

Abstract
Newborn infants who have hereditary spherocytosis (HS) can develop anemia and hyperbilirubinemia. Bilirubin-induced neurologic dysfunction is less likely in these neonates if the diagnosis of HS is recognized and appropriate treatment provided. Among neonates listed in the USA Kernicterus Registry, HS was the third most common underlying hemolytic condition after glucose-6-phosphate dehydrogenase deficiency and ABO hemolytic disease. HS is the leading cause of direct antiglobulin test (direct Coombs) negative hemolytic anemia requiring erythrocyte transfusion in the first months of life. We anticipate that as physicians become more familiar with diagnosing HS in the newborn period, fewer neonates with HS will develop hazardous hyperbilirubinemia or present to emergency departments with unanticipated symptomatic anemia. We predict that early suspicion, prompt diagnosis and treatment, and anticipatory guidance will prevent adverse outcomes in neonates with HS. The purpose of this article was to review the neonatal presentation of HS and to provide practical and up-to-date means of diagnosing and treating HS in neonates.
Hereditary spherocytosis (HS) is a heterogeneous disorder in which abnormalities of red blood cell structural proteins lead to loss of erythrocyte membrane surface area, resulting in spherical-shaped, hyperdense, poorly deformable red blood cells (Fig 1) with a shortened life span.1–5 HS occurs worldwide and affects individuals from all racial and ethnic groups. Individual pediatricians encounter HS uncommonly, but hospitals and health care systems with large delivery services regularly deal with this condition, particularly in white neonates of northern European ancestry, in whom the condition can be as frequent as 1 in 1000 to 2000 births. Early suspicion and diagnosis of HS allow appropriate management, including provision of anticipatory guidance to parents, which can reduce the risk of adverse outcomes.
FIGURE 1.
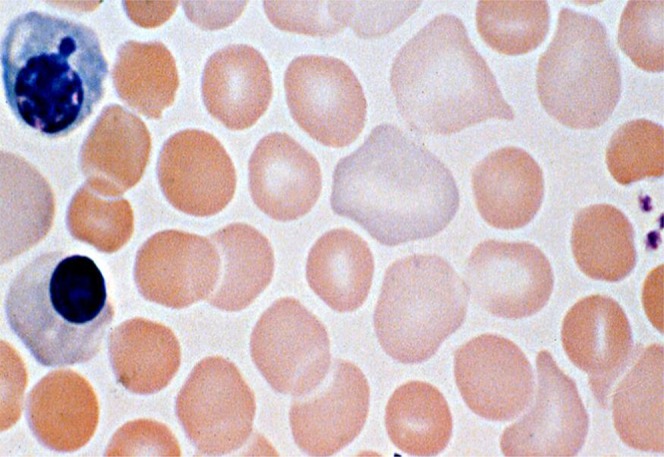
Photomicrograph of a stained blood film from a neonate with hereditary spherocytosis (band 3 mutation). Several spherocytes are present in this field, along with polychromatophilic erythrocytes and 2 nucleated red blood cells.
The purpose of the present article was to review the neonatal presentation of HS and to provide practical and up-to-date means of diagnosing and treating HS in neonates.
Pathogenesis
The loss of membrane surface area in HS erythrocytes is due to defects in various erythrocyte membrane proteins, ankyrin-1, band 3, β-spectrin, α-spectrin, and protein 4.2 (Table 1, Fig 2).1–5 Numerous mutations in the genes encoding these membrane proteins have been described. Destruction of poorly deformable HS erythrocytes in the spleen is the primary cause of hemolysis in patients with HS.
TABLE 1.
Erythrocyte Membrane Proteins Involved in HS
| Protein | Gene | Chromosomal Location | Percentage of HS Cases | Typical Severity | Inheritance |
|---|---|---|---|---|---|
| Ankyrin-1 | ANK1 | 8p11.2 | 40–50 | Mild to moderate | Autosomal dominant |
| Band 3 | SLC4A1 | 17q21 | 20–35 | Mild to moderate | Autosomal dominant |
| β-spectrin | SPTB | 14q23-24.1 | 15–30 | Mild to moderate | Autosomal dominant |
| α-spectrin | SPTA1 | 1q22-23 | <5 | Severe | Autosomal recessive |
| Protein 4.2 | EPB42 | 15q15-21 | <5 | Mild to moderate | Autosomal recessive |
FIGURE 2.

