.83 ± 2.14 | 55.52 ± 1.86 | 67.61 ± 1.41 | 77.53 ± 2.15 | 88.37 ± 1.75 | 92.27 ± 2.45 | 96.91 ± 2.01 | 99.07 ± 1.09 |
| F14 | 53.62 ± 1.71 | 58.37 ± 2.02 | 70.28 ± 2.12 | 79.96 ± 2.01 | 89.43 ± 2.20 | 94.37 ± 1.99 | 99.39 ± 1.41 | 100.62 ± 1.03 |
| F15 | 55.11 ± 1.80 | 59.86 ± 1.41 | 71.77 ± 1.44 | 81.45 ± 1.88 | 90.92 ± 1.21 | 95.86 ± 1.04 | 100.88 ± 0.96 | 102.11 ± 0.98 |
| Stilnoct® | 53.68 ± 1.42 | 63.51 ± 1.55 | 72.35 ± 1.11 | 80.93 ± 1.23 | 86.88 ± 1.08 | 93.09 ± 0.97 | 98.09 ± 0.98 | 100.23 ± 0.94 |
n = 3
Table III.
In Vitro Dissolution Profile of Zolpidem tartrate from Core Tablets of Formulations F1–F15
| Time (min) | 30 (mean cumulative percent ± SD drug released) | 60 (mean cumulative percent ± SD drug released) | 90 (mean cumulative percent ± SD drug released) | 120 (mean cumulative percent ± SD drug released) | 180 (mean cumulative percent ± SD drug released) | 240 (mean cumulative percent ± SD drug released) | 300 (mean cumulative percent ± SD drug released) | 360 (mean cumulative percent ± SD drug released) |
|---|---|---|---|---|---|---|---|---|
| F1 | 2.03 ± 1.23 | 5.79 ± 1.88 | 19.09 ± 2.09 | 27.53 ± 1.98 | 34.50 ± 2.12 | 42.47 ± 0.99 | 47.54 ± 0.98 | 50.95 ± 0.85 |
| F2 | 0.98 ± 1.89 | 2.87 ± 1.67 | 9.12 ± 2.09 | 12.75 ± 3.12 | 18.95 ± 1.89 | 25.01 ± 2.16 | 33.82 ± 3.11 | 43.31 ± 2.09 |
| F3 | 1.52 ± 1.94 | 6.48 ± 2.98 | 12.50 ± 1.97 | 23.17 ± 2.1 | 27.72 ± 3.13 | 35.23 ± 1.98 | 42.16 ± 0.99 | 48.08 ± 1.07 |
| F4 | 1.51 ± 1.72 | 7.03 ± 1.99 | 13.18 ± 1.98 | 23.71 ± 3.04 | 33.34 ± 2.19 | 40.78 ± 1.66 | 47.05 ± 0.97 | 49.61 ± 1.01 |
| F5 | 1.51 ± 1.72 | 7.03 ± 1.99 | 13.18 ± 1.98 | 23.71 ± 3.04 | 33.34 ± 2.19 | 40.78 ± 1.66 | 47.05 ± 0.97 | 49.61 ± 1.01 |
| F6 | 2.31 ± 1.32 | 7.04 ± 1.76 | 20.37 ± 2.17 | 29.35 ± 1.98 | 39.26 ± 0.92 | 48.95 ± 2.15 | 51.37 ± 0.74 | 51.94 ± 0.85 |
| F7 | 1.32 ± 1.98 | 5.44 ± 1.48 | 12.12 ± 2.11 | 19.75 ± 3.12 | 26.01 ± 1.35 | 30.61 ± 1.78 | 38.72 ± 2.34 | 45.96 ± 2.14 |
| F8 | 1.94 ± 2.15 | 6.36 ± 1.84 | 16.71 ± 1.43 | 25.65 ± 2.16 | 35.59 ± 2.44 | 41.96 ± 1.66 | 47.52 ± 1.99 | 49.77 ± 1.17 |
| F9 | 1.86 ± 1.68 | 6.27 ± 2.03 | 15.98 ± 1.65 | 24.97 ± 1.29 | 34.86 ± 1.97 | 41.02 ± 2.19 | 47.11 ± 1.37 | 50.24 ± 1.03 |
| F10 | 1.86 ± 1.68 | 6.27 ± 2.03 | 15.98 ± 1.65 | 24.97 ± 1.29 | 34.86 ± 1.97 | 41.02 ± 2.19 | 47.11 ± 1.37 | 50.24 ± 1.03 |
| F11 | 3.68 ± 1.43 | 8.27 ± 1.54 | 24.98 ± 0.96 | 33.35 ± 1.11 | 45.26 ± 1.32 | 49.95 ± 1.01 | 51.23 ± 0.24 | 51.74 ± 0.55 |
