

Abstract
The first two authors contributed equally to this work.Silence of p120-catenin has shown promise in inducing proliferation in human corneal endothelial cells (HCECs), but there is concern regarding off-target effects in potential clinical applications. We aimed to develop ex vivo expansion of HCECs using natural compounds, and we hypothesized that lysophosphatidic acid (LPA) can unlock the mitotic block in contact-inhibited HCECs via enhancing nuclear translocation of yes-associated protein (YAP). Firstly, we verified that exogenous YAP could induce cell proliferation in contact-inhibited HCEC monolayers and postconfluent B4G12 cells. In B4G12 cells, enhanced cyclin D1 expression, reduced p27KIP1/p21CIP1 levels, and the G1/S transition were detected upon transfection with YAP. Secondly, we confirmed that LPA induced nuclear expression of YAP and promoted cell proliferation. Moreover, PI3K and ROCK, but not ERK or p38, were required for LPA-induced YAP nuclear translocation. Finally, cells treated with LPA or transfected with YAP remained hexagonal in shape, in addition to unchanged expression of ZO-1, Na/K-ATPase, and smooth muscle actin (SMA), suggestive of a preserved phenotype, without endothelial–mesenchymal transition. Collectively, our findings indicate an innovative strategy for ex vivo cultivation of HCECs for transplantation and cell therapy.
Introduction
Facing the aqueous humor–containing anterior chamber, the corneal endothelium regulates stromal hydration and subsequent corneal transparency through the expression of the tight junction component ZO-1, which forms barriers,1 and partly through the expression of Na/K-ATPases, which act as pumps.2 In contrast to the situations in other species, human corneal endothelial cells (HCECs) retain only a very limited proliferative potential both in vivo3 and in vitro,4 which is related to the arrest of cell cycle in G1 phase due to contact inhibition mediated by intercellular junctions. Thus, loss of HCECs caused by surgery, disease, or aging may potentially lead to persistent corneal endothelial dysfunction.5 To meet the increasing needs of thin lamellar grafts for endothelial keratoplasty,6 the issue of transplantation using ex vivo-expanded HCECs for transplantation has been a focus of research for the past three decades.7,8
As derivatives of the neural crest or neuroectoderm, such as corneal epithelial cells, HCECs undergo endothelial–mesenchymal transition (EnMT) under pathological conditions or after stimulation by certain factors, such as TGF-β.9,10 To stimulate ex vivo expansion of HCECs, growth factors such as bFGF can be used11; however, EnMT is often activated.10 On the other hand, downregulation of p120-catenin using siRNA in both contact-inhibited HCECs10 and retinal pigment epithelial cells12 uniquely promotes proliferation by activating trafficking of p120-catenin to the nucleus, thus relieving the repression of the cell cycle by nuclear Kaiso without inducing EnMT.10 This nuclear p120/Kaiso signaling is associated with activation of the RhoA/ROCK signaling and inhibition of the Hippo pathway, but without activation of the Wnt/β-catenin signaling.10,13,14 To prevent potential biohazards related to off-target effects induced by RNA silencing, we aimed to develop an alternative strategy for ex vivo expansion of HCECs for clinical applications.
The Hippo pathway was identified through genetic screens of Drosophila melanogaster and is highly conserved in mammals. This pathway is involved in controlling organ size and regulating embryonic development15,16 and is also a regulator of contact inhibition,17 which plays crucial roles in regulating cell proliferation and apoptosis.18,19 The transcriptional coactivator yes-associated protein (YAP) is an important mediator of the Hippo pathway. Upon formation of cellular contacts, i.e., activation of the Hippo pathway, YAP is phosphorylated by Lats1/2 and then sequestered in the cytoplasm. In contrast, blocking the Hippo pathway results in dephosphorylation and nuclear translocation of YAP, further heterodimerization of YAP with the transcription factor TEAD1, and subsequent upregulation of related gene expression.20 Evidence has shown beneficial roles of YAP in stimulating tissue repair and regeneration following injury, such as in the intestine,21 skin,22 or heart.23 Previous studies have also indicated that YAP overexpression abolishes contact inhibition and promotes cellular growth,24 and such regulatory mechanisms have been substantiated in various cell types. For instance, YAP was shown to enhance the transcription of proliferation-related genes, such as Ki-67, c-myc, and SOX4 in murine livers,25 Cdk6 in human fetal lung fibroblasts,26 and CCND1 in malignant mesotheliomas.27 In contrast, in esophageal squamous cell carcinomas, silencing of YAP was shown to enhance the transcription of p21CIP1 (a CDK inhibitor).28 Taken together, these results suggest that YAP manipulates cellular proliferation through regulating the activity of cell cycle mediators. However, the detailed mechanisms associated with YAP in HCECs have not yet been explored.
Recently, various upstream modulators of YAP have been identified,29 including lysophosphatidic acid (LPA).30 LPA, recognized as a natural component originating from the cell membrane, has long been known to affect cell adhesion, migration, and proliferation,31,32 and this compound might show potential for clinical applications. LPA has been demonstrated to promote wound healing in corneal epithelial cells via the PI3K/AKT pathway and to regulate cell proliferation via transactivation of the EGF-R pathway.33 Moreover, YAP was shown to be involved in the LPA-induced proliferation of HEK293A cells.20 Upon activation of PI3K by LPA, components of several signaling pathway cascades may be activated, such as Erk1/2, p38,34 and cdc42.35 More recently, it has been confirmed that LPA inhibits the Hippo pathway kinases Lats1/2 via G protein-coupled receptor through the RhoA/ROCK signaling pathway and activates downstream gene transcripts via increased nuclear translocation of YAP.36 The hypothesis that LPA induces nuclear translocation of YAP via PI3K/AKT or RhoA/ROCK signaling to regulate cellular proliferation is collectively supported by the aforementioned studies. Although LPA receptors have been shown to be expressed in HCECs,37 whether they act as regulators of YAP and their mechanisms of action remain undetermined.
In this study, we demonstrated that transfected YAP induces proliferation in contact-inhibited HCECs via promotion of cyclin D1 and inhibition of p27KIP1/p21CIP1. We also showed that exogenous LPA enhances nuclear translocation of YAP and promotes proliferation in contact-inhibited HCECs and, at the same time, maintains a normal phenotype without induction of EnMT. Importantly, we further verified that LPA unlocks the mitotic block in contact-inhibited HCEC monolayers through nuclear translocation of YAP, which is related to activation of the PI3K/AKT and RhoA/ROCK pathways.
Results
After being stripped from the inner surface of the cornea and treated with collagenase overnight, the endothelial layer attached to Descemet’s membrane was digested into HCEC aggregates. In serum-containing medium, HCEC aggregates adhered to fibronectin collagen–coated plastic dishes and expanded to monolayers after ex vivo culture for 7 days (Figure 1a). In the HCEC monolayers, close cell–cell contacts and a polygonal cell morphology were established and preserved, mimicking those observed in vivo. Additionally, immunofluorescence revealed expression of Na/K-ATPase and ZO-1, suggesting that ion pumps and intercellular tight junctions are maintained in the HCEC monolayers (Figure 1a,b). Next, cell proliferation was assayed via BrdU labeling. In the HCEC monolayer group treated with collagenase, BrdU-labeled nuclei were scarce, indicating that proliferation was inhibited by contact inhibition. In contrast, significantly more proliferating cells labeled with BrdU were observed in the HCEC suspension culture group treated with trypsin/EDTA (Figure 1c). Despite the increase in proliferating HCECs in the suspension culture, fibroblast-like cells resulting from EnMT were observed by phase-contrast microscopy as well as positive immunostaining of SMA fiber but weak expression of ATPase and ZO-1 in the margin of cells (Figure 1c,d). Consequently, we sought to identify alternative methods, similar to p120-siRNA treatment,10 to induce proliferation in contact-inhibited HCECs while preserving their normal phenotype.