Schematic diagram of the erythrocyte membrane. The upper portion represents the erythrocyte’s external surface, separated from the internal structures by the lipid bilayer. Proteins shown where mutations result in HS include band 3, ankyrin-1, α-spectrin, β-spectrin, and protein 4.2. (Reprinted with permission from Gallagher PG. Abnormalities of the erythrocyte membrane. Pediatr Clin North Am. 2013;60(6):1358).
The clinical spectrum of HS during the perinatal period ranges from severe fetal anemia with hydrops fetalis to the asymptomatic neonate. This wide range is due, in part, to the various genes and specific mutations involved, as well as to the presence or absence of coinherited conditions. For instance, HS coinherited with mutations or polymorphisms of genes involved in bilirubin uptake into hepatocytes (SLC01B1) or intrahepatic bilirubin conjugation (UGT1A1) can increase the risk of hazardous hyperbilirubinemia and kernicterus.6–9
Making the Diagnosis of HS in a Neonate
As with any relatively uncommon neonatal disorder, the first step in making the diagnosis of HS is considering it in the differential diagnosis. Thus, a basic understanding of HS is useful to physicians caring for neonates. We have found that the triad of anemia, splenomegaly, and jaundice, which is found in older children and adults with HS, is rare in neonates. More than one-half of neonates with HS are not anemic in the first week of life, and splenomegaly is rarely detected.10 Jaundice is the most common presenting feature of HS in neonates.11 In addition, the typically sluggish erythropoietic response of neonates often renders the reticulocyte count low relative to the degree of anemia10; spherocytes are less often observed on the blood smear of neonates; and other markers of hemolysis seen in older patients, such as low haptoglobin levels,12 may be poor indicators of hemolysis in the neonate.
Severe neonatal jaundice can be the result of various underlying causes.13–16 Unfortunately, as illustrated by Johnson et al,13 even when hyperbilirubinemia is severe enough to cause kernicterus, the underlying cause of the jaundice frequently remains unidentified. Perhaps the intense focus on reducing the elevated bilirubin level into a safe range sometimes results in neglecting evaluation of how the bilirubin became so high in the first place.14
Approximately 65% of neonates with HS have a parent with HS.1,3,5 When a parent has HS, it is important that this information be placed prominently in the prenatal record and communicated verbally, before birth, to the physicians and the hospital staff who will be providing neonatal care. All parents with HS should be encouraged to communicate this information to their infant’s physician before delivery. Failure to do so sometimes occurs when the affected parent has been asymptomatic since undergoing splenectomy as a child and has all but forgotten about the condition, and fails to consider that it might be problematic for the newborn infant. It can be helpful to specifically inquire of parents of anemic and/or jaundiced neonates about a family history of anemia, jaundice, splenectomy, or early gallstones.
One way to suspect HS in a jaundiced neonate is to obtain a complete blood cell count for interpretation of the red blood cell indices, in particular the mean corpuscular hemoglobin concentration (MCHC) and the mean corpuscular volume (MCV), and to examine the peripheral blood smear for the presence of spherocytes and polychromasia.17 Typically, a neonate with HS will have an elevated MCHC. Figure 3 displays the histograms of MCHC measurements in 3 groups of jaundiced neonates: (1) direct antiglobulin test (DAT)-negative jaundice; (2) DAT-positive jaundice (primarily ABO hemolytic disease); and (3) HS. The histogram reveals some overlap in MCHC, but, as a general rule, if a neonate’s MCHC is elevated (>36.5 to 37 g/dL), HS is likely.11 Another way to estimate whether a neonate has HS takes advantage of the fact that in most neonates with HS, the MCV is low. Based on this scenario, the neonatal HS ratio can be calculated by dividing the MCHC by the MCV. Using this ratio, little overlap is seen between normal neonates and those later proven to have HS. In fact, in the Intermountain Healthcare database, a neonatal HS ratio >0.36 indicates that HS is present with 97% sensitivity, >99% specificity, and >99% negative predictive value.18
FIGURE 3.