| F12 | 1.78 ± 2.76 | 6.04 ± 2.09 | 15.38 ± 1.27 | 26.92 ± 2.17 | 34.26 ± 1.98 | 38.61 ± 2.16 | 41.82 ± 1.75 | 44.24 ± 1.21 |
| F13 | 2.44 ± 1.94 | 7.13 ± 1.80 | 19.22 ± 1.04 | 29.16 ± 1.65 | 39.98 ± 1.24 | 43.87 ± 1.97 | 48.52 ± 1.01 | 50.68 ± 0.98 |
| F14 | 2.32 ± 1.83 | 7.07 ± 1.31 | 18.98 ± 1.47 | 28.66 ± 1.29 | 38.13 ± 1.39 | 43.06 ± 1.32 | 48.09 ± 0.76 | 49.32 ± 0.87 |
| F15 | 2.32 ± 1.83 | 7.07 ± 1.31 | 18.98 ± 1.47 | 28.66 ± 1.29 | 38.13 ± 1.39 | 43.06 ± 1.32 | 48.09 ± 0.76 | 49.32 ± 0.87 |
n = 3
Fig. 2.
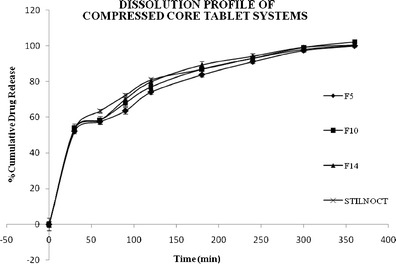
Dissolution profiles of formulations F5, F10, F14, and F15 and Stilnoct ER 12.5 mg
Fig. 3.
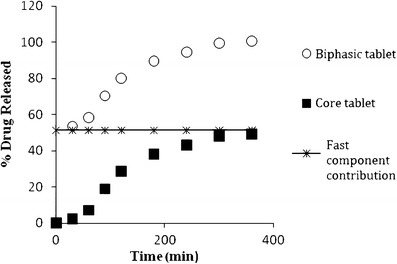
Dissolution profiles from compressed core tablets, fast component, and core tablets of formulation F14
It was found that the cumulative percentage of drug release decreases with increasing the polymer concentration as well as with the increase in viscosity grade of the polymer. Similarity factor was calculated and the formulation F14 was found to have f2 = 78. Thus, F14 was considered as optimized formula.
Drug Released from Biphasic Delivery System Tablets
The results for the fitting of the kinetic model for drug release from core and biphasic delivery system are shown in Table IV. The values for the release rate constants (K0, K1, KH, KK, and Ks), the correlation coefficients (R2), and the release exponent (n) are considered. The correlation coefficient (R2) was used as an indication of the best fit, for each of the models considered.
Table IV.
Modeling of Dissolution Profiles
| Tablet system | Zero-order | First-order | Higuchi model | Korsmeyer-Peppas model | Hixson-Crowell model | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R 2 | K | R 2 | K | R 2 | K | R 2 | n | K | R 2 | K | |
| Core + outer layer | 0.892 | 0.145 | 0.914 | −0.006 | 0.958 | 3.773 | 0.971 | 0.281 | 20.04 | 0.944 | −0.012 |
| Core | 0.892 | 0.145 | 0.933 | −0.001 | 0.958 | 3.773 | 0.917 | 1.23 | 0.05 | 0.920 | −0.002 |
For the optimized formulation F14, Korsmeyer-Peppas plot showed linearity with correlation coefficient (R2) 0.971, and the n value was found to be 0.281, indicating Fickian diffusion. The results for the cores (R2 slightly higher for the Higuchi model, 0.958, than for the first-order model, 0.933, and n = 1.23) indicate super case II transport.
