

Abstract
Cyclotides are head-to-tail cyclized peptides comprising a stabilizing cystine-knot motif. To date, they are well known for their diverse bioactivities such as anti-HIV and immunosuppressive properties. Yet little is known about specific molecular mechanisms, in particular the interaction of cyclotides with cellular protein targets. Native and synthetic cyclotide-like peptides from Momordica plants are potent and selective inhibitors of different serine-type proteinases such as trypsin, chymotrypsin, matriptase, and tryptase-beta. This study describes the bioactivity-guided isolation of a cyclotide from Psychotria solitudinum as an inhibitor of another serine-type protease, namely, the human prolyl oligopeptidase (POP). Analysis of the inhibitory potency of Psychotria extracts and subsequent fractionation by liquid chromatography yielded the isolated peptide psysol 2 (1), which exhibited an IC50 of 25 μM. In addition the prototypical cyclotide kalata B1 inhibited POP activity with an IC50 of 5.6 μM. The inhibitory activity appeared to be selective for POP, since neither psysol 2 nor kalata B1 were able to inhibit the proteolytic activity of trypsin or chymotrypsin. The enzyme POP is well known for its role in memory and learning processes, and it is currently being considered as a promising therapeutic target for the cognitive deficits associated with several psychiatric and neurodegenerative diseases, such as schizophrenia and Parkinson’s disease. In the context of discovery and development of POP inhibitors with beneficial ADME properties, cyclotides may be suitable starting points considering their stability in biological fluids and possible oral bioavailability.
Natural products are considered to be a rich source for drug discovery.1,2 In particular ribosomal synthesized and post-translational modified peptides (RiPP) are regarded as good starting points for pharmacological screening due to their biological and chemical diversity. Many RiPPs contain modifications, for instance C-terminal amidation, cyclization, side-chain methylation or pyroglutamate formation. These modifications are thought to be beneficial for improving ADME properties (absorption, distribution, metabolism, and excretion), such as oral bioavailability and blood-brain-barrier passage, which are still considered as major challenges of peptide-based drug development.3−5 The distribution of RiPPs is widespread in nature, and they have been previously described in isolates derived from fungi and bacteria (e.g., cyanobactins, thiopeptides, microcins, and lasso peptides), plants (e.g., cyclolinopeptides and cyclotides), and animals (e.g., scorpion toxins and conopeptides).5,6
As one representative class of RiPPs, the plant peptide family of cyclotides are considered as potential drug lead molecules due to their diverse bioactivities, intrinsic stability, and possible oral bioavailability.4 Furthermore, cyclotides have been recently described as natural templates for G protein-coupled receptor ligand design,7 one of the most important classes of transmembrane receptors.8 Cyclotides are expressed in many plant species all around the world such as Violaceae, Rubiaceae, Solanaceae, Fabaceae, and Poaceae, but their phylogenetic distribution is still under investigation.9 The distribution of cyclotides in the coffee-plant family (Rubiaceae) has been extensively studied using a combined peptidomics and transcriptomics discovery approach, and several members of the Psychotria alliance have been identified to produce cyclotides, i.e., Psychotria brachiata Sw., P. capitata, P. deflexa DC., P. poeppigiana Müll. Arg., P. solitudinum Standl., and P. suerensis Donn. Sm., as well as Carapichea ipecacuanha (Brot.) L. Andersson, Chassalia curviflora (Wall.) Thwaites, Notopleura capacifolia (Dwyer) C.M. Taylor, and Palicourea tetragona Ruiz&Pav.10 Cyclotides comprise a head-to-tail cyclized peptide backbone and three conserved disulfide bonds, which together form the so-called cyclic cystine-knot (CCK) motif.11 These post-translational modifications confer them remarkable stability in biological fluids,12 and therefore they have been considered as templates for peptide drug engineering.11 Utilizing the structural plasticity of the CCK framework,13 epitope grafting of bioactive peptide sequences onto the stabilizing scaffold of cyclotides has been successfully established and this methodology has already provided a number of synthetic peptide drug leads.14,15 Grafted cyclotide probes targeting the chemokine receptor CXCR4,16 the melanocortin-4 receptor,17 the vascular endothelial growth factor-A18 or a p53 tumor suppressor ligand19 have been developed. Importantly, a recent study has emphasized the potential of grafted cyclotides as oral bioactive peptide drugs,20 and hence cyclotides are gaining interest for preclinical drug development.
Within the family of circular and cysteine-rich plant peptides cyclotide-like peptides have been isolated from the seeds of Momordica plants.21,22 These cyclic squash trypsin inhibitors (TIs) share the structural CCK motif with cyclotides, but are more similar in sequence to the acyclic squash TI peptide family.23 On the basis of their intrinsic activity as potent inhibitors of the serine protease trypsin and their stabilizing CCK motif,24Momordica-type cyclotides have been used as templates for the design of selective proteinase inhibitors.25 For example, development of inhibitors has been reported for the human mast cell tryptase-beta,26 the human leukocyte elastase,27 and the type-II transmembrane serine protease matriptase.28,29
In summary, cyclotides including the cyclic squash TIs are promising peptides for ligand design and drug development due to their stability in biological fluids and their repertoire of biological activities. Therefore, this research aims to identify protein targets of cyclotides. The cytosolic enzyme prolyl oligopeptidase (POP; EC 3.4.21.26), also known as prolyl endopeptidase or postproline cleaving enzyme, is a serine protease that cleaves peptide bonds at the C-terminal side of proline within short peptides.30 Human POP is a 81 kDa protein and appeared to be a promising candidate as a target of cyclotides due to the reported inhibition of different serine-type proteases of cyclotide-like peptides isolated from Momordica plants. Much attention is currently focused on the implication of POP as a therapeutic target; it has been shown that inhibitors of this protease have neuroprotective, antiamnesic, and cognition-enhancing properties. These findings stimulated the development of several families of POP inhibitors as therapeutic agents for the treatment of the cognitive deficits associated with central nervous system disorders and neurodegenerative diseases.31,32 In the present study a bioassay-guided fractionation approach using three Psychotria and one Viola plant species to identify and characterize cyclotides as a novel class of POP inhibitors has been performed. This approach aims to support the concept of using natural products as a rich source of bioactive compounds for drug discovery and in particular to exemplify that cyclotides constitute a natural combinatorial library of circular peptides with enormous potential for pharmacological applications.