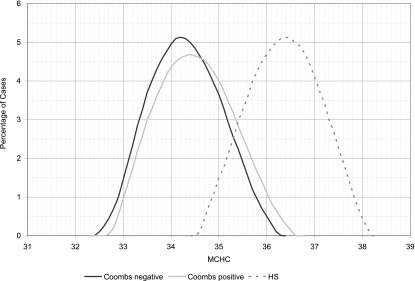
Frequency distribution of MCHC (grams per deciliter) values in 3 groups of jaundiced neonates: (1) DAT negative (Coombs negative); (2) DAT positive (Coombs positive); and (3) HS. (Reprinted with permission from Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics. 2010;125(1):124).
It is likely that different makes and models of automated cell counters generate slight differences in erythrocyte indices, such as the MCV and MCHC. Validation of the neonatal HS ratio on all models of counters would be useful. The mean hemoglobin content of all the measured red blood cells as well as reticulocyte parameters can sometimes be helpful in suggesting HS,19 but we are unaware of reports of their use in newborn infants.
Figure 4 reviews our recommendations for how to evaluate a neonate for HS when a parent is known to have HS. Because the common forms of HS are inherited in an autosomal dominant manner, each child born to a parent with HS has a 50% chance of inheriting the disorder. The key element in the algorithm is to treat the neonate as if he or she has HS until proven otherwise. In addition to obtaining erythrocyte indices, this approach includes peripheral smear examination and reticulocyte count, adhering to the American Academy of Pediatrics’ guidelines for bilirubin monitoring in the birth hospital, aggressive phototherapy when indicated, and a follow-up bilirubin assessment no later than 24 hours after the hospital discharge.20,21 Figure 5 reviews our recommendations for evaluating a jaundiced neonate for HS when neither parent has HS. There is no need to wait until the neonate is several months old, or until significant anemia develops, to begin the evaluation. By following the algorithm in Fig 4 (parent has HS) or Fig 5 (parent does not have HS), the diagnosis can often be made during the birth hospitalization.
FIGURE 4.

Evaluating a neonate during the birth hospitalization whose parent has HS. The evaluation includes the following: (1) observe the serum bilirubin level and treat according to the American Academy of Pediatrics’ guidelines during and for several days after the birth hospitalization as if this were a known case of hemolysis; (2) during the birth hospitalization initiate the evaluation as given in the graphic or as suggested by hematology consultation; and (3) consider that the neonate could have more severe jaundice than the parent did as a neonate, particularly if there has been coinheritance of a polymorphism-retarding bilirubin uptake or conjugation. CBC, complete blood cell count.
FIGURE 5.

Evaluating a neonate with problematic jaundice in whom the etiology is unclear. Not all neonates who receive phototherapy for ≥2 days have hemolytic jaundice. However, if hemolytic jaundice is suspected, the algorithm given here for stepwise evaluation of the etiology might be useful. CBC, complete blood cell count; G6PD, glucose-6-phosphate dehydrogenase deficiency.
The presence of spherocytes on peripheral blood smear is helpful when considering the diagnosis of HS (Fig 1),14 but up to one-third of neonates with HS do not have spherocytes identified prominently on their blood smear. Some neonates with band 3 deficiency have pincered red blood cells.22 A subset of neonates with DAT-negative ABO incompatibility severe enough to produce spherocytes on peripheral smear have been described. In most cases, the differentiation from HS is clear (eg, family history, maternal/infant blood group analysis), but additional testing such as elution of anti-A or anti-B from neonatal erythrocytes or detection of free anti-A or anti-B immunoglobulin G antibody in neonatal serum (indirect Coombs test) can clarify the diagnosis.