Figure 1.

Contact inhibition develops in aggregates from an expansion culture of human corneal endothelial cells (HCECs) with a normal phenotype, but without endothelial–mesenchymal transition (EnMT). (a) Following stripping from Descemet’s membrane, the corneal endothelial layers formed HCEC aggregates after 16 hours of digestion by collagenase A at 37 °C. After 1 day of culture in serum-containing medium, the HCEC aggregates adhered to the dish surface (left panel). After 7 days in culture, the HCEC aggregates expanded into HCEC monolayers (right panel). (b) HCEC monolayers cultured until day 7 showed a hexagonal morphology under phase microscopy and a normal immunostaining pattern for Na/K-ATPase (ATPase) and ZO-1, but no expression of α-smooth muscle actin (SMA), indicating no evidence of EnMT. (c) The proliferation of HCECs digested with trypsin/EDTA (suspension culture), as assayed via BrdU labeling (green), was significantly higher than in HCEC monolayers (expansion culture), suggestive of contact inhibition in the HCEC monolayers (n = 3; **P < 0.01). (d) The suspension culture of HCECs showed a fibroblast-like morphology and expression of SMA fiber, but weak expression of ATPase and ZO-1 in the margin of cells, demonstrating the specificity of antibodies and an EnMT phenotype. The cell nuclei were counterstained with Hoechst 33342 (blue).
Exogenous expression of YAP promoted proliferation in contact-inhibited HCECs
YAP has been reported to promote proliferation in miscellaneous types of cells.25–28 To understand the effect of YAP on inducing proliferation in HCECs, HCEC monolayers were transfected with the pCMV6-YAP vector (pCMV6-YAP) for 72 hours, and monolayers transfected with the pCMV6-AC-GFP vector (pCMV6-control) served as controls. Subsequently, immunofluorescence revealed expression of YAP and BrdU-labeling, showing colocalization in cells transfected with pCMV6-YAP, suggesting an induction of proliferation by YAP in contact-inhibited HCEC monolayers (Figure 2a). On the other hand, EnMT was not induced in the HCEC monolayers, as there was positive immunostaining for Na/K-ATPase and ZO-1, whereas SMA staining was negative (Figure 2b).
Figure 2.

Overexpression of YAP leads to proliferation in contact-inhibited human corneal endothelial cells (HCECs). (a) HCEC monolayers were transfected with either the pCMV6-YAP vector (pCMV6-YAP) or the pCMV6-AC-GFP vector (pCMV6-control), as a control. After transfection, the HCEC monolayers were further cultured in HCEC growth medium for 2 days. The cultures were first starved for 2 hours, then fixed and immunostained with YAP (green) and BrdU (red; smaller figure in the right panel). Immunofluorescence images of YAP, BrdU, and nuclei (Hoechst 33342) were merged, and the colocalization of YAP and BrdU appeared as white color. Expansion culture of HCEC aggregates exhibited confluent monolayer cells under DIC microscopy (lower panel). In the pCMV6-YAP group, BrdU labeling was significantly increased. Colocalization of YAP and BrdU indicated that proliferation in contact-inhibited HCECs was promoted by YAP (n = 3; **P < 0.01). (b) HCEC monolayers transfected with the pCMV6-YAP vector for 2 days showed a hexagonal morphology under phase contrast microscopy, with normal immunostaining patterns of ATPase and ZO-1, but without expression of SMA, indicating no evidence of endothelial–mesenchymal transition. SMA, smooth muscle actin; YAP, yes-associated protein.
Exogenous expression of YAP promoted proliferation in B4G12 cells by activating cell cycle mediators
Due to the scarce availability of HCECs, we used a human corneal endothelial cell line (B4G12 cells) for further mechanistic studies. B4G12 cells are a clonal subpopulation from the parental cell line HCEC-12, established from normal cells of the cornea endothelium,38 described as representing differentiated corneal endothelial cells and retaining the expression of Na/K-ATPase and ZO-1 (Figure 3a).
Figure 3.

Enhanced nuclear levels of YAP promote proliferation in B4G12 cells. (a) B4G12 cells (human corneal endothelial cell line) cultured until day 3 after confluence showed a hexagonal morphology under phase microscopy and normal immunostaining patterns of ATPase and ZO-1, but no expression of smooth muscle actin (SMA), suggesting a phenotype similar to HCECs. The cell nuclei were counterstained with Hoechst 33342 (blue). (b) B4G12 cells were serum starved for 24 hours and were transfected with either the pCMV6-YAP vector (pCMV6-YAP) or the pCMV6-AC-GFP vector (pCMV6-control), as a control. After transfection, from day 2 to day 6, cell growth was analyzed via cell counting. (c) Flow cytometry analysis showed that transfection of pCMV6-YAP results in a decrease in the G0-G1 population and a concomitant increase in the S-phase population compared with control cultures (n = 3; *P < 0.05; **P < 0.01). (d) The expression of transfected YAP was determined by western blotting, which indicated that the levels of exogenous YAP protein were significantly higher. The effects of YAP overexpression on the expression of proliferation-related proteins were examined by western blotting, which indicated that transfected YAP leads to upregulation of cyclin D1 expression and concomitant downregulation of p27KIP1 and p21CIP1 (CDK inhibitors) expression. YAP, yes-associated protein.
According to the results of cell counting, while the proliferation rate was significantly higher in B4G12 cells than in HCECs, the proliferation rate was even higher in pCMV6-YAP-transfected B4G12 cells than that in pCMV6-Control-transfected B4G12 cells from day 2 to day 5 (Figure 3b). To better understand how YAP promotes cell proliferation, the possible effect of YAP on cell cycle progression was analyzed via flow cytometry. As shown in Figure 3c, the G0-G1 population of the total cells in the control cultures was estimated to be 61.2%; upon transfection with YAP, the G0-G1 population was significantly reduced to 55.7%. In conjunction with a reduced G0-G1 cell population, the S-phase population significantly increased from 29.1% to 33.1% upon transfection of YAP. To elucidate the regulatory role of YAP in cell proliferation, pCMV6-YAP (with a GFP-tag)-transfected B4G12 cells were examined using western blot analysis to determine the effect of YAP on cell cycle-modulating proteins. As shown in Figure 3d, the pCMV6-YAP-transfected cells expressed a GFP-tagged YAP protein band, which was not present in the pCMV6-control-transfected cells. Upon observing the increased trend of G1/S phase transition, we proceeded to investigate the cell cycle mediator cyclin D and its regulators p27KIP1/p21CIP1. Importantly, YAP overexpression led to the upregulation of cyclin D1 and the concomitant downregulation of p27KIP1/p21CIP1, implying that YAP could inhibit CDK inhibitors and activate CDKs. Accordingly, G1/S phase transition proceeded, promoting cell proliferation, which has also been observed in various cell types.24,27,28
LPA enhanced nuclear accumulation of YAP and promoted proliferation in HCECs and B4G12 cells
As noted above, LPA has been demonstrated to induce nuclear translocation of YAP in numerous cell types.20,29,30 To confirm that LPA induces nuclear localization of YAP in HCECs in a dose-dependent manner, different concentrations of LPA were added to the medium for 4 hours, after HCEC monolayers had been starved for 24 hours. Next, nuclear expression of YAP was compared through western blot and immunofluorescence analyses. Substantial nuclear localization of YAP was identified in cells treated with 20 μmol/l LPA (Figure 4a,b). Likewise, a BrdU-labeling assay indicated proliferation in contact-inhibited HCECs treated with LPA (Figure 4c). Similar to what was observed in HCECs transfected with YAP, immunostaining for Na/K-ATPase, ZO-1, and SMA also indicated the absence of EnMT during LPA-induced HCEC proliferation (Figure 4d).