Fourier Transform Infrared Spectroscopy
The interaction between the drug and the excipients often leads to identifiable changes in the infrared (IR) spectra of the drug excipient mixture in the formulation. The IR spectra of drug excipient mixture were compared with the standard spectrum of zolpidem tartrate.
The IR spectrum of pure drug showed a peak at 2,924.09 cm−1 indicating aromatic =C–H stretching, a peak at 1,643.35 cm−1 due to C=O amide stretching. A tertiary amine stretching gave a doublet at 1,265.30 and 1,303.88 cm−1. The IR spectra of drug with HPMC K100M CR showed peaks at 2,916.93 and 1,648.87 cm−1 and a doublet at 1,260.32 and 1,308.87 cm−1; spectra of drug with HPMC K15M CR showed small peak shifts to 2,917.42 cm−1, 1,648.51 cm−1, and a singlet at 1,260.36 cm−1 and that of drug with HPMC K4M CR showed small peak shifts to 2,918.29 cm−1, 1,649.06 cm−1, and a singlet at 1,260.53 cm−1. The spectrum reveals the characteristic peaks for the important functional groups in the drug structure are retained, indicating no significant interaction between the drug and the excipients used in the formulations. The overlay of IR spectra is shown in Fig. 4.
Fig. 4.
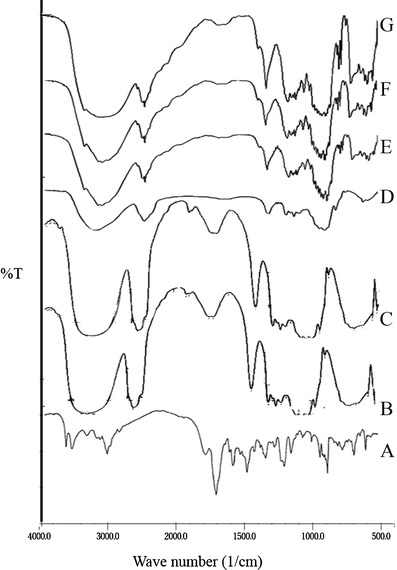
The overlay of IR spectra of A zolpidem tartrate, B HPMC K4M CR, C HPMC K15M CR, D HPMC K100M CR, E zolpidem tartrate + HPMC K4M CR blend, F zolpidem tartrate + HPMC K15M CR blend, and G zolpidem tartrate + HPMC K100M CR blend
Differential Scanning Calorimetry
Differential scanning calorimetry enables the quantitative detection of all processes in which energy is utilized or produced (endothermic or exothermic phase transformations). The DSC thermogram of pure drug, pure polymers, and blends of drug and polymers overlay is shown in Fig. 5. The DSC thermogram of zolpidem tartrate showed sharp endothermic peak at 190°C corresponding to its melting point. The DSC thermograms of zolpidem tartrate with different grades of polymers used in formulations showed the endothermic peaks at 90°C to 100°C. The DSC thermograms of zolpidem tartrate with polymer HPMC K100M CR showed a marked decrease in endothermic peak height. With polymers HPMC K15M CR and HPMC K4M CR also, a decrease in the endothermic peak is observed, indicating the drug is homogeneously distributed in the polymer or the drug may have dissolved in the melted excipients and this reduced the drug endotherm.
Fig. 5.
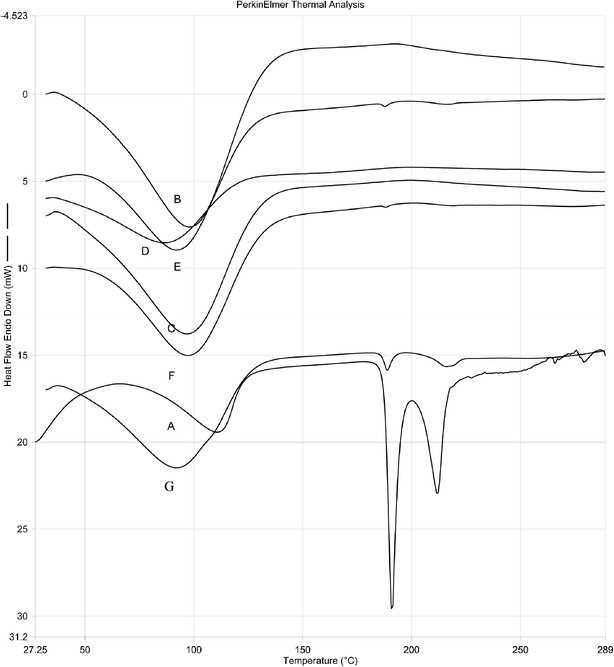
The overlay of DSC thermograms of A zolpidem tartrate, B HPMC K4M CR, C HPMC K15M CR, D HPMC K100M CR, E zolpidem tartrate + HPMC K4M CR blend, F zolpidem tartrate + HPMC K15M CR blend, and G zolpidem tartrate + HPMC K100M CR blend
Stability Studies
Accelerated stability studies performed on the final optimized formulation (F14) showed that the parameters like general appearance, hardness, thickness, assay, and in vitro dissolution study were within the specified limits as shown in Table V.