When the diagnosis of HS is uncertain, eosin-5-maleimide (EMA) binding or osmotic fragility testing can be helpful23–25 (Fig 6). EMA binding is a flow cytometry–based test that measures the relative amount of fluorescently labeled EMA dye bound to band 3 and Rh-related proteins in the erythrocyte membranes. In HS, the reduction in band 3 and other membrane proteins leads to decreased fluorescence intensity. In the Primary Children’s Hospital hematology clinic, EMA flow has outperformed other diagnostic tests for HS in newborn infants.24 Neonatal erythrocytes exhibit altered response to osmotic stress compared with adult erythrocytes. However, after incubation, osmotic fragility has been successfully used to diagnose HS in neonates.26,27 New flow cytometric methods of assessing erythrocyte osmotic fragility are promising,28–30 but we do not have experience with these methods for diagnosing HS in newborn infants nor do we know of published studies in which these methods were proven effective with the blood of neonates. Sodium dodecyl sulfate polyacrylamide gel electrophoresis has been used as well but has generally been replaced by other methods.31
FIGURE 6.

A, Results of Osmotic Fragility testing of a neonate with HS. The diagnosis is supported by finding that red blood cells are more fragile to osmotic lysis than normal (the normal range is shown by the 2 dotted lines). HS cells undergo more hemolysis than normal as the sodium chloride (NaCl) concentration is increased. B, Results of Eosin-5-Maleimide Fluorescent Cytometric (EMA-FITC) testing of the same neonate with HS shown in panel (A). The diagnosis is supported by finding that a large fraction of the red blood cells have reduced EMA binding, with a leftward displacement of events, compared with the normal range (footprint). Side scatter (SSC) reflects cell size.
Table 2 lists laboratory evaluations we consider to be helpful in diagnosing HS in a neonate. Sequencing of the relevant genes can be performed as a confirmatory test (available in a few reference laboratories) when desired.32 We reserve genetic sequencing for cases with no family history and a severe HS phenotype. The goal would be to make a clear diagnosis, allow for appropriate therapy, and to provide parents and family members with genetic information for counseling regarding risk of recurrence.
TABLE 2.
Laboratory Evaluation for HS in a Jaundiced Neonate
| HS Ratio (MCHC/MCV) | EMA Flowa | Incubated Osmotic Fragility | DNA Sequencingb |
|---|---|---|---|
| Neonates with HS will generally have a high MCHC and a low MCV, producing an elevated ratio (>0.36) | EMA dye binds stoichiometrically to band 3 and Rh-related membrane proteins. Decreased fluorescence intensity of EMA-tagged erythrocytes due to loss of membrane proteins is seen in HS. Decreased EMA binding is also seen in other disorders such as hereditary pyropoikilocytosis and congenital dyserythropoietic anemia II | HS erythrocytes are more susceptible to osmotic lysis than normal erythrocytes due to decreased membrane surface area. Incubation overnight stresses the already fragile HS erythrocyte, accentuating the defect. Spherocytes from any cause, including ABO incompatibility, will yield a positive result on osmotic fragility testing | Not needed to diagnosis most cases of HS. However, it can establish the diagnosis in difficult cases. Consider sequencing of relevant genes when family history is negative and severe DAT-negative hemolysis is idiopathic |
Available in many reference laboratories.
Available in selected hematology/genetics reference laboratories. For example: Blood Disease Reference Hematology Laboratory, http://www.yaleblooddiseaselab.org/, 310 Cedar Street, CB 541a, New Haven, CT 06520, Tel. (203)737-1349, Fax (203)785-3896; ARUP Laboratories, http://www.aruplab.com/testing, 500 Chipeta Way, Salt Lake City, UT 84108, Tel. (800)522-2787, Fax (800)522-2706; GeneDx, http://www.genedx.com, 207 Perry Parkway Gaithersburg, MD 20877, Tel. (301)519-2100, Fax (301)519-2892; and Mayo Medical Laboratories, http://www.mayomedicallaboratories.com/, 3050 Superior Drive NW, Rochester, MN 55901, Tel. (800)553-1710, Fax (507)284-1759.