Figure 4.

LPA enhances nuclear levels of YAP and promotes proliferation in contact-inhibited HCECs. (a) The stimulatory effects of LPA on nuclear levels of YAP in contacted-inhibited HCECs were evaluated by western blotting assays. HCEC monolayers were serum starved for 24 hours and incubated in LPA at different doses for an additional 4 hours. The nuclear proteins were extracted from each experimental group and fractionated on 10% SDS–PAGE gels (5 μg/lane). After transfer, the membranes were blotted with an antibody against either YAP or histone as a loading control. (b) The distribution of nuclear YAP (green) was detected in immunofluorescein assays in contacted-inhibited HCECs 4 hours after the addition of 20 μmol/l LPA. (c) Cell proliferation was examined via BrdU labeling, and the BrdU labeling (green) in HCEC monolayers treated with 20 μmol/l LPA was significantly higher than in HCEC monolayers treated with phosphate-buffered saline (n = 3; **P < 0.01). (d) HCECs treated with LPA (20 μmol/l) showed a hexagonal morphology under phase microscopy, with a normal immunostaining pattern for ATPase and ZO-1, but without any expression of SMA, indicating no evidence of endothelial–mesenchymal transition. The cell nuclei were counterstained with Hoechst 33342 (blue). LPA, lysophosphatidic acid; SMA, smooth muscle actin; YAP, yes-associated protein.
To further clarify the signaling pathways involved in the enhanced proliferation induced by LPA, B4G12 cells were used. First, nuclear translocation of YAP was confirmed to be induced by LPA in a dose-dependent manner. Postconfluent B4G12 cells were starved for 24 hours, and different concentrations of LPA were then added to the medium for 4 hours. Subsequently, nuclear expression of YAP was compared by western blot and immunofluorescence analyses. Cell proliferation was also evaluated based on cell counts. Similar to what was observed in HCEC monolayers, nuclear localization of YAP and cell proliferation were significantly increased in LPA-treated B4G12 cells (Figure 5a–c). Moreover, EnMT was not induced in postconfluent B4G12 cells treated with LPA according to the immunostaining results for Na/K-ATPase, ZO-1, and SMA (Figure 5d).
Figure 5.

LPA also enhances nuclear levels of YAP and promotes proliferation in B4G12 cells. (a) The dose-dependent stimulation of nuclear YAP in postconfluent day 4 B4G12 cells by LPA was measured by western blotting. B4G12 cells were serum starved for 24 hours and incubated in the presence of LPA at different doses for an additional 4 hours. Nuclear proteins (Nu) were extracted from each experimental group and fractionated in 10% SDS–PAGE gels (5 μg/lane). After being transferred to membranes, the proteins were subjected to blotting with an antibody against either YAP or histone H3, as a loading control. (b) The distribution of nuclear YAP (green) was detected via an immunofluorescein assay in postconfluent day 4 B4G12 cells 4 hours after 20 μmol/l LPA was added. (c) B4G12 cells were serum starved for 24 hours and treated with LPA (20 μmol/l). After treatment, from day 2 to day 6, cell growth was analyzed through cell counting (n = 3; **P < 0.01). (d) B4G12 cells treated with LPA (20 μmol/l) showed a hexagonal morphology under phase microscopy, with a normal immunostaining pattern of ATPase and ZO-1, but without any expression of SMA, indicating no evidence of endothelial–mesenchymal transition. The cell nuclei were counterstained with Hoechst 33342 (blue). LPA, lysophosphatidic acid; SMA, smooth muscle actin; YAP, yes-associated protein.
Nuclear translocation of YAP in the B4G12 cell line was enhanced by LPA via the Rho/ROCK and PI3K pathway
PI3K is one of characterized downstream effectors of LPA signaling.33 We therefore used specific inhibitors of PI3K or its possible downstream effectors (ERK1/2, p38, and ROCK) to clarify the potential pathways underlying the effect of LPA on YAP nuclear translocation. For this purpose, B4G12 cells were initially treated with desired inhibitors for 2 hours, followed by LPA treatment for 4 hours, and the cells were finally used for western blotting assays to investigate YAP nuclear translocation. As shown in Figure 6a (upper panel), Y27632 (ROCK inhibitor) effectively suppressed LPA-induced nuclear translocation of YAP, and LY294002 (PI3K inhibitor) partially suppressed LPA-induced nuclear translocation of YAP, suggesting their roles in regulating YAP. Moreover, LPA-induced nuclear translocation of YAP was not inhibited in cells treated with PD98059 and SB203580 (ERK and p38 inhibitors, respectively), suggesting that neither pathway is involved in the regulatory effect of LPA on YAP. Additionally, cell cycle mediators (cyclin D1, p27KIP1/p21CIP1) were investigated. As shown in Supplementary Figure S2, similar to what was observed in the YAP-transfected group (Figure 4d), LPA treatment also upregulated cyclin D1 expression and downregulated p27KIP1/p21CIP1 expression, while Y27632 treatment restored these changes to the baseline levels of the control group. Interestingly, the effects of LY294002 treatment on cyclin D1 and cell proliferation were more prominent than those of Y27632 treatment (Supplementary Figures S1 and S2).
Figure 6.