Results and Discussion
Prolyl oligopeptidase is known to play an important role in many cognitive disorders such as Parkinson’s disease,33 as well as depression and schizophrenia.34 Peptidomimetics recently attracted attention as possible inhibitors of POP.35 Nevertheless clinical applications of peptides have been limited so far, for instance by their low systemic stability or lack of oral bioavailability.36 To bridge the gap of suitable peptide leads and optimized peptide candidates for preclinical studies,32,37 the potential of cyclotides as POP inhibitors has been investigated in the present study.
Preparation of Plant Extracts and Analytical Characterization
Plants of three Rubiaceae species, i.e., Psychotria solitudinum, P. poeppigiana, and P. capitata, were collected in the field in Costa Rica. The Violaceae plant Viola tricolor L. was purchased from a commercial distributor. Plant extracts were prepared as previously described.38 Briefly, the dried and pulverized plant material was extracted with DCM/methanol, and the resulting filtrate was further treated with reversed-phase (RP) solid-phase extraction. This yielded an enriched portion of hydrophobic compounds such as cyclotides present in those extracts. These extracts were characterized by mass spectrometry (MS) and high-performance liquid chromatography (HPLC) (Figure 1). This peptidomics-based analytical workflow has been previously established10,38 and appeared to be a rapid tool for the identification of cyclotides in the Psychotria extracts. Molecular weight signals in the range 2500–3500 Da that typically correspond to the presence of cyclotides were recorded by MALDI-TOF MS in all three Psychotria extracts (Figure 1A–C), and previously reported cyclotides psysol 1, psypoe 1, and psycap 1 could be identified.10 Furthermore, in agreement with an earlier study analytical HPLC analysis of these extracts revealed late-eluting peak patterns in RP gradients−typical for cyclotide-containing samples−indicating the presence of cyclotides in these Psychotria extracts.10 The prototypic cyclotide plant V. tricolor has been known as a rich source of cyclotides, and accordingly numerous cyclotides have been identified in the Viola extract,38,39 for example, kalata S, varv C, varv D, varv E, varv F, vaby B, kalata B1, vigno 3, cycloviolacin O12, and cycloviolacin O20 (Figure 1D).
Figure 1.

Analytical characterization of crude cyclotide extracts. Cyclotide extracts of plant species, i.e., Psychotria solitudinum (A), Psychotria poeppigiana (B), Psychotria capitata (C), and Viola tricolor (D), were analyzed using MALDI-TOF MS (left panels) and RP-HPLC (right panels). MALDI-TOF spectra are presented in the range 2500–3500 Da of each plant extract, indicating signals in a m/z range typical for cyclotides. The molecular weights of major monoisotopic [M + H]+ signals were compared to those of known cyclotides (www.cybase.org.au or Koehbach et al.10), identified, and labeled with names and their corresponding molecular weight. HPLC analysis was performed using linear gradients as described in the Experimental Section, and A280 traces are presented; the gray boxes indicate HPLC fractions that have been used for POP inhibition screening (see Supplementary Table S1).
Plants of the Psychotria alliance within the Rubiaceae family offer a rich source of cyclotides.40 Indeed, Psychotria cyclotides comprise prototypic features of the cyclotide family, which are in particular (i) the presence of the CCK motif, which confers them with the frequently reported stability,12 and (ii) the sequence diversity of cyclotide loop residues, which makes them an attractive peptide library for bioactivity screening. Apart from cyclic Momordica TIs,23 cyclotides have to the best of our knowledge never been characterized for their potential as inhibitors of proteolytic enzymes. Specifically it was of interest to study their inhibitory effects toward human POP, an enzyme that has been identified as a promising pharmaceutical target.35
Effects of Psychotria Plant Extracts on the Activity of Human Prolyl Oligopeptidase
Cyclotide extracts of P. solitudinum, P. poeppigiana, P. capitata, and V. tricolor were characterized for their effect toward the inhibitory activity of purified human POP in a classical enzyme inhibition assay setup similar to that described earlier by Toide and colleagues.41 Using a high-throughput 96-well plate assay the activity of the enzyme was determined in the presence of different concentrations of the plant extracts. POP activity was measured by fluorimetry using Z-Gly-Pro-AMC as substrate following the fluorescent signal upon the enzymatic release of AMC. The amount of fluorescence of the control samples containing only POP enzyme and buffer was defined as 100% activity. The activity of the corresponding cyclotide extracts was measured and normalized to the activity of the control (eq 1, Experimental Section). The cyclotide extracts exhibited POP inhibition at all tested concentrations. Extracts from Psychotria species exhibited strong inhibitory effects: at 100 μg/mL of cyclotide extract (the lowest concentration tested), POP had a remaining activity of 32%, 23%, and 20%, respectively, in the presence of P. poeppigiana, P. capitata, and P. solitudinum extract, respectively (Figure 2). Almost full inhibition was observed when applying 400 μg/mL of cyclotide extract; that is, the remaining POP activities were 15% for P. poeppigiana, 9% for P. capitata, and 10% for P. solitudinum. The Viola plant extract also exhibited POP inhibitory effects ranging from 2% to 17% remaining POP activity (Figure 2). For all extracts tested, the POP inhibition was concentration-dependent, and therefore it was reasonable to assume that the observed effects were due to a molecular interaction of POP with cyclotides of the plant extracts.
Figure 2.

Inhibition of prolyl oligopeptidase activity by cyclotide plant extracts. Cyclotide extracts (as shown in Figure 1) of Psychotria solitudinum, Psychotria poeppigiana, Psychotria capitata, and Viola tricolor were dissolved in ddH2O for the POP activity assay at concentrations of 100–400 μg/mL and tested for their potential to inhibit POP. The measurements were performed in triplicate, and the data are presented as mean ± STDEV.
This hypothesis is in agreement with previous observations that squash TIs isolated from the seeds of Momordica species inhibit the activity of other serine proteinases.21,22,29 Due to the importance of POP in human disease42 and knowing that POP substrates and cyclotides are similar in molecular size, we attempted to isolate and identify cyclotides that were able to inhibit the activity of human POP using a bioassay-guided fractionation approach.