Table V.
Accelerated Stability Study Results
| Sr. no | Test | Specification | 40 ± 2°C, 75 ± 5% RH | |
|---|---|---|---|---|
| Initial | 12th week | |||
| 1 | Color | Off-white tablet | No color change | No color change |
| 2 | Hardness (kp) | 6.5 ± 0.5 | 6.5 ± 0.5 | 6.5 ± 0.5 |
| 3 | Thickness (mm) | 4.00 ± 0.5 | 4.09 ± 0.09 | 4.08 ± 0.08 |
| 4 | In vitro dissolution (%) | 30 min (50–70%) | 53.62 ± 1.71 | 51.24 ± 1.93 |
| 90 min (70–85%) | 70.28 ± 2.12 | 70.07 ± 1.08 | ||
| 240 min (NLT 90%) | 94.37 ± 1.99 | 93.66 ± 1.99 | ||
| 5 | Assay (%) | 90–110 | 99.36 ± 1.54 | 98.72 ± 2.64 |
R H relative humidity
DISCUSSIONS
The physical properties (weight, thickness, hardness, and friability) of the core tablets and compressed core tablet systems for all the formulations were within the acceptable limits. From the dissolution study, the release profiles are characterized by a burst release within 30 min, followed by a slow-release period, typical of a biphasic quick/slow delivery system. For all formulations, upon contact with the dissolution media, the modified-release tablets rapidly disintegrated into the fast-releasing phase and the matrix core tablets. The prompt tablet disintegration was due to the presence of sodium starch glycolate, which swells very quickly on contact with the dissolution medium. After the initial phase, the release was dependent on the composition of the matrix core, in particular, the grade and concentration of HPMC. The ability of the HPMC particles to hydrate and form a gel layer around a core is well-known and is essential to sustaining and controlling the release of a drug from the matrix (34). Throughout the dissolution test, a continuous gel layer formed in the HPMC matrix core was responsible for guiding the release of the drug.
In the biphasic delivery system developed by Maggi et al. (35), the in vitro dissolution tests showed that the drugs (ketoprofen and praziquantel) contained in the fast-release layer dissolved within 15 min because of the presence of sodium starch glycolate, while the drug contained in the HPMC-prolonged release layer was released at different times, depending on the percentage and viscosity grade of the HPMC.
It was found that the cumulative percentage of drug release decreases with increasing the polymer concentration as well as with the increase in viscosity grade of the polymer. F14 was considered as optimized formula by calculating the similarity factor. This formulation showed that the drug release was as per USP limits for zolpidem tartrate extended-release tablets by releasing 50–70% drug within 30 min, followed by 70–90% release in 90 min and not less than 90% at the end of 4 h (17).
For the optimized formulation F14, Korsmeyer-Peppas plot indicated that Fickian diffusion is an important mechanism. The results for the cores indicate super case II transport. However, the analysis of the results applying these mathematical models is purely empirical, and no definitive conclusion can be drawn concerning the dominant mass transport mechanisms.
Compatibility studies reveal that the drug is compatible with the all excipients used in the formulation. The stability studies concluded that the formulation can withstand to the general stress conditions of temperature and humidity.