LPA-induced nuclear translocation of YAP is mediated by the PI3K and ROCK pathways in B4G12 cells. (a) The effects of ROCK and PI3K phosphatase inhibitors on LPA-induced YAP nuclear translocation were measured by western blotting. Postconfluent day 4 B4G12 cells were serum starved for 24 hours, followed by pretreatment with LY294002 (PI3K inhibitor, 50 μmol/l), PD98059 (ERK1/2 inhibitor, 20 μmol/l), SB203580 (p38 inhibitor, 20 μmol/l), or Y27632 (ROCK inhibitor, 20 μmol/l) for 2 hours; the cells were then treated with LPA for 4 hours. Cell nuclear extracts (Nu) were subsequently prepared from each experimental group and fractionated in 10% SDS–PAGE gels (5 μg/lane). After transfer to membranes, the proteins were subjected to blotting with an antibody against either YAP or histone H3 as a loading control. (b) LPA stimulated the nuclear translocation of YAP, phosphorylation of AKT (p-AKT), and GTP binding of RhoA (GTP-RhoA) in a dose-dependent manner. Postconfluent day 4 B4G12 cells were serum starved for 24 hours, followed by LPA treatment for 4 hours. Nuclear extracts (Nu) or total lysates were subjected to immunoblotting with YAP, p-AKT and GTP-RhoA antibodies. Histone H3 and GAPDH were also detected by blotting as loading controls. (c) To examine whether LPA-induced translocation of YAP results in activation of the AKT or RhoA pathway, postconfluent day 4 B4G12 cells were pretransfected with YAP siRNA for 48 hours, serum starved for an additional 24 hours, and treated with LPA (20 µmol/l) for another four hours. Nuclear extracts (Nu) or total lysates were subjected to immunoblotting with YAP, AKT, and RhoA antibodies. Histone H3 and GAPDH were also detected by blotting as loading controls. (d,e) The effects of PI3K and ROCK inhibitors on LPA-dependent YAP translocation were examined via western blotting. Postconfluent day 4 B4G12 cells were serum starved for 24 hours, followed by pretreatment with various concentrations of LY294002 or Y27632 for an additional 2 hours and treatment with LPA (20 µmol/l) for another 4 hours. Total cell lysates were prepared and probed with YAP and pathway-related antibodies through western blotting. LPA, lysophosphatidic acid; YAP, yes-associated protein.
Further investigation showed that LPA stimulated nuclear translocation of YAP in a dose-dependent manner and led to concomitant upregulation of phosphorylated AKT (p-AKT, a PI3K downstream effector) and a GTP-bound RhoA status (RhoA-GTP, a ROCK upstream effector) according to western blotting (Figure 6b), indicating that the kinase activity of AKT and RhoA is governed by LPA. Subsequently, to illustrate the up- and downstream relationships of the kinases and YAP within the signaling pathway, the effect of LPA on YAP-silenced B4G12 cells was examined, while nontargeting siRNA served as a control. The fact that p-AKT and RhoA-GTP were not influenced by YAP silencing excludes YAP from being an upstream effector of PI3K and ROCK (Figure 6c).
To understand the roles of the PIK3 and ROCK signaling pathways in LPA-induced nuclear translocation of YAP, B4G12 cells were pretreated with LY294002 or Y27632 for 2 hours, followed by LPA treatment for 4 hours. The cells were then observed to determine the effects of kinase inhibitors on nuclear translocation of YAP and its associated kinases. As shown in Figure 6d, LY294002 treatment partially suppressed LPA-induced nuclear translocation of YAP, accompanied by a reduction in p-AKT levels (Figure 6e), suggesting involvement of the PI3K/AKT pathway in LPA-induced nuclear translocation of YAP. However, given that the levels of RhoA-GTP were not altered by LY294002, the involvement of the PI3K/AKT pathway in nuclear translocation of YAP was likely mediated by pathways other than the Rho-ROCK pathway. Besides, Y27632 effectively suppressed LPA-induced YAP nuclear translocation in a dose-dependent manner, suggestive of a distinct role of ROCK signaling in controlling YAP by LPA. Moreover, p-AKT levels were not influenced by Y27632, indicating that PI3K did not play a pivotal role in the ROCK pathway during the 4-hour LPA treatment. In summary, the results support the notion that the induction of YAP nuclear translocation by LPA is mediated by either the PI3K/AKT or RhoA/ROCK pathway, independently (Figure 7).
Figure 7.
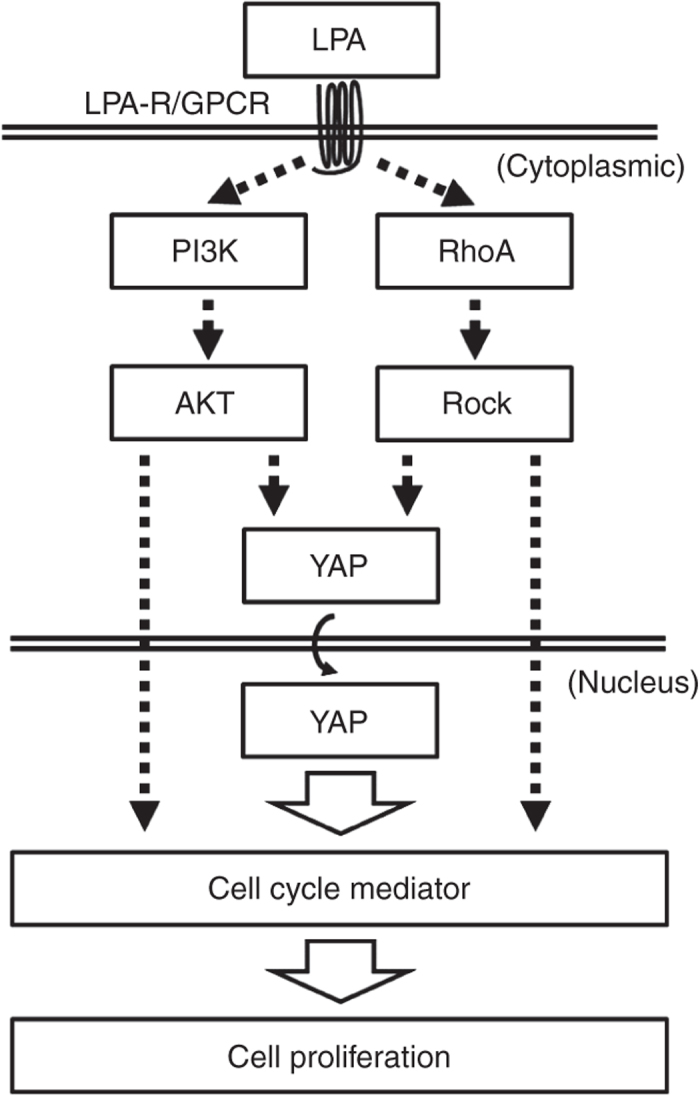
Schematic pathways of proliferation in human corneal endothelial cells regulated by lysophosphatidic acid. AKT, protein kinase B; GPCR, protein-couple receptor; LPA-R, lysophosphatidic acid receptor; PI3K, phosphoinositide-3 kinase; RhoA, Ras homolog gene family, member A; ROCK, Rho-kinase; YAP, yes-associated protein.
Discussion
With the advantages of smaller wounds, faster visual recovery, and lower rejection rates, endothelial keratoplasty (EK) is evolving as the lamellar keratoplasty of choice for corneal endothelial disorders. On the other hand, for countries with limited corneal donors, cultivated EK (c-EK) is emerging as a promising technology with clinical implications. To develop c-EK, successful ex vivo expansion of HCECs is fundamental. Although suspension culture can be used for the ex vivo cultivation of HCECs,39 EnMT occurs as an adverse effect.10 We previously attempted to reverse EnMT to a normal phenotype through the temporary use of serum-free culture media, with only partial success compared with the in vivo morphology (unpublished data). Upon fabrication of engineered grafts for c-EK, EnMT could be effectively inhibited by cellular contacts on carriers seeded with HCECs at 100% confluency (unpublished data). However, in such grafts, cellular proliferation is suppressed by contact inhibition, resulting in reduced cell density after the c-EK procedure. Therefore, adding LPA to the culture medium may be able to relieve contact inhibition-induced cell growth arrest, and this partially regained proliferation (~6%) of HCECs is likely to compensate the cell loss during the preparation of the c-EK grafts. Although proliferation can be induced with a retained in vivo HCEC morphology through the transfection of p120-siRNA,10 the fact that RNA interference involves collateral inhibition of other genes restrains its clinical eligibility. In addition to administration of LPA, after completion of safety validation, adenovirus vector-carried exogenous expression of YAP for c-EK is supposed to provide better effects to unlock mitotic block through transient but high transfection efficiency.