Bioactivity-Guided Fractionation of Cyclotide Extracts
The four cyclotide extracts that exhibited potent inhibition of POP activity were fractionated by RP-HPLC as previously described.38 The crude extracts, containing numerous cyclotides, were separated by preparative RP-HPLC in three to four fractions, as indicated by the gray shading in the chromatograms of Figure 1. Those fractions were forwarded to the POP inhibition assay and tested again in three concentrations of 100, 200, and 400 μg/μL for each fraction (Supplementary Table S1). All fractions of each plant extract were capable of inhibiting the activity of POP to a certain percentage; for example, fraction 3 of P. solitudinum (Psysol-F3) inhibited POP activity by 48–78%, fraction 4 of P. poeppigiana (Psypoe-F4) by 47–83%, and fraction 2 of V. tricolor (Vitri-F2) by 51–86%, respectively. According to MALDI-TOF MS analysis, fraction Vitri-F2 does not appear to contain many cyclotides, but mainly other small organic plant compounds that may be responsible for its POP inhibitory activity (data not shown).
Three fractions of P. solitudinum were further characterized to determine quantitative POP inhibition data (IC50) and to purify an active cyclotide. Consequently, for all three fractions Psysol-F1–F3 a concentration-dependent inhibition assay of POP was performed to confirm the initial results. Psysol-F3 yielded the highest potency (IC50 = 100.4 μg/mL), whereas fraction Psysol-F1 (IC50 = 237.2 μg/mL) and fraction Psysol-F2 exhibited about 3-fold less potency (IC50 = 285.2 μg/mL) to inhibit the enzymatic activity of human POP (Table 1, Figure 3A). In addition we tested POP inhibition of a subfraction of Vitri-F3, i.e., the most potent cyclotide-containing V. tricolor fraction. This fraction comprised a coeluting cyclotide mixture of kalata S and kalata B1 (Supplementary Figure S1), which exhibited a concentration-dependent inhibition of POP activity with an IC50 of 28.5 μg/mL (Table 1, Figure 3B).
Table 1. POP Enzyme Inhibition Potency of Cyclotides.
| fraction/compound | IC50 [μg/mL] |
|---|---|
| Psysol-F1 | 237.2 ± 23.4a |
| Psysol-F2 | 285.2 ± 58.9a |
| pPsysol-F3 | 104.3 ± 16.0a |
| Vitri-F3 (subfraction) | 28.5 ± 0.3a |
| IC50 [μM] | |
| psysol 2 (1) | 25.0 ± 0.3;a (27.8b,c) |
| kalata B1 | 5.6c |
IC50 values are calculated using nonlinear regression analysis and are presented as mean ± STDEV of at least two independent experiments (as described in the Experimental Section).
Potency of synthetic cyclotide is shown in parentheses.
Values are presented as the mean of three replicates.
Figure 3.

Concentration-dependent inhibition of human prolyl oligopeptidase by cyclotides. (A) Concentration-response inhibition curves of three Psychotria solitudinum fractions (Psysol-F1–F3) after RP-HPLC fractionation of the cyclotide extract. Each data point is presented as the mean ± STDEV of two independent experiments or as the mean of three replicates, respectively (see Table 1). The inhibitory potency was quantified using nonlinear regression analysis, yielding IC50 values of 237.2 μg/mL (Psysol-F1), 285.2 μg/mL (Psysol-F2), and 100.4 μg/mL (Psysol-F3). Similarly, Viola tricolor fraction (Vitri-F3) was purified by RP-HPLC to yield sample sub-Vitri-F3. This subfraction comprises mainly the two coeluting cyclotides kalata S and kalata B1 (Supplementary Figure S1). (B) The fraction exhibited a POP inhibitory potency (IC50) of 28.5 μg/mL. The most potent fraction of Psychotria solitudinum (Psysol-F3) was further purified to obtain the isolated cyclotide psysol 2 (1), which inhibited human POP activity in a concentration-dependent manner with an IC50 of 25 μM (C, solid line). To confirm the inhibition of human POP by plant-extracted cyclotides, psysol 2 (1) and kalata B1 were synthesized and analyzed; synthetic psysol 2 (1) (C, dashed line) inhibits human POP activity in a concentration-dependent manner with an IC50 of 27.8 μM, and synthetic kalata B1 yielded an IC50 of 5.6 μM (D).
Isolation and Structural Characterization of Psysol 2 from Psychotria solitudinum
The most potent P. solitudinum fraction, Psysol-F3, contained one major compound (1), as determined by RP-HPLC (Figure 4A), with a molecular weight of 2904.12 Da (Figure 4B). This peptide was purified using semipreparative RP-HPLC, and its purity of ≥95% and its molecular weight were confirmed by analytical RP-HPLC (Figure 4C) and MALDI-TOF MS (Figure 4D), respectively. To determine the peptide sequence of compound 1, which was named psysol 2 (Psychotria solitudinum cyclotide 2), a previously optimized MALDI-based peptidomics approach has been utilized.43 Initially the cysteine content of the peptide was determined by a combination of reduction and alkylation. Purified psysol 2 (1) was treated with dithiothreitol (DTT), and the reduced sulfhydryl groups were modified with iodoacetamide. This chemical derivatization resulted in an addition of 348.1 Da, which corresponds to the presence of six cysteine residues (Figure 5A,B). Subsequently the reduced and S-alkylated aliquots of psysol 2 (1) were digested with trypsin or endoproteinase GluC to confirm the presence of a circular backbone and to elucidate the peptide’s primary sequence by de novo peptide sequencing. Upon trypsin and endoproteinase GluC digest of the sulfhydryl-reduced and S-alkylated peptide, its molecular weight increased by 18 Da as a result of the addition of H2O during “ring-opening” of the backbone-cyclized peptide (Figure 5C,D); this confirmed the cyclic nature of psysol 2 (1), and it indicated the presence of one Lys or Arg residue and one Glu residue in the peptides’ sequence, respectively. Manual interpretation of the MS/MS peptide fragmentation pattern of each digest revealed the sequence of psysol 2 (1) (Figure 5C,D). Since the two isobaric residues Leu and Ile cannot be resolved by MS/MS fragmentation, homology alignment analysis10 (www.cybase.org.au) of psysol 2 (1) to other known cyclotides, in particular to closely related Psychotria cyclotides, has been performed; hence this combined approach led to the identification of the psysol 2 (1) sequence as cyclo-GLPICGESCVGGTCNTPGCTCTWPVCTRN (Figure 6A).