Treatment
Phototherapy should reduce the bilirubin level of jaundiced neonates with HS, and it is the mainstay of treatment in the first days after birth.33–35 When a bilirubin level is found to be in the high or high to intermediate risk zone (>75th percentile reference interval), phototherapy should be provided immediately. An exchange transfusion followed by intensive phototherapy should follow American Academy of Pediatrics’ guidelines.20,21,36
When signs of anemia appear, packed erythrocyte transfusions are helpful. Longitudinal studies indicate that transfusion requirements abate in most patients by 1 year of age. The few patients who remain transfusion dependent (typically those with severe anemia in utero or immediately after birth) experience severe HS. In a few reports, recombinant erythropoietin (rEPO, or long-acting darbepoetin rEPO) therapy has been used as an alternative or adjunct to transfusion.37–39 The rationale for rEPO treatment relates to the relative hypoplastic phase of erythropoiesis during the first weeks to months after birth. This phase might be associated with the abrupt decrease after birth from the highly stimulated erythropoiesis during fetal life, the switch of erythropoietin (EPO) production from the liver to the kidney, the switch from fetal to adult hemoglobin, or a lower serum level of EPO in infants compared with older children. Patients with moderate or severe HS should receive folate supplementation to prevent complications of folic acid deficiency. Splenectomy is rarely undertaken in the first year of life. Because hemolysis abates in most patients, careful symptomatic management is prudent, with transfusion therapy as indicated.
Natural History of HS During Infancy
This topic has received relatively little attention in published series or reviews.40–45 Delhommeau et al10 reported on 34 infants with HS during their first year of life, and neonatal jaundice was present in virtually all of the infants. Twenty-seven infants were treated with phototherapy, and 3 received exchange transfusions in the first week due to hyperbilirubinemia. Thirty-one of the 34 infants had pallor and dyspnea during the first month. Twenty-six (76%) required ≥1 red blood cell transfusion during the first year; 12 had a single transfusion, and 14 had ≥2 transfusions. The authors’ practice was to order transfusions if the hemoglobin concentration fell to <10 g/dL in the first week and <8.5 g/dL thereafter. Transfusions were rarely needed in the first week of life but were commonly given in month 2 of life. Only 8 (24%) of those with HS continued to receive transfusions after 6 months of age. Six underwent splenectomies at 2 to 5 years of age.
The natural history of HS during the first week of life can involve hyperbilirubinemia, which is sometimes severe, leading to bilirubin-induced neurologic dysfunction.6,7,13,43 In the USA Kernicterus Registry, 23 of the 125 patients with kernicterus had a hemolytic condition, of whom 3 had HS and severe icteric sequelae.13
There should be close observation for hematologic decompensation during acute illnesses. Infants with HS may experience hemolytic or aplastic crises, similar to children and adults with HS. This outcome is particularly true after 6 months of age when maternally derived immunoglobulin G antibodies to microbes have waned. Parvovirus B19–mediated aplasia is a common occurrence in children with HS.45 Parents should be provided anticipatory guidance regarding the occurrence of hematologic decompensation accompanying systemic illness. The wide spectrum of disease severity requires individualized plans for frequency of follow-up visits. These visits should assess the degree of anemia and monitor growth and development. Children with HS who continue to require blood transfusions should be closely monitored for iron overload.
Conclusions
A working knowledge of the presentation, diagnosis, and treatment of HS is essential for those providing care to neonates. When a newborn infant has significant hyperbilirubinemia, a careful review of the family history, evaluation of the red blood cell indices, and interpretation of the peripheral blood smear may be all that is needed to make a diagnosis of HS. In selected cases measuring erythrocyte EMA binding, incubated osmotic fragility or flow cytometric detection of erythrocyte osmotic fragility may be helpful. In problematic cases, hematologic consultation and DNA sequencing can provide the diagnosis. In any newborn infant with hemolytic jaundice, rigorous bilirubin monitoring and treatment are needed, particularly during the first weeks of life, and monitoring of the hemoglobin level is required during the first months of life. If health care providers develop an early suspicion of HS, establish the diagnosis, provide appropriate treatment, and offer anticipatory guidance to home caregivers, adverse outcomes can be avoided.