Upon transfection of p120-siRNA, cell proliferation is observed, accompanied by nuclear translocation of YAP,10 which is also linked to the regulation of cell proliferation.25–28 Hence, in this study, we first demonstrated that YAP indeed promotes cell proliferation in contact-inhibited HCECs (Figure 2a). Then, we determined that exogenous expression of YAP results in increased expression of cyclin D1 protein, but decreased expression of p27KIP1/p21CIP1 proteins in the B4G12 human corneal endothelial cell line (Figure 3d). Cell cycle analysis (Figure 3c) indicated that the induced G1/S transition originated from the regulatory effects of transfected YAP. Apart from directly regulating cell cycle mediators, nuclear YAP was observed to exhibit crosstalk with the β-catenin/Wnt pathway, which is also involved in cellular contacts and the regulation of proliferation.40 Therefore, it is plausible to investigate relationships between the cadherin/catenin complex and the YAP signaling pathway in the regulation of cell proliferation.
To facilitate clinical application, we attempted to replace p120-siRNA with LPA to induce the nuclear translocation of YAP. In this study, we demonstrated that LPA induces nuclear translocation of YAP in contact-inhibited HCECs, and we subsequently explored the possible underlying mechanisms using B4G12 cells. Phospholipids such as LPA and sphingosine-1-phosphate41 were shown to act as regulators of YAP, although sphingosine-1-phosphate did not influence nuclear translocation of YAP in our study (unpublished data). Although our data indicate a better growth-promoting effect of overexpressed YAP than LPA (Figures 3b and 5c) and the role of LPA in stimulating cell proliferation therefore remains clinically undetermined, the current mechanistic investigation of the release of contact inhibition is still beneficial for improving cell culture procedures.
LPA plays various roles in physiology, such as promoting cell proliferation, migration, and survival,42 as well as in the pathophysiology of cancer.43 Because LPA is only used as an additive in the culture medium during ex vivo expansion, our protocol design is highlighted by a lack of concern regarding tumorigenesis. Despite the rare reports of EnMT being induced by LPA,44 EnMT was not detected in the HCEC monolayers treated with LPA in our study (Figure 4d). On the other hand, although YAP may be considered a candidate oncogene, transfected YAP did not induce EnMT in HCEC monolayers in the present study (Figure 2b). Moreover, to exclude the possibility of carcinogenesis, proliferation is only induced at the stage of ex vivo cultivation, and future transplantation will only be performed after cessation of induced proliferation and verification of a normal physiological distribution of YAP.
The cellular effects of LPA have been reported to be mediated by receptor stimulation of AKT,33 ERK,45 Rho,46 and p38.47 However, in the LPA-induced nuclear translocation of YAP in ovarian cancer cells, only ROCK is involved, while PI3K, ERK, and p38 are not.30 Our results were predominantly compatible with previous reports, except for the role of PI3K, which was possibly attributed to different lineages, types, or passages of the target cells.
Given that LPA-induced nuclear translocation of YAP was influenced by both PI3K and ROCK (Figure 6a) and that silencing of YAP did not alter AKT phosphorylation at T308 or modulate RhoA-GTP bounding, we have demonstrated that the observed phenomenon of nuclear translocation is not due to feedback regulation to AKT and RhoA. Next, to elucidate the cause–effect relationships of PI3K and ROCK, we adopted kinase inhibitors of PI3K and ROCK, to observe their effects on pathway-related mediators. Because the PI3K inhibitor did not modulate the enhancement of GTP-bound RhoA by LPA, and in turn, the ROCK inhibitor did not alter AKT, we speculated that during LPA-induced nuclear translocation of YAP, the PI3K and ROCK pathways are mutually independent. It has been documented that LPA directly affects Rho signaling in neuronal and nonneuronal cell lines46 and that the Rho/ROCK pathway is involved in the LPA-induced nuclear translocation of YAP,30 suggesting the existence of a PI3K-independent LPA/ROCK/YAP pathway.
Although the inhibitory effect of PI3K on LPA-induced nuclear translocation of YAP was only partial (Figure 6a), it was demonstrated that LPA activates the PI3K pathway to facilitate corneal epithelial wound healing.33 We also observed that the effects of LY294002 on cyclin D1 and cell proliferation were more significant than those of Y27632 (Supplementary Figures S1 and S2), indicating that LPA-activated PI3K, likely through YAP-independent regulation, also plays a pivotal role in proliferative regulation. Furthermore, it is likely that LPA-related factors, including PI3K, ROCK, and YAP, act on cell proliferation with multipathway effects. After demonstrating the provocative effects of YAP and LPA on proliferation, we focused on delineating the roles of the PI3K and ROCK pathways in the LPA-induced nuclear translocation of YAP. Concerning the roles of the PI3K and ROCK pathways in LPA-induced proliferation, given the different effects of kinase inhibitors of the two pathways on both nuclear translocation of YAP and expression of cell cycle mediators (Figure 6a and Supplementary Figure S2), we postulate that there are YAP-independent pathways involved in the process of LPA-induced cell proliferation (Figure 7), which merits future research.
The ROCK inhibitor Y27632 was found to inhibit apoptosis and enhance the survival of monkey corneal endothelial cells,48 while the effects of Y27632 on the proliferation of HCECs remain under discussion.49 Interestingly, in recent reports, under a postconfluent status, Y27632 failed to promote proliferation in HCECs,50 and during wound healing, Y27632 promoted proliferation in corneal endothelial cells.51 Here, we hypothesize that this discrepancy may originate from difference in donors age, definition of confluence status, or the status of cellular contacts. In our experience, a hexagonal morphology is retained in Y27632-treated ex vivo-cultivated HCEC monolayers for a prolonged interval, indicating the ability of this inhibitor to maintain cell survival (unpublished data). In postconfluent B4G12 cells, Y27632 inhibited nuclear translocation of YAP and attenuated the effects of LPA on cell cycle mediators (Figure 6a and Supplementary Figure S2). However, because the proliferation regulation of cell lines may be significantly different from that of normal cells, the regulatory role of Y27632 in HCECs proliferation still need to be further examined.
When exploring possible signaling pathways, we chose AKT phosphorylation at T308 to evaluate the effect of the PI3K inhibitor and attempted to locate a downstream factor of ROCK for evaluation. To date, no published work has evaluated the kinase inhibition effect of the ROCK inhibitor Y27632 in HCECs. It has been reported that Y27632 suppresses p-MLC in human umbilical vein endothelial cells and that Y27632 suppresses the phosphorylation of MYPT-1 in aortic endothelial cells. Thus, we measured the levels of p-MLC in Y27632-treated B4G12 cells but did not detect any significant changes (unpublished data). However, based on the observation of significant nuclear translocation of YAP and unaltered levels of p-AKT, we believe that Y27632 exerts effects other than those related to cytotoxicity.