Figure 4.

Purification and analysis of psysol 2 (1). The analytical A280 HPLC trace of Psychotria solitudinum fraction 3 (Psysol-F3) (A) and its corresponding MALDI-TOF spectrum (B) are shown, indicating the presence of one main compound in this fraction. Using RP-HPLC separation a single compound, 1, could be isolated from this fraction. This purified compound 1 was characterized by RP-HPLC (C) and MALDI-TOF MS (D). Inset in (D) shows the isotope pattern of psysol 2 (1). HPLC indicated a purity of >95%.
Figure 5.

De novo sequencing of the cyclotide psysol 2 (1). The isolated active compound 1 of Psychotria solitudinum was characterized by chemical derivatization and MS. Using sulfhydryl reduction by DTT a mass shift of 6 Da (2911.12 m/z) compared to the native mass signal of 2905.12 m/z was observed, indicating the presence of six cysteine residues (A). Subsequently, iodoacetamide derivatization yielded the mass of 3253.22 m/z, corresponding to S-carbamidomethylation of the six cysteines (B). De novo amino acid sequencing was performed by interpretation of MS/MS fragmentation spectra using trypsin (C) and endoproteinase Glu C (D) digests. The sequence was determined by manual assignment of the N-terminal b-ion and C-terminal y-ion series and the ion fragmentation calculator tool (Data Explorer AB Sciex). The disulfide connectivity of Cys I–IV, Cys II–V, and Cys III–VI and the isobaric amino acids Leu and Ile were assigned based on homology with known sequences.10
Figure 6.
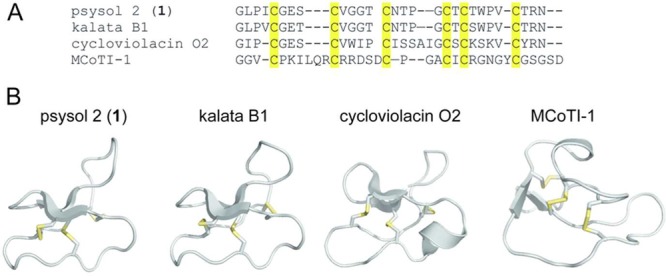
Sequence alignments and homology model of psysol 2 (1). (A) The amino acid sequence of psysol 2 (1) was used for alignment based on the conserved CCK motif with the prototypic cyclotides kalata B1 (Möbius type), cycloviolacin O2 (bracelet), and MCoTI-1 (cyclic TI). The structural model of the novel cyclotide psysol 2 (1) was obtained with the CycloMod tool (www.cybase.org), and the structural features were compared to kalata B1 (pdb code: 1NB1), cycloviolacin O2 (2KNM), and MCoTI-1 (1IB9). All structures are presented as ribbon cartoons, which were prepared using PyMol. β-sheet motifs are indicated by flat arrows, and the disulfide bonds are highlighted in yellow.
Since psysol 2 (1) contains six cysteine residues and a circular peptide backbone, it is likely to form the typical CCK motif.10 The presence of a Pro residue in loop 5 allowed classification of psysol 2 (1) as a Möbius-type cyclotide due to sequence homology to other cyclotides of this subfamily (Figure 6 A).11 In addition to the similar molecular sequence of psysol 2 (1) and known cyclotides, the structural similarity has been confirmed by modeling of psysol 2 (1) using the CycloMod tool of CyBase44 (Figure 6B). The sequence alignment of prototypic cyclotides with psysol 2 (1), namely, kalata B1 (Möbius), cycloviolacin O2 (bracelet), and MCoTI-1 (cyclic TI) clearly demonstrated that psysol 2 (1) has the highest homology with kalata B1, with only three residues differing, in positions 4, 7, and 22. Compared to cycloviolacin O2, psysol 2 (1) lacks the cationic residues in loop 5 and the hydrophobic residue in loop 3. As mentioned above, the Momordica TIs and cyclotides share only the CCK motif,45 and hence psysol 2 (1) and MCoTI-1 have little sequence homology. The psysol 2 (1) model further predicted a triple-stranded β-sheet as a secondary structure element, which is typical for Möbius-type cyclotides.
Structure–Activity and Specificity of Cyclotide POP Inhibitors
Plant purified psysol 2 (1) was tested for POP inhibitory activity, and as expected from bioassay-guided fractionation, it inhibited human POP activity with an IC50 of 25 ± 0.3 μM. The activity was confirmed with a synthetic psysol 2 cyclotide (Supplementary Figure S2), which exhibited an IC50 of 27.8 μM (Table 1, Figure 3C). Since psysol 2 (1) and kalata B1 share high sequence homology and knowing that the active V. tricolor fraction Vitri-F3 contains kalata B1, we synthesized this cyclotide (Supplementary Figure S2) and analyzed its POP inhibitory activity. Kalata B1 was slightly more potent and inhibited human POP activity with an IC50 of 5.6 μM (Table 1, Figure 3D). These observed differences in activity may be due to the amino acid differences and/or minor structural variation between the two cyclotides.
Similar plant peptides, namely, the family of cyclic and acyclic squash TI peptides, are well-known inhibitors of trypsin and chymotrypsin. To determine inhibitor specificity of cyclotides, we therefore tested the ability of kalata B1 and psysol 2 (1) to inhibit the activity of those two pancreatic enzymes (Supplementary Figure S3). Interestingly, cyclotides were not capable of inhibiting the activity of trypsin or chymotrypsin at concentrations of 25 and 75 μM, respectively. This suggests that cyclotides, and in particular kalata B1, are at least an order of magnitude more selective toward inhibition of POP over trypsin and chymotrypsin.
It is noteworthy that psysol 2 (1) and kalata B1 contain three proline residues each (Figure 6), and prolyl groups are thought to be a key feature of many POP substrates.42 This is also the case for other active cyclotides that have been identified in the POP inhibitory fractions of the other plant extracts (Figure 1); most of them contain three proline residues, in loops 3, 5, and 6, respectively (Supplementary Figure S4). It will be interesting in future studies to determine the importance of those Pro residues for the inhibitory activity of cyclotides. In summary the observed bioactive properties and structural features of cyclotides warrant further investigations for applications of these interesting circular peptides as human POP inhibitors.