Footnotes
Dr Christensen conceptualized and drafted the initial manuscript; Drs Yaish and Gallagher authored the manuscript; and all authors approved the final version as submitted.
FINANCIAL DISCLOSURE: The authors have indicated they have no financial relationships relevant to this article to disclose.
FUNDING: This work was supported by NIH grant RO1-HL65448. Funded by the National Institutes of Health.
POTENTIAL CONFLICT OF INTEREST: The authors have indicated they have no potential conflicts of interest to disclose.
References
- 1.Gallagher PG. Abnormalities of the erythrocyte membrane. Pediatr Clin North Am. 2013;60(6):1349–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sangerman J, Maksimova Y, Edelman EJ, Morrow JS, Forget BG, Gallagher PG. Ankyrin-linked hereditary spherocytosis in an African-American kindred. Am J Hematol. 2008;83(10):789–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheffield MJ, Christensen RD. Evaluating neonatal hyperbilirubinemia in late preterm Hispanic twins led to the diagnosis of hereditary spherocytosis in them, and in their sibling and in their mother. J Perinatol. 2011;31(9):625–627 [DOI] [PubMed] [Google Scholar]
- 4.Crisp RL, Solari L, Vota D, et al. A prospective study to assess the predictive value for hereditary spherocytosis using five laboratory tests (cryohemolysis test, eosin-5′-maleimide flow cytometry, osmotic fragility test, autohemolysis test, and SDS-PAGE) on 50 hereditary spherocytosis families in Argentina. Ann Hematol. 2011;90(6):625–634 [DOI] [PubMed] [Google Scholar]
- 5.Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411–1426 [DOI] [PubMed] [Google Scholar]
- 6.Iolascon A, Faienza MF, Moretti A, Perrotta S, Miraglia del Giudice E. UGT1 promoter polymorphism accounts for increased neonatal appearance of hereditary spherocytosis. Blood. 1998;91(3):1093. [PubMed] [Google Scholar]
- 7.Berardi A, Lugli L, Ferrari F, et al. Kernicterus associated with hereditary spherocytosis and UGT1A1 promoter polymorphism. Biol Neonate. 2006;90(4):243–246 [DOI] [PubMed] [Google Scholar]
- 8.Qader A, Ismail AQ, Gandhi A, El-Shimy N. Intractable neonatal jaundice due to hereditary spherocytosis and Gilbert's syndrome. BMJ Case Rep. 2011 Jul 28;2011 [DOI] [PMC free article] [PubMed]
- 9.Korkmaz U, Duman AE, Oğütmen Koç D, et al. Severe jaundice due to coexistence of Dubin-Johnson syndrome and hereditary spherocytosis: a case report. Turk J Gastroenterol. 2011;22(4):422–425 [DOI] [PubMed] [Google Scholar]
- 10.Delhommeau F, Cynober T, Schischmanoff PO, et al. Natural history of hereditary spherocytosis during the first year of life. Blood. 2000;95(2):393–397 [PubMed] [Google Scholar]
- 11.Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics. 2010;125(1):120–125 [DOI] [PubMed] [Google Scholar]
- 12.Chavez-Bueno S, Beasley JA, Goldbeck JM, et al. ‘Haptoglobin concentrations in preterm and term newborns’. J Perinatol. 2011;31(7):500–503 [DOI] [PubMed] [Google Scholar]
- 13.Johnson L, Bhutani VK, Karp K, Sivieri EM, Shapiro SM. Clinical report from the pilot USA Kernicterus Registry (1992 to 2004). J Perinatol. 2009;29(suppl 1):S25–S45 [DOI] [PubMed] [Google Scholar]
- 14.Christensen RD, Lambert DK, Henry E, et al. Unexplained extreme hyperbilirubinemia among neonates in a multihospital healthcare system. Blood Cells Mol Dis. 2013;50(2):105–109 [DOI] [PubMed] [Google Scholar]
- 15.Sgro M, Campbell D, Shah V. Incidence and causes of severe neonatal hyperbilirubinemia in Canada. CMAJ. 2006;175(6):587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saada V, Cynober T, Brossard Y, et al. Incidence of hereditary spherocytosis in a population of jaundiced neonates. Pediatr Hematol Oncol. 2006;23(5):387–397 [DOI] [PubMed] [Google Scholar]
- 17.Christensen RD, Yaish HM, Lemons RS. Neonatal hemolytic jaundice: morphologic features of erythrocytes that will help you diagnose the underlying condition. Neonatology. 2014;105(4):243–249 [DOI] [PubMed] [Google Scholar]
- 18.Yaish HM, Christensen RD, Henry E, Baer VL, Bennett ST. A simple method of screening newborn infants for hereditary spherocytosis. J Applied Hematol. 2013:27–32 [Google Scholar]
- 19.Mullier F, Lainey E, Fenneteau O, et al. Additional erythrocytic and reticulocytic parameters helpful for diagnosis of hereditary spherocytosis: results of a multicentre study. Ann Hematol. 2011;90(7):759–768 [DOI] [PubMed] [Google Scholar]
- 20.American Academy of Pediatrics Subcommittee on Hyperbilirubinemia . Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114(1):297–316 [DOI] [PubMed] [Google Scholar]
- 21.Maisels MJ, Bhutani VK, Bogen D, Newman TB, Stark AR, Watchko JF. Hyperbilirubinemia in the newborn infant > or =35 weeks’ gestation: an update with clarifications. Pediatrics. 2009;124(4):1193–1198 [DOI] [PubMed] [Google Scholar]
- 22.Jarolim P, Murray JL, Rubin HL, et al. Characterization of 13 novel band 3 gene defects in hereditary spherocytosis with band 3 deficiency. Blood. 1996;88(11):4366–4374 [PubMed] [Google Scholar]
- 23.King MJ, Zanella A. Hereditary red cell membrane disorders and laboratory diagnostic testing. Int J Lab Hematol. 2013;35(3):237–243 [DOI] [PubMed] [Google Scholar]
- 24.Christensen RD, Agarwal AM, Nussenzveig RH, Heikal N, Liew MA, Yaish HM. Evaluating eosin-5-maleimide binding as a diagnostic test for hereditary spherocytosis in newborn infants [published online ahead of print November 6, 2014]. J Perinatol. doi:10.1038/jp.2014.202 [DOI] [PubMed] [Google Scholar]
- 25.Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ, General Haematology Task Force of the British Committee for Standards in Haematology . Guidelines for the diagnosis and management of hereditary spherocytosis—2011 update. Br J Haematol. 2012;156(1):37–49 [DOI] [PubMed] [Google Scholar]
- 26.Schröter W, Kahsnitz E. Diagnosis of hereditary spherocytosis in newborn infants. J Pediatr. 1983;103(3):460–463 [DOI] [PubMed] [Google Scholar]
- 27.Trucco JI, Brown AK. Neonatal manifestations of hereditary spherocytosis. Am J Dis Child. 1967;113(2):263–270 [DOI] [PubMed] [Google Scholar]
- 28.Won DI, Suh JS. Flow cytometric detection of erythrocyte osmotic fragility. Cytometry B Clin Cytom. 2009;76(2):135–141 [DOI] [PubMed] [Google Scholar]
- 29.Warang P, Gupta M, Kedar P, Ghosh K, Colah R. Flow cytometric osmotic fragility—an effective screening approach for red cell membranopathies. Cytometry B Clin Cytom. 2011;80(3):186–190 [DOI] [PubMed] [Google Scholar]
- 30.Crisp RL, Solari L, Gammella D, Schvartzman GA, Rapetti MC, Donato H. Use of capillary blood to diagnose hereditary spherocytosis. Pediatr Blood Cancer. 2012;59(7):1299–1301 [DOI] [PubMed] [Google Scholar]
- 31.Bianchi P, Fermo E, Vercellati C, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012;97(4):516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christensen RD, Nussenzveig RH, Yaish HM, Henry E, Eggert LD, Agarwal AM. Causes of hemolysis in neonates with extreme hyperbilirubinemia. J Perinatol. 2014;34(8):616–619 [DOI] [PubMed] [Google Scholar]
- 33.Maisels MJ, McDonagh AF. Phototherapy for neonatal jaundice. N Engl J Med. 2008;358(9):920–928 [DOI] [PubMed] [Google Scholar]
- 34.Johnson L, Bhutani VK. The clinical syndrome of bilirubin-induced neurologic dysfunction. Semin Perinatol. 2011;35(3):101–113 [DOI] [PubMed] [Google Scholar]
- 35.Dennery PA, Seidman DS, Stevenson DK. Neonatal hyperbilirubinemia. N Engl J Med. 2001;344(8):581–590 [DOI] [PubMed] [Google Scholar]
- 36.Flaherman VJ, Kuzniewicz MW, Escobar GJ, Newman TB. Total serum bilirubin exceeding exchange transfusion thresholds in the setting of universal screening. J Pediatr. 2012;160(5):796–800.e1 [DOI] [PubMed]
- 37.Tchernia G, Delhommeau F, Perrotta S, et al. ESPHI working group on hemolytic anemias . Recombinant erythropoietin therapy as an alternative to blood transfusions in infants with hereditary spherocytosis. Hematol J. 2000;1(3):146–152 [DOI] [PubMed] [Google Scholar]
- 38.Neuman-Łaniec M, Wierzba J, Irga N, Wasilewska E, Balcerska A. Recombinant erythropoietin—an alternative therapy to red cell blood transfusions in infants with hereditary spherocytosis [in Polish]. Przegl Lek. 2002;59(10):871–872 [PubMed] [Google Scholar]
- 39.Schiff M, Haÿs S, Sann L, Putet G. Recombinant human erythropoietin (r-HuEPO) therapy in a newborn with hereditary spherocytosis [in French]. Arch Pediatr. 2003;10(4):333–336 [DOI] [PubMed] [Google Scholar]
- 40.Delaunay J, Nouyrigat V, Proust A, et al. Different impacts of alleles alphaLEPRA and alphaLELY as assessed versus a novel, virtually null allele of the SPTA1 gene in trans. Br J Haematol. 2004;127(1):118–122 [DOI] [PubMed] [Google Scholar]
- 41.Shapiro CM, Josephson AM, Rozengvaig S, Kauffman A. Hereditary spherocytosis in the neonatal period: diagnosis, incidence, and treatment. J Pediatr. 1957;50(3):308–314 [DOI] [PubMed] [Google Scholar]
- 42.Christensen RD, Yaish HM, Nussenzveig RH, et al. Acute kernicterus in a neonate with O/B blood group incompatibility and a mutation in SLC4A1. Pediatrics. 2013;132(2). Available at: www.pediatrics.org/cgi/content/full/132/2/e531 [DOI] [PubMed] [Google Scholar]
- 43.Bogardus HH, Maksimova YD, Forget BG, Gallagher PG. A de novo band 3 mutation in hereditary spherocytosis. Pediatr Blood Cancer. 2012;58(6):1004. [DOI] [PubMed] [Google Scholar]
- 44.Gundel F, Eber S, Heep A. A new ankyrin mutation (ANK1 EXON E9X) causing severe hereditary spherocytosis in the neonatal period. Ann Hematol. 2011;90(2):231–232 [DOI] [PubMed] [Google Scholar]
- 45.Forde DG, Cope A, Stone B. Acute parvovirus B19 infection in identical twins unmasking previously unidentified hereditary spherocytosis. BMJ Case Rep. 2014 Jul 29;2014 [DOI] [PMC free article] [PubMed]