Collectively, our data support a role for the LPA-induced nuclear translocation of YAP in inducing proliferation in postmitotic HCECs, and this effect is associated with the PI3K and ROCK pathways. As a recognized natural component of the cell membrane, we believe that LPA represents a promising cell culture supplement for use in tissue engineering. Hopefully, in the future, advanced tissue engineering technology for treating disorders of postmitotic tissues or organs will be realized based on the accumulating knowledge regarding the regulation of the HCEC proliferative capacity.
Materials and Methods
Materials
Human endothelium serum-free medium (HESFM), Opti-MEM medium, trypsin-EDTA, fetal bovine serum (FBS), phosphate-buffered saline (PBS), gentamicin, amphotericin B, Lipofectamine 2000, and an Alexa-Fluor-conjugated secondary IgG antibody were purchased from Invitrogen (Carlsbad, CA). LPA, RPMI 1640 vitamin solution, dimethyl sulfoxide (DMSO), Hoechst 33342 dye, methanol, Triton X-100, Y-27632, PD98059, and SB203580 were purchased from Sigma-Aldrich (St Louis, MO). LY294002 was purchased from Cell Signaling (Beverly, MA). Collagenase A was purchased from Roche Applied Science (Indianapolis, IN). Recombinant Human FGF-basic was purchased from Peprotech (London, UK). Recombinant Human EGF was purchased from Upstate, Millipore (Billerica, MA). YAP siRNA and nontargeting control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A green fluorescent protein-tagged, full-length open reading frame clone (pCMV6-YAP) of human YAP (NM_01130145) and its control vector (pCMV6-AC-GFP) were purchased from Origene Technologies (Rockville, MD).
Antibodies against YAP (rabbit polyclonal), histone H3 (rabbit polyclonal), and p-AKT (Thr308; rabbit antibody clone 244F9) were purchased from Cell Signaling. Rabbit polyclonal p27KIP1 and p21CIP1 antibodies were purchased from Santa Cruz Biotechnology. A mouse cyclin D1 antibody (clone CD1.1) was purchased from Abcam (La Jolla, CA), and a mouse BrdU antibody (RPN20Ab) was purchased from Amersham, GE Healthcare (Chalfont St Giles, UK). A mouse ZO-1 antibody (clone ZO1-1A12) was purchased from Invitrogen, and a mouse Na/K-ATPase antibody (clone C464.6) was purchased from Upstate, Millipore. A mouse GAPDH antibody (clone 6C5) was purchased from Chemicon, Millipore (Billerica, MA), and a rabbit polyclonal RhoA antibody was purchased from Thermo Scientific (Rockford, IL). All plastic cell culture wares were obtained from Corning Incorporated Life Sciences (Acton, MA).
Source of tissue donors
This study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board of Chang Gung Memorial Hospital, which ruled that consent forms were not required from donors because the human tissues used in this study were delinked, postoperative residual specimens. A total of 34 corneas from donors aged 28 to 59 years (mean: 49.4 ± 8.7) were used in this study. After removing central part for transplantation, the remaining human corneoscleral tissues were procured within 5 days and maintained at 4 °C in Optisol (Chiron Vision, Irvine, CA) following corneal transplantation surgeries at the Department of Ophthalmology, Chang Gung Memorial Hospital, Linkou, Taiwan.
Cell preparation
The isolation and culture of HCECs followed previous methods.52 Briefly, after the central corneas had been used for corneal transplantation, the remaining corneoscleral tissues were rinsed three times with wash medium (containing Opti-MEM, 200 μg/ml gentamicin, and 10 μg/ml amphotericin B). Under a dissecting microscope, the trabecular meshwork was cleaned, and Descemet’s membranes containing HCECs were stripped using forceps. Following digestion at 37 °C for 24 hours with 0.5 mg/ml collagenase A in wash medium, HCEC aggregates were collected via centrifugation at 495 g for 8 minutes to remove the digestion solution and then cultured in 8-well Lab-Tek II chamber slides (Nalge Nunc International, Rochester, NY) coated with a fibronectin-collagen coating mix (AthenaES, Baltimore, MD) in HCEC growth medium, composed of Opti-MEM supplemented with 10% FBS, 20 ng/ml hEGF, 10 ng/ml FGF-basic, RPMI 1640 vitamin (1×) solution (Sigma-Aldrich), 25 µg/ml gentamicin, and 1.25 µg/ml amphotericin B.
The human corneal endothelial cell line B4G12 (Creative Bioarray, NY) was cultured in B4G12 medium, composed of HESFM supplemented with 2% FBS and 10 ng/ml bFGF. To reduce the proliferation rate for the purpose of acclimatization, B4G12 cells were subcultured with equal volumes of B4G12 medium and HESFM until the final concentrations of FBS and bFGF in the medium reached 0.25% and 1.25 ng/ml, respectively (LS-B4G12 medium). B4G12 cells were cryopreserved in liquid nitrogen after acclimatization and were cultured in LS-B4G12 medium. Cells that had undergone less than five passages were used for experiments.
YAP expression vector and siRNA transfection
HCEC monolayers were cultured until day 7 and then preincubated with Opti-MEM overnight. B4G12 cells were cultured until postconfluence day 4 and then preincubated with Opti-MEM overnight. Both cell types were subsequently transfected with the pCMV6-YAP vector (5 μg/ml) or YAP siRNA (50 nmol/l) using lipofectamine 2000 for 6 hours. The control vector or nontargeting siRNA was used as a control to minimize nonspecific effects. After transfection, the two cell types were cultured in HCEC growth medium or LS-B4G12 medium for further investigations.
Immunofluorescence staining
HCEC monolayers or B4G12 cells were cultured in 8-well chamber slides. The cells were subsequently fixed in 4% formaldehyde for 15 minutes at room temperature, rinsed with PBS, permeabilized with 0.2% Triton X-100 for 15 minutes, and rinsed again with PBS. After incubation with 2% BSA to block nonspecific staining for 30 minutes, the slides were incubated with the primary antibodies (all at 1:100 dilution) for 24 hours at 4 °C. After three washes with PBS, the slides were incubated with the corresponding Alexa-Fluor-conjugated secondary IgG for 60 minutes at room temperature. The samples were then counterstained with Hoechst 33342. Sections were mounted with Gel Mount (Biomeda, Foster City, CA) and examined under a Zeiss fluorescence microscope (Oberkochen, Germany) or a confocal microscope (Leica, Deerfield, IL).
Measurement of cell proliferation
HCECs were examined using a cell proliferation kit (GE Healthcare) according to the manufacturer’s instructions. Briefly, HCEC monolayers were cultured until day 7 and then transfected with YAP or treated with LPA (20 µmol/l). After 24 hours, BrdU was added at a final concentration of 10 µmol/l in the HCEC growth medium for 24 hours. The BrdU-labeled cells were detected using a BrdU antibody, and the number of BrdU-positive cells was counted in five randomized fields (×200).