Drug Development Potential of Cyclotide-Based POP Inhibitors
The vast majority of previously known POP inhibitors are small-molecule peptidomimetics based on systemic modifications of the canonical compound benzyloxycarbonyl-prolyl-prolinal (Z-Pro-prolinal). This compound acts as a transition-state analogue due to the presence of a covalent group acting as a “warhead”.31,32 Although many peptidomimetics, for example, S-17092, Z-321, JTP-4819, and ONO-1603, have been described as POP inhibitors with micromolar to nanomolar potencies, the presence of a warhead moiety is an important parameter to obtain inhibitory potencies in the low nanomolar range.31,32 Despite some initial success as potential therapeutics of cognitive deficits associated with aging and Alzheimer’s disease, the development of these peptidomimetics was discontinued during clinical phase I and II, respectively.35 Due to a lack of public information, it is not clear why these compounds failed, but most known POP inhibitors comprise the narrow family of pyrrolidinyl analogues.35 To our knowledge there are currently no “classical peptide” POP inhibitors available. It has been repeatedly highlighted that cyclotides are an emerging peptide class with enormous potential in medicinal chemistry and for pharmaceutical applications.4,7,12,14,15,20 Therefore, cyclotides may be promising tools for the development of novel peptide-based POP inhibitor drugs.
The most interesting features of cyclotides are their circular peptide backbone and the cystine-knot fold constituting the CCK motif. Together this improves the stability of these peptides, and hence cyclotides are known to be heat-stable, resistant to proteolytic degradation, and unaffected by the acidic pH conditions in the gastrointestinal tract,12 all of which are prerequisites for conferring peptides with oral bioavailability.20 Another feature of cyclotides is their enormous diversity and sequence variation. It has been well documented that the majority of the amino acids within the intercysteine loops are amenable to variation without affecting CCK topology,13 and indeed hundreds of cyclotides displaying unique amino acid sequences have been isolated from plants.46
This natural variation together with the structural plasticity of cyclotides has been recognized by peptide chemists to engineer novel cyclotide-based peptide therapeutics,12,16−19,26−29 by grafting of linear and otherwise unstable bioactive amino acid sequences onto the intercysteine loops of cyclotides to “protect” them from degradation within the stabilized CCK framework.15
Although peptide grafting is a promising tool in drug development,47 there are examples of naturally occurring cyclic or disulfide-knotted peptides, which appeared to be potent protease inhibitors. For example a cyclic peptide stabilized by one disulfide bond is the sunflower-trypsin inhibitor SFTI-1, which inhibits serine proteases. SFTI-1 is considered to be the smallest known peptide trypsin inhibitor (14-mer) and belongs to the Bowman-Birk inhibitor family. As mentioned above, squash TIs are also potent inhibitors of trypsin and chymotrypsin,48 and this includes in particular the cyclotide-like cyclic squash TIs isolated from Momordica plants.21 The present study has confirmed these previous examples that cyclic or disulfide-knotted peptides, and in particular cyclotides, are naturally occurring inhibitors of serine proteases. In addition, we were able to demonstrate that Möbius-type cyclotides do not inhibit the enzymatic activity of trypsin and chymotrypsin, but appear to be specific for inhibition of prolyl-oligopeptidase. The combination of having a natural peptide template with intrinsic inhibitory potency against proteases and knowing that the CCK-fold is amenable to peptide engineering for optimization of this given activity makes the family of cyclotides interesting natural molecules for future investigations regarding design and development of human POP inhibitors.
To conclude, the present study described the identification of the cyclotides kalata B1 and psysol 2 (1) to inhibit human POP activity in vitro. This may have provided lead compounds for further investigation in the field of POP therapeutics. There are currently no POP inhibitors available as approved drugs, and consequently there is a need for discovery and development of suitable and improved POP inhibitors. Cyclotides can be regarded as the first of a novel class of circular peptide POP inhibitors. Due to their known potential in peptide drug development,4,7,12,14,15,20 cyclotides appear to be an interesting class of natural products for future studies to design and develop human POP inhibitors.
Experimental Section
General Experimental Methods
RP-HPLC was performed using a Dionex Ultimate 3000 station (Dionex, Amsterdam, The Netherlands).38,49 The device was equipped with a binary pump, autosampler, column oven, multiwavelength detector, and fraction collector. Absorbance wavelengths of 214, 254, and 280 nm were recorded of all analytical and preparative separations.
Plant Material
Psychotria plant material (Psychotria solitudinum, P. capitata, and P. poeppigiana) was collected in Costa Rica at the tropical research station La Gamba, and species were identified by H. Greger and A. Berger (University of Vienna, Austria).10 Herbarium accession numbers are HG-2607083, HG-24070811, and HG-3007081, respectively. Collection and export of plant material were kindly permitted by the Costa Rican Ministry of Ambient and Energy under permit numbers 050-2013-SWAC, 217-2012-SWAC, and DGVS-109-2013, respectively. Samples were dried, stored at 23 °C, and protected from moisture and UV irradiation until extraction. Pulverized Viola tricolor L. (Violaceae) plant material (Herba Violae Tricoloris plv.) was purchased from Kottas Pharma GmbH (Vienna, Austria).
Extraction and Isolation
Cyclotide extracts were prepared as previously described.38 Briefly, dry plant material was ground using a coffee grinder and extracted for 24 h in 100–200 mL of dichloromethane/methanol, 1:1 (v/v), by continuous agitation at 23 °C. After filtration, 0.5 vol of ddH2O was added, and the methanol/water phase, which contained the enriched cyclotide mixture, was obtained by liquid/liquid phase separation. This aqueous extract was treated with RP C18 solid-phase extraction. First, the methanol content was reduced to less than 10% vol by dilution with ddH2O. Solid-phase extraction was performed using ZEOprep 60 Å, C18 irregular 40–63 μm material (Zeochem, Uetikon, Switzerland) that has been activated with methanol and equilibrated using 0.1% trifluoroacetic acid (TFA). The aqueous extract was applied to the C18 material, washed with ddH2O/CH3CN/TFA, 90/10/0.1% (v/v/v), and eluted with ddH2O/CH3CN/TFA, 20/80/0.1% (v/v/v). These eluates were lyophilized and reconstituted in ddH2O buffer prior to the POP inhibition tests or in 0.1% TFA for mass spectrometry based analysis or HPLC separation.