CONCLUSIONS
A biphasic release system was achieved by a quick/slow delivery, characterized by an initial rapid release phase, followed by a period of slow release. The results obtained with the dissolution tests showed that the release profile is dependent on both the grade and amount of polymer in the core tablet. The developed formulations were matched with Stilnoct ER for drug release profile. The similarity factor (f2) and dissimilarity factor (f1) were calculated, and it was concluded that the formulation F14 of this modified-release biphasic delivery system tablets would be a promising formulation for the treatment of chronic insomnia by supporting sleep maintenance. This formulation followed the USP limits for drug release of zolpidem tartrate extended-release tablets. Thus, the optimized formula F14, containing 4% sodium starch glycolate in immediate-release coat and 15% w/w of HPMC K4M CR in extended-release core, can show improved efficacy and better patient compliance. The biphasic release systems of selective drugs rationally can meet the benefits of pharmacotherapy.
ACKNOWLEDGMENTS
The authors are grateful to M/s. Anglo-French Drugs and Industries Ltd. (Bangalore, India) for providing necessary facilities to carry out this research work.
REFERENCES
- 1.Ellis JG, Perlis ML, Neale LF, Espie CA, Bastien CH. The natural history of insomnia: focus on prevalence and incidence of acute insomnia. J Psychiatr Res. 2012;46:1278–85. doi: 10.1016/j.jpsychires.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Lacks P, Morin CM. Recent advances in the assessment and treatment of insomnia. J Consult Clin Psychol. 1992;60(4):586–94. doi: 10.1037/0022-006X.60.4.586. [DOI] [PubMed] [Google Scholar]
- 3.Bogan RK. Treatment options for insomnia—pharmacodynamics of zolpidem extended-release to benefit next-day performance. Postgrad Med. 2008;120(3):161–71. doi: 10.3810/pgm.2008.09.1916. [DOI] [PubMed] [Google Scholar]
- 4.Zolpidem—compound summary. http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=5732. Accessed 11 November 2013.
- 5.Zolpidem tartrate: highlights of prescribing information. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=11a7fa3f-5dd3-4ba6-8a5b-0be321c58dfa. Accessed 11 November 2013.
- 6.Colombo P. Swelling-controlled release in hydrogel matrixes for oral route. Adv Drug Delv Rev. 1993;11:37–57. doi: 10.1016/0169-409X(93)90026-Z. [DOI] [Google Scholar]
- 7.Martindale WH. The extra pharmacopeia. 30. London: Pharmaceutical Press; 1993. p. 1783. [Google Scholar]
- 8.Sahoo J, Murthy PN, Biswal S, Sahoo SK, Mahapatra AK. Comparative study of propranolol hydrochloride release from matrix tablets with Kollidon®SR or hydroxy propyl methyl cellulose. AAPS Pharm Sci Tech. 2008;9(2):577–82. doi: 10.1208/s12249-008-9092-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bin C, Joshi SC, Lam YC. Bio-fluid uptake and release of Indomethacin of direct-compressed HPMC tablets. Carbohydr Polym. 2009;75(2):282–6. doi: 10.1016/j.carbpol.2008.07.025. [DOI] [Google Scholar]
- 10.Conte U, Maggi L. A flexible technology for the linear, pulsative and delayed release drugs, allowing for easy accommodation of difficult in vitro targets. J Control Release. 2000;64:263–8. doi: 10.1016/S0168-3659(99)00147-9. [DOI] [PubMed] [Google Scholar]
- 11.Patel GM, Patel MM. Compressed matrix dual-component vaginal drug delivery system containing metoclopramide hydrochloride. Acta Pharm. 2009;59:273–88. doi: 10.2478/v10007-009-0029-4. [DOI] [PubMed] [Google Scholar]
- 12.Doghramji PP. Insomnia: zolpidem extended-release for the treatment of sleep induction and sleep maintenance symptoms. Med Gen Med. 2007;9(1):11. [PMC free article] [PubMed] [Google Scholar]
- 13.Monti JM, Spence DW, Perumal SRP, Langer SZ, Hardeland R. Pharmacotherapy of insomnia: focus on zolpidem extended release. Cli Med Ther. 2009;1123–40.
- 14.Kirkwood C, Neill J, Breden E. Zolpidem modified-release in insomnia. Neuropsychiatr Dis Treat. 2007;3(5):521–6. [PMC free article] [PubMed] [Google Scholar]
- 15.Van Dalen F, Jansen KA, Dorkoosh FA; Synthon Bv. Zolpidem tablets. United States patent 8148393 B2. 2012.