The proliferation rate of B4G12 cells was measured through cell counting. B4G12 cells were seeded onto 35-mm culture dishes at a density of 8.8 × 105 cells per dish. The next day, the cells were transfected with YAP or treated with LPA (20 µmol/l). In the YAP transfection experiments, 24 hours after YAP transfection, the cells were serum starved in HESFM for another 24 hours, and the medium was replaced daily. In the LPA treatment experiments, 20 µmol/l LPA was added in HESFM, and the medium was replaced daily. Cell counts were obtained daily with three repeats from day 1 to day 5 using a Coulter Counter (Beckman-Coulter, Brea, CA).
Cell cycle analysis
B4G12 cells were seeded into 35-mm culture dishes at a density of 8.8 × 105 cells per dish. The next day, the cells were transfected with pCMV6-YAP or the corresponding control vector, as described above. Twenty-four hours after transfection, the cells were serum starved in HESFM for 24 hours. The cells were then harvested using trypsin-EDTA and pelleted at 600 g for 5 minutes. The cell pellets were resuspended in 300 μl of PBS, and 700 μl of ice-cold 99.9% ethanol was added and mixed well, followed by fixation at −20 °C overnight. The following day, the cells were pelleted and resuspended in 1 ml of PBS containing 10 µg/ml propidium iodide (Invitrogen) and 1 mg/ml RNase A (US Biological, Salem, MA). The cells were subsequently incubated at 37 °C for 1 hour and analyzed using a FACStar flow cytometer (Becton–Dickinson, Mountain View, CA), with excitation at 488 nm. Approximately 10,000 cells were examined in each sample. The cell cycle was analyzed using CELLQuest software (BD Biosciences, San Jose, CA).
Protein extraction and western blotting
Total cell lysates were prepared in RIPA buffer supplemented with 10 mmol/l sodium fluoride, 10 mmol/l sodium orthovanadate, and 1× protease inhibitor cocktail (Sigma-Aldrich). The suspension was transferred to a microfuge tube on ice, sonicated to disrupt the cells, and centrifuged for 15 minutes at 4 °C at maximum speed. The supernatants were then pooled as the total protein extract.
Cellular nuclear proteins were extracted using a nuclear extraction kit (Affymetrix, Santa Clara, CA) according to the manufacturer’s instructions. Briefly, the cultures were washed with cold PBS twice, then incubated in a 10× volume of a cytosolic extraction buffer (supplemented with 1 mmol/l dithiothreitol, 1× protease and phosphatase inhibitor) on ice for 10 minutes. The nuclei were collected via scraping, and the nuclear clump was disrupted through repetitive pipetting. The suspension was then centrifuged at 14,000g for 3 minutes at 4 °C, and the pellets were resuspended in 150 ml of nuclear extraction buffer supplemented with 1 mmol/l dithiothreitol and 1× protease and phosphatase inhibitor and subsequently vortexed at maximum speed for 10 seconds. The sample was placed on ice for 2 hours, with shaking every 20 minutes, and eventually centrifuged at 14,000g for 5 minutes at 4 °C. The supernatant was collected as the nuclear protein extract. Protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA).
The protein extracts were resolved in 10% acrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore), which were then blocked with 5% (w/v) fat-free milk in TBST (50 mmol/l Tris-HCl, pH 7.5, 150 mmol/l NaCl, 0.05% (v/v) Tween-20), and probed with the desired primary antibodies at a 1:1,000 dilution for all antibodies except GAPDH (1:10,000 dilution) at 4 °C overnight, followed by reaction with appropriate horseradish peroxidase–conjugated secondary antibodies. The immunoreactive protein bands were visualized via enhanced chemiluminescence (ECL; GE Healthcare).
GTP-bound RhoA assay
Rho activation was assessed in 1 mg of cell lysate using the Active RhoA Pull-Down and Detection Kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. Briefly, to pull down the GTP-bound form of RhoA, a GST-rhotekin-RBD fusion protein and glutathione resin were used. The amount of pulled-down GTP-bound RhoA was determined in a western blot assay using a RhoA antibody.
Statistics
All data are reported as the mean ± SD for each group, and at least three independent experiments were performed. The data were compared using Student’s unpaired t-test with Microsoft Excel version 2010 (Microsoft, Redmont, WA). Test results were reported as two-tailed P values, where P < 0.05* and P < 0.01** were considered significant.
Acknowledgments
This study was supported by the Chang Gung Memorial Hospital (CMRPG3C0521, CMRPD1C0251, and CMRPG3D0901) to D.H.-K.M., J.-K.C., and H.-C.C. The authors also thank the assistance from the Microscope Core Laboratory, Chang Gung Memorial Hospital, Linkou.
The authors declare no conflict of interest.
References
- Srinivas SP. Dynamic regulation of barrier integrity of the corneal endothelium. Optom Vis Sci. 2010;87:E239–E254. doi: 10.1097/OPX.0b013e3181d39464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanno JA. Identity and regulation of ion transport mechanisms in the corneal endothelium. Prog Retin Eye Res. 2003;22:69–94. doi: 10.1016/s1350-9462(02)00059-9. [DOI] [PubMed] [Google Scholar]
- Laing RA, Neubauer L, Oak SS, Kayne HL, Leibowitz HM. Evidence for mitosis in the adult corneal endothelium. Ophthalmology. 1984;91:1129–1134. doi: 10.1016/s0161-6420(84)34176-8. [DOI] [PubMed] [Google Scholar]
- Joyce NC. Proliferative capacity of corneal endothelial cells. Exp Eye Res. 2012;95:16–23. doi: 10.1016/j.exer.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima S. Clinical investigations on the corneal endothelium. Ophthalmology. 1982;89:525–530. doi: 10.1016/s0161-6420(82)34755-7. [DOI] [PubMed] [Google Scholar]
- Anshu A, Price MO, Tan DT, Price FW., Jr Endothelial keratoplasty: a revolution in evolution. Surv Ophthalmol. 2012;57:236–252. doi: 10.1016/j.survophthal.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Gospodarowicz D, Greenburg G, Alvarado J. Transplantation of cultured bovine corneal endothelial cells to rabbit cornea: clinical implications for human studies. Proc Natl Acad Sci USA. 1979;76:464–468. doi: 10.1073/pnas.76.1.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumblatt MM, Maurice DM, McCulley JP. Transplantation of tissue-cultured corneal endothelium. Invest Ophthalmol Vis Sci. 1978;17:1135–1141. [PubMed] [Google Scholar]
- Saika S, Yamanaka O, Okada Y, Tanaka S, Miyamoto T, Sumioka T. TGF beta in fibroproliferative diseases in the eye. Front Biosci (Schol Ed) 2009;1:376–390. doi: 10.2741/S32. [DOI] [PubMed] [Google Scholar]
- Zhu YT, Chen HC, Chen SY, Tseng SC. Nuclear p120 catenin unlocks mitotic block of contact-inhibited human corneal endothelial monolayers without disrupting adherent junctions. J Cell Sci. 2012;125 Pt 15:3636–3648. doi: 10.1242/jcs.103267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguère L, Cheng J, Gospodarowicz D. Factors involved in the control of proliferation of bovine corneal endothelial cells maintained in serum-free medium. J Cell Physiol. 1982;110:72–80. doi: 10.1002/jcp.1041100112. [DOI] [PubMed] [Google Scholar]