Further cyclotide purification was achieved by preparative chromatography with a Phenomenex Jupiter C18 column (250 × 20.2 mm, 10 μm), and semipreparative separation was carried out with a Dichrom Kromasil C18 column (250 × 10 mm, 5 μm). Flow rates were set to 8 and 3 mL/min, respectively, and the solvents consisted of 0.1% TFA (solvent A) or ddH2O/CH3CN/TFA, 10/90/0.1% (v/v/v) (solvent B). Linear gradients from 5% to 65% solvent B (1% per min) were applied to achieve separation of extracts. Resulting fractions were freeze-dried and analyzed by analytical HPLC using a Phenomenex Kinetex (150 × 3 mm, 2.1 μm) column. Purified cyclotide psysol 2 (1) was further structurally characterized by de novo peptide sequencing using MALDI-TOF/TOF and homology modeling using CyBase tools.
Peptide Sequencing
Cyclotide structural elucidation of the cyclotide psysol 2 (1) was performed by MS using a MALDI-TOF/TOF 4800 analyzer from AB Sciex (Framingham, MA, USA) as previously described.10 Peptide mapping of plant extracts or HPLC fractions was performed by dissolving dried sample material in 0.1% TFA. The samples were mixed at a ratio of 1:6 (v/v) with a matrix solution consisting of saturated α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich, St. Louis, MO, USA) in ddH2O/CH3CN/TFA, 50/50/0.1% (v/v/v). A 0.5 μL aliquot of the mixture was directly spotted onto the MALDI target plate, and the droplet was allowed to air-dry. Mass spectra were obtained by combining 2500 shots in the spectral range 2500 to 4500 m/z using the MS reflector positive ionization mode. For de novo sequencing peptides were dissolved at a concentration of 5 mg/mL in 0.1 M NH4HCO3 (pH 8.0), and disulfide bonds were reduced using DTT in a final concentration of 10 mM for 60 min at 37 °C. The sample was S-alkylated using iodoacetamide (100 mM) for 10 min in the dark; remaining alkylation reagent was quenched by addition of DTT (5 mM). Prior to MS/MS fragmentation the peptide was digested with 0.2 μg of trypsin “proteomics grade” (Sigma-Aldrich) or 0.4 μg of endoproteinase GluC “proteomics grade” (New England Biolabs, Ipswich, MA, USA). The digest was performed at 37 °C for 16–18 h, and the peptide mixture was quenched using 0.5% TFA followed by ZipTip desalting (Millipore, Billerica, MA, USA). Precursor fragmentation was obtained in the MS/MS positive 1 kV reflector mode by acquiring approximately 5000 spectra using optimized laser intensity and digitizer enhancement settings. All MS/MS spectra were recorded using the metastable ion suppressor function. The cyclotide amino acid sequence was obtained by manual assignment of N-terminal b-ion and C-terminal y-ion series and automated sequence analysis using the DataExplorer software (AB Sciex). The disulfide connectivity of CysI-IV, CysII-V, and CysIII-VI and the isobaric amino acids Leu and Ile were assigned based on homology with known sequences.10
Cyclotide Homology Modeling
The three-dimensional structure of the cyclotide psysol 2 (1) was performed by homology modeling using the CycloMod tool on CyBase (www.cybase.org.au). Structural images were prepared using PyMol from the pdb files for kalata B1 (1NB1), cycloviolacin O2 (2KNM), and MCoTI-1 (1IB9).10
Cyclotide Synthesis
The cyclotides psysol 2 (1) and kalata B1 have been synthesized by Fmoc-based solid-phase peptide synthesis, folded, and analyzed using previously established protocols.7,50
Prolyl Oligopeptidase Inhibition Assays
Samples were prepared by dissolving freeze-dried material in ddH2O buffer. Several concentrations of cyclotide extracts or fractions between 100 and 400 μg/μL were used for the measurement. The purity of isolated or synthesized psysol 2 (1) and kalata B1 was evaluated with RP-HPLC at A280, and their concentrations were determined using the Beer–Lambert law with the molar absorption coefficients of 5875 cm–1 M–1 (psysol 2) and 6410 cm–1 M–1 (kalata B1), respectively. For the POP inhibition assay the reactions were performed in triplicate in 96-well microtiter plates. Human POP (2 μL of a 6 μM solution; prepared by recombinant expression according to Tarrago et al.51) was preincubated for 15 min at 30 °C with buffer (137 μL of a 0.1 M Na2HPO4 and KH2PO4 (1:1, w/w) solution, pH 7.4) and the corresponding cyclotide extract solution (3 μL) or ddH2O (3 μL, controls). After preincubation, POP substrate (Z-Gly-Pro-AMC, 10 μL of a 3 mM solution prepared in 40% 1,4-dioxane; Bachem, Bubendorf, Switzeland) was added, and the reaction mixture was incubated while shaking (90 rpm) for 1 h at 37 °C. The formation of AMC was measured by fluorimetry. The excitation and emission wavelengths were 360 ± 40 and 485 ± 20 nm, respectively. The percentage of inhibition was calculated as
| 1 |
where X is the activity of POP in the absence of the inhibitor and Y is the activity in the presence of the cyclotide solution. The IC50 value was defined as the concentration of compound required to inhibit 50% of POP activity under these assay conditions. The inhibition data were analyzed using GraphPad Prism5 v 5.04. Inhibition curves were generated by plotting the logarithmic concentrations vs enzyme activity, and IC50 values were calculated by fitting the data to a three-parameter Hill equation using nonlinear regression analysis. Each inhibitory experiment was repeated independently twice, unless otherwise stated, and IC50 values were calculated from each biological repeat and are presented as mean ± STDEV.
