

Abstract
Accumulation of the nerve growth factor precursor (proNGF) and its receptor p75NTR have been associated with several neurodegenerative diseases in both brain and retina. However, whether proNGF contributes to microvascular degeneration remain unexplored. This study seeks to investigate the mechanism by which proNGF/p75NTR induce endothelial cell (EC) death and development of acellular capillaries, a surrogate marker of retinal ischemia. Stable overexpression of the cleavage-resistant proNGF and molecular silencing of p75NTR were utilized in human retinal EC and rat retinas in vivo. Stable overexpression of proNGF decreased NGF levels and induced retinal vascular cell death evident by 1.9-fold increase in acellular capillaries and activation of JNK and cleaved-PARP that were mitigated by p75NTRshRNA. In vitro, overexpression of proNGF did not alter TNF-α level, reduced NGF, however induced EC apoptosis evident by activation of JNK and p38 MAPK, cleaved-PARP. Silencing p75NTR using siRNA restored expression of NGF and TrkA activation and prevented EC apoptosis. Treatment of EC with human-mutant proNGF induced apoptosis that coincided with marked protein interaction and nuclear translocation of p75NTR and the neurotrophin receptor interacting factor. These effects were abolished by a selective p75NTR antagonist