- 16.Guo M, Nandi I, Patel A, Wu Ch. Compression coated tablets. United States patent 20040068000 A1. 2004.
- 17.Zolpidem tartrate extended release tablets. The United States Pharmacopeial Convention, Revision Bulletin 2012. http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/revisions/m2061-zolpidem_tartrate_er_tablets.pdf. Accessed: 11 November 2013.
- 18.Morre JW, Flanner HH. Mathematical comparison of dissolution profiles. Pharm Technol. 1996;20:64–74. [Google Scholar]
- 19.CDER. Center for Drug Evaluation and Research. Guidance for industry, dissolution testing of immediate release solid oral dosage. 1997. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070237.pdf. Accessed: 11 November 2013.
- 20.EMEA. European Agency for the Evaluation of Medicinal Products. Human medicines evaluation unit, note for guidance on quality of modified release products: (A) oral dosage forms; (B) transdermal dosage forms; section I (quality), CPMP/QWP/604/96. 1999. http://www.tga.gov.au/pdf/euguide/qwp060496en.pdf. Accessed: 11 November 2013.
- 21.Costa P, Sausa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–33. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 22.Ritger PL, Peppas NA. A simple equation for description of solute release. I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5:23–36. doi: 10.1016/0168-3659(87)90034-4. [DOI] [PubMed] [Google Scholar]
- 23.Donbrow M, Samuelov Y. Zero order drug delivery from double-layered porous films: release rate profiles from ethylcellulose, hydroxypropylcellulose and polyethylene glycol mixtures. J Pharm Pharmacol. 1980;32:463–70. doi: 10.1111/j.2042-7158.1980.tb12970.x. [DOI] [PubMed] [Google Scholar]
- 24.Narashimhan B, Mallapragada SK, Peppas NA. Release kinetics, data interpretation. In: Mathiowitz, editor. Encyclopedia of controlled drug delivery. New York: John Wiley and Sons, Inc; 1999. p. 921. [Google Scholar]
- 25.Silvina A, Bravo M, Lamas C, Claudio J. In-vitro studies of diclofenac sodium controlled-release from biopolymeric hydrophilic matrices. J Pharm Pharmaceut Sci. 2002;5(3):213–9. [PubMed] [Google Scholar]
- 26.Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50:874–5. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 27.Higuchi T. Mechanism of sustained-action medication: theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–9. doi: 10.1002/jps.2600521210. [DOI] [PubMed] [Google Scholar]
- 28.Korsmeyer RW, Gurny R, Doelker EM, Buri P, Peppas NA. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1016/0378-5173(83)90064-9. [DOI] [PubMed] [Google Scholar]
- 29.Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60:110–1. [PubMed] [Google Scholar]
- 30.Cox PJ, Khan KA, Munday DL, Sujjareevath J. Development and evaluation of a multiple-unit oral sustained release dosage form for S (+)-ibuprofen: preparation and release kinetics. Int J Pharm. 1999;193:73–84. doi: 10.1016/S0378-5173(99)00320-8. [DOI] [PubMed] [Google Scholar]
- 31.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Poloniae Pharm Drug Res. 2010;67(3):217–23. [PubMed] [Google Scholar]
- 32.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Adv Drug Delv Rev. 2001;48:139–57. doi: 10.1016/S0169-409X(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 33.Jacques CHM, Hopfenberg HB, Stannett V. Super case II transport of organic vapors in glassy polymers. In: Hopfenberger HB, editor. Permeability of plastic films and coatings to gases, vapors, and liquids. New York: Plenum Press; 1974. pp. 73–86. [Google Scholar]
- 34.Colombo P, Bettini R, Santi P, Peppas NA. Swellable matrices for controlled drug delivery: gel-layer behavior, mechanisms and optimal performance. Pharm Sci Technol Today. 2000;3:198–204. doi: 10.1016/S1461-5347(00)00269-8. [DOI] [PubMed] [Google Scholar]
- 35.Maggi L, Machiste EO, Torre ML, Conte U. Formulation of biphasic release tablets containing slightly soluble drugs. Eur J Pharm Biopharm. 1999;48:37–42. doi: 10.1016/S0939-6411(99)00019-3. [DOI] [PubMed] [Google Scholar]