Trypsin and Chymotrypsin Inhibition Assays
Inhibitory activity was performed in triplicate in 96-well microtiter plates. Bovine trypsin (10 μL of a 10 ng/μL solution; Roche, Basel, Switzerland) or bovine α-chymotrypsin (10 μL of a 2.5 ng/μL solution; Sigma-Aldrich) was preincubated for 15 min at 37 °C with buffer (125 μL of a 20 mM Tris-HCl solution, pH 8.0) and with 3 μL of the cyclotide solution or ddH2O as negative control. After preincubation, trypsin substrate (Bz-Arg-AMC-HCl, 10 μL of a 3 mM solution prepared in 40% 1,4-dioxane; Bachem, Bubendorf, Switzerland) or chymotrypsin substrate (Ala-Ala-Phe-AMC, 10 μL of a 3 mM solution prepared in 40% 1,4-dioxane; Sigma-Aldrich), respectively, was added, and the reaction was incubated while shaking (90 rpm) for 1 h at 37 °C. The reaction was stopped with sodium acetate (150 μL, 1 M, pH 4), and the formation of AMC was measured by fluorimetry. The excitation and emission wavelengths were 360 ± 40 and 485 ± 20 nm, respectively. Phenylmethane-sulfonylfluoride (PMSF; 3 μL of a 5 mM solution in DMSO) was used as a positive control of inhibition; additional controls with DMSO solution only were also performed. Data were calculated as percentage of inhibition and are presented as mean ± STDEV (of three replicates).
Acknowledgments
The authors would like to thank M. Auer for assistance with peptide purification. This work was financially supported by the Austrian Science Fund (FWF P-24743) to C.W.G., by MICIN-FEDER (BIO2013-40716R) to E.G., and the Generalitat de Catalunya (XRB and 2014SGR-521) to E.G. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supporting Information Available
Supplementary Table S1: Concentration-dependent POP inhibitory activity of Psychotria solitudinum, P. capitata, P. poeppigiana, and Viola tricolor HPLC fractions. Supplementary Figure S1: Analysis of purified fraction Vitri-F3 from Viola tricolor. Supplementary Figure S2: Analytical characterization of synthesized psysol 2 and kalata B1. Supplementary Figure S3: Inhibitor specificity as determined by trypsin and chymotrypsin inhibition assays. Supplementary Figure S4: Sequence alignment of cyclotides identified in Psychotria species and Viola tricolor. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ School of Biomedical Sciences, The University of Queensland, Brisbane, QLD 4072, Australia.
The authors declare no competing financial interest.
Supplementary Material
References
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2012, 75, 311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlin L.; Göransson U.; Alsmark C.; Weden C.; Backlund A. Phytochem. Rev. 2010, 9, 279–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlieghe P.; Lisowski V.; Martinez J.; Khrestchatisky M. Drug Discovery Today 2010, 15, 40–56. [DOI] [PubMed] [Google Scholar]
- Thell K.; Hellinger R.; Schabbauer G.; Gruber C. W. Drug Discovery Today 2014, 19, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K. D.; Fischbach M. A.; Garavelli J. S.; Göransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. N.; Moore B. S.; Muller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Piel J.; Reaney M. J. T.; Rebuffat S.; Ross R. P.; Sahl H. G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Sussmuth R. D.; Tagg J. R.; Tang G. L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. Nat. Prod. Rep. 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman R.; Gunasekera S.; Stromstedt A. A.; Göransson U. J. Nat. Prod. 2014, 77, 724–736. [DOI] [PubMed] [Google Scholar]
- Koehbach J.; O’Brien M.; Muttenthaler M.; Miazzo M.; Akcan M.; Elliott A. G.; Daly N. L.; Harvey P. J.; Arrowsmith S.; Gunasekera S.; Smith T. J.; Wray S.; Göransson U.; Dawson P. E.; Craik D. J.; Freissmuth M.; Gruber C. W. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 21183–21188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber C. W.; Muttenthaler M.; Freissmuth M. Curr. Pharm. Des. 2010, 16, 3071–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber C. W.; Elliott A. G.; Ireland D. C.; Delprete P. G.; Dessein S.; Göransson U.; Trabi M.; Wang C. K.; Kinghorn A. B.; Robbrecht E.; Craik D. J. Plant Cell 2008, 20, 2471–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehbach J.; Attah A. F.; Berger A.; Hellinger R.; Kutchan T. M.; Carpenter E. J.; Rolf M.; Sonibare M. A.; Moody J. O.; Wong G. K.; Dessein S.; Greger H.; Gruber C. W. Biopolymers 2013, 100, 438–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craik D. J.; Daly N. L.; Bond T.; Waine C. J. Mol. Biol. 1999, 294, 1327–1336. [DOI] [PubMed] [Google Scholar]
- Wang C. K.; Gruber C. W.; Cemazar M.; Siatskas C.; Tagore P.; Payne N.; Sun G. Z.; Wang S. H.; Bernard C. C.; Craik D. J. ACS Chem. Biol. 2014, 9, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R. J.; Daly N. L.; Craik D. J. Biochem. J. 2006, 394, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poth A. G.; Chan L. Y.; Craik D. J. Biopolymers 2013, 100, 480–491. [DOI] [PubMed] [Google Scholar]
- Schroeder C. I.; Swedberg J. E.; Craik D. J. Curr. Protein Peptide Sci. 2013, 14, 532–542. [DOI] [PubMed] [Google Scholar]