- Chen HC, Zhu YT, Chen SY, Tseng SC. Selective activation of p120ctn-Kaiso signaling to unlock contact inhibition of ARPE-19 cells without epithelial-mesenchymal transition. PLoS One. 2012;7:e36864. doi: 10.1371/journal.pone.0036864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YT, Han B, Li F, Chen SY, Tighe S, Zhang S. Knockdown of both p120 catenin and Kaiso promotes expansion of human corneal endothelial monolayers via RhoA-ROCK-noncanonical BMP-NFκB pathway. Invest Ophthalmol Vis Sci. 2014;55:1509–1518. doi: 10.1167/iovs.13-13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YT, Li F, Han B, Tighe S, Zhang S, Chen SY. Activation of RhoA-ROCK-BMP signaling reprograms adult human corneal endothelial cells. J Cell Biol. 2014;206:799–811. doi: 10.1083/jcb.201404032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell. 2008;13:188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS One. 2013;8:e65539. doi: 10.1371/journal.pone.0065539. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010;24:2383–2388. doi: 10.1101/gad.1978810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Ran Byun M, Furutani-Seiki M, Hong JH, Jung HS. YAP and TAZ regulate skin wound healing. J Invest Dermatol. 2014;134:518–525. doi: 10.1038/jid.2013.339. [DOI] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA. 2013;110:13839–13844. doi: 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry ER, Camargo FD. The Hippo superhighway: signaling crossroads converging on the Hippo/Yap pathway in stem cells and development. Curr Opin Cell Biol. 2013;25:247–253. doi: 10.1016/j.ceb.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Xie Q, Chen J, Feng H, Peng S, Adams U, Bai Y. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013;73:3615–3624. doi: 10.1158/0008-5472.CAN-12-3793. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31:5117–5122. doi: 10.1038/onc.2012.5. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Imoto I, Matsui T, Kozaki K, Haruki S, Sudol M. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 2011;32:389–398. doi: 10.1093/carcin/bgq254. [DOI] [PubMed] [Google Scholar]
- Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR. Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol. 2012;23:770–784. doi: 10.1016/j.semcdb.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Xu Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal. 2013;11:31. doi: 10.1186/1478-811X-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalink K, Hordijk PL, Moolenaar WH. Growth factor-like effects of lysophosphatidic acid, a novel lipid mediator. Biochim Biophys Acta. 1994;1198:185–196. doi: 10.1016/0304-419x(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Tigyi G. Physiological responses to lysophosphatidic acid and related glycero-phospholipids. Prostaglandins. 2001;64:47–62. doi: 10.1016/s0090-6980(01)00107-1. [DOI] [PubMed] [Google Scholar]
- Xu KP, Yin J, Yu FS. Lysophosphatidic acid promoting corneal epithelial wound healing by transactivation of epidermal growth factor receptor. Invest Ophthalmol Vis Sci. 2007;48:636–643. doi: 10.1167/iovs.06-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malchinkhuu E, Sato K, Horiuchi Y, Mogi C, Ohwada S, Ishiuchi S. Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene. 2005;24:6676–6688. doi: 10.1038/sj.onc.1208805. [DOI] [PubMed] [Google Scholar]
- Cechin SR, Dunkley PR, Rodnight R. Signal transduction mechanisms involved in the proliferation of C6 glioma cells induced by lysophosphatidic acid. Neurochem Res. 2005;30:603–611. doi: 10.1007/s11064-005-2747-4. [DOI] [PubMed] [Google Scholar]
- Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DA, Du H, Jaggar JH, Brindley DN, Tigyi GJ, Watsky MA. Injury-elicited differential transcriptional regulation of phospholipid growth factor receptors in the cornea. Am J Physiol Cell Physiol. 2002;283:C1646–C1654. doi: 10.1152/ajpcell.00323.2002. [DOI] [PubMed] [Google Scholar]
- Valtink M, Gruschwitz R, Funk RH, Engelmann K. Two clonal cell lines of immortalized human corneal endothelial cells show either differentiated or precursor cell characteristics. Cells Tissues Organs. 2008;187:286–294. doi: 10.1159/000113406. [DOI] [PubMed] [Google Scholar]
- Senoo T, Obara Y, Joyce NC. EDTA: a promoter of proliferation in human corneal endothelium. Invest Ophthalmol Vis Sci. 2000;41:2930–2935. [PubMed] [Google Scholar]
- Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell. 2014;158:157–170. doi: 10.1016/j.cell.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol. 2012;19:955–962. doi: 10.1016/j.chembiol.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–881. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- Mills GB, Eder A, Fang X, Hasegawa Y, Mao M, Lu Y. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat Res. 2002;107:259–283. doi: 10.1007/978-1-4757-3587-1_12. [DOI] [PubMed] [Google Scholar]
- Ochiai S, Furuta D, Sugita K, Taniura H, Fujita N. GPR87 mediates lysophosphatidic acid-induced colony dispersal in A431 cells. Eur J Pharmacol. 2013;715:15–20. doi: 10.1016/j.ejphar.2013.06.029. [DOI] [PubMed] [Google Scholar]
- Meyer zu Heringdorf D, Jakobs KH. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta. 2007;1768:923–940. doi: 10.1016/j.bbamem.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Fukushima N, Kimura Y, Chun J. A single receptor encoded by vzg-1/lpA1/edg-2 couples to G proteins and mediates multiple cellular responses to lysophosphatidic acid. Proc Natl Acad Sci USA. 1998;95:6151–6156. doi: 10.1073/pnas.95.11.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong GO, Shin SH, Seo EJ, Kwon YW, Heo SC, Kim KH. TAZ mediates lysophosphatidic acid-induced migration and proliferation of epithelial ovarian cancer cells. Cell Physiol Biochem. 2013;32:253–263. doi: 10.1159/000354434. [DOI] [PubMed] [Google Scholar]
- Okumura N, Ueno M, Koizumi N, Sakamoto Y, Hirata K, Hamuro J. Enhancement on primate corneal endothelial cell survival in vitro by a ROCK inhibitor. Invest Ophthalmol Vis Sci. 2009;50:3680–3687. doi: 10.1167/iovs.08-2634. [DOI] [PubMed] [Google Scholar]
- Koizumi N, Okumura N, Kinoshita S. Author response: human corneal endothelium regeneration: effect of ROCK Inhibitor. Invest Ophthalmol Vis Sci. 2013;54:5594–5595. doi: 10.1167/iovs.13-12813. [DOI] [PubMed] [Google Scholar]
- Pipparelli A, Arsenijevic Y, Thuret G, Gain P, Nicolas M, Majo F. ROCK inhibitor enhances adhesion and wound healing of human corneal endothelial cells. PLoS One. 2013;8:e62095. doi: 10.1371/journal.pone.0062095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura N, Nakano S, Kay EP, Numata R, Ota A, Sowa Y. Involvement of cyclin D and p27 in cell proliferation mediated by ROCK inhibitors Y-27632 and Y-39983 during corneal endothelium wound healing. Invest Ophthalmol Vis Sci. 2014;55:318–329. doi: 10.1167/iovs.13-12225. [DOI] [PubMed] [Google Scholar]
- Li W, Sabater AL, Chen YT, Hayashida Y, Chen SY, He H. A novel method of isolation, preservation, and expansion of human corneal endothelial cells. Invest Ophthalmol Vis Sci. 2007;48:614–620. doi: 10.1167/iovs.06-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.