- Aboye T. L.; Ha H.; Majumder S.; Christ F.; Debyser Z.; Shekhtman A.; Neamati N.; Camarero J. A. J. Med. Chem. 2012, 55, 10729–10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasen R.; Daly N. L.; Wulff B. S.; Andresen T. L.; Conde-Frieboes K. W.; Craik D. J. J. Biol. Chem. 2012, 287, 40493–40501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekera S.; Foley F. M.; Clark R. J.; Sando L.; Fabri L. J.; Craik D. J.; Daly N. L. J. Med. Chem. 2008, 51, 7697–7704. [DOI] [PubMed] [Google Scholar]
- Ji Y. B.; Majumder S.; Millard M.; Borra R.; Bi T.; Elnagar A. Y.; Neamati N.; Shekhtman A.; Camarero J. A. J. Am. Chem. Soc. 2013, 135, 11623–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. T. T.; Rowlands D. K.; Wong C. H.; Lo T. W. C.; Nguyen G. K. T.; Li H. Y.; Tam J. P. Angew. Chem., Int. Ed. 2012, 51, 5620–5624. [DOI] [PubMed] [Google Scholar]
- Hernandez J. F.; Gagnon J.; Chiche L.; Nguyen T. M.; Andrieu J. P.; Heitz A.; Hong T. T.; Pham T. T. C.; Nguyen D. L. Biochemistry 2000, 39, 5722–5730. [DOI] [PubMed] [Google Scholar]
- Huang B.; Ng T. B.; Fong W. P.; Wan C. C.; Yeung H. W. Int. J. Biochem. Cell Biol. 1999, 31, 707–715. [DOI] [PubMed] [Google Scholar]
- Mylne J. S.; Chan L. Y.; Chanson A. H.; Daly N. L.; Schaefer H.; Bailey T. L.; Nguyencong P.; Cascales L.; Craik D. J. Plant Cell 2012, 24, 2765–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan L. Y.; Wang C. K. L.; Major J. M.; Greenwood K. P.; Lewis R. J.; Craik D. J.; Daly N. L. J. Nat. Prod. 2009, 72, 1453–1458. [DOI] [PubMed] [Google Scholar]
- Daly N. L.; Thorstholm L.; Greenwood K. P.; King G. J.; Rosengren K. J.; Heras B.; Martin J. L.; Craik D. J. J. Biol. Chem. 2013, 288, 36141–36148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerhoff C. P.; Avrutina O.; Schmoldt H. U.; Gabrijelcic-Geiger D.; Diederichsen U.; Kolmar H. J. Mol. Biol. 2010, 395, 167–175. [DOI] [PubMed] [Google Scholar]
- Thongyoo P.; Bonomelli C.; Leatherbarrow R. J.; Tate E. W. J. Med. Chem. 2009, 52, 6197–6200. [DOI] [PubMed] [Google Scholar]
- Quimbar P.; Malik U.; Sommerhoff C. P.; Kaas Q.; Chan L. Y.; Huang Y. H.; Grundhuber M.; Dunse K.; Craik D. J.; Anderson M. A.; Daly N. L. J. Biol. Chem. 2013, 288, 13885–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray K.; Elghadban S.; Thongyoo P.; Owen K. A.; Szabo R.; Bugge T. H.; Tate E. W.; Leatherbarrow R. J.; Ellis V. Thromb. Haemostasis 2014, 112, 402–411. [DOI] [PubMed] [Google Scholar]
- Fulop V.; Bocskei Z.; Polgar L. Cell 1998, 94, 161–170. [DOI] [PubMed] [Google Scholar]
- Lawandi J.; Gerber-Lemaire S.; Juillerat-Jeanneret L.; Moitessier N. J. Med. Chem. 2010, 53, 3423–3438. [DOI] [PubMed] [Google Scholar]
- Lopez A.; Tarrago T.; Giralt E. Expert Opin. Ther. Pat. 2011, 21, 1023–1044. [DOI] [PubMed] [Google Scholar]
- Dokleja L.; Hannula M. J.; Myohanen T. T. Neurosci. Lett. 2014, 583C, 37–42. [DOI] [PubMed] [Google Scholar]
- Maes M.; Goossens F.; Scharpe S.; Calabrese J.; Desnyder R.; Meltzer H. Y. Psychiatry Res. 1995, 58, 217–225. [DOI] [PubMed] [Google Scholar]
- Lopez A.; Mendieta L.; Prades R.; Royo S.; Tarrago T.; Giralt E. Future Med. Chem. 2013, 5, 1509–1523. [DOI] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. Chem. Biol. Drug Des. 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- Tenorio-Laranga J.; Coret-Ferrer F.; Casanova-Estruch B.; Burgal M.; Garcia-Horsman J. A. J. Neuroinflamm. 2010, 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellinger R.; Koehbach J.; Fedchuk H.; Sauer B.; Huber R.; Gruber C. W.; Gründemann C. J. Ethnopharmacol. 2014, 151, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabi M.; Svangard E.; Herrmann A.; Göransson U.; Claeson P.; Craik D. J.; Bohlin L. J. Nat. Prod. 2004, 67, 806–810. [DOI] [PubMed] [Google Scholar]
- Gerlach S. L.; Burman R.; Bohlin L.; Mondal D.; Göransson U. J. Nat. Prod. 2010, 73, 1207–1213. [DOI] [PubMed] [Google Scholar]
- Toide K.; Iwamoto Y.; Fujiwara T.; Abe H. J. Pharmacol. Exp. Ther. 1995, 274, 1370–1378. [PubMed] [Google Scholar]
- Garcia-Horsman J. A.; Mannisto P. T.; Venalainen J. I. Neuropeptides 2007, 41, 1–24. [DOI] [PubMed] [Google Scholar]
- Hashempour H.; Koehbach J.; Daly N. L.; Ghassempour A.; Gruber C. W. Amino Acids 2013, 44, 581–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. K.; Kaas Q.; Chiche L.; Craik D. J. Nucleic Acids Res. 2008, 36, D206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly N. L.; Rosengren K. J.; Craik D. J. Adv. Drug Delivery Rev. 2009, 61, 918–930. [DOI] [PubMed] [Google Scholar]
- Ireland D. C.; Clark R. J.; Daly N. L.; Craik D. J. J. Nat. Prod. 2010, 73, 1610–1622. [DOI] [PubMed] [Google Scholar]
- Northfield S. E.; Wang C. K.; Schroeder C. I.; Durek T.; Kan M. W.; Swedberg J. E.; Craik D. J. Eur. J. Med. Chem. 2014, 77, 248–257. [DOI] [PubMed] [Google Scholar]
- Chiche L.; Heitz A.; Gelly J. C.; Gracy J.; Chau P. T.; Ha P. T.; Hernandez J. F.; Le-Nguyen D. Curr. Protein Peptide Sci. 2004, 5, 341–349. [DOI] [PubMed] [Google Scholar]
- Gründemann C.; Koehbach J.; Huber R.; Gruber C. W. J. Nat. Prod. 2012, 75, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheneval O.; Schroeder C. I.; Durek T.; Walsh P.; Huang Y. H.; Liras S.; Price D. A.; Craik D. J. J. Org. Chem. 2014, 79, 5538–5544. [DOI] [PubMed] [Google Scholar]
- Tarrago T.; Frutos S.; Rodriguez-Mias R. A.; Giralt E. ChemBioChem 2006, 7, 827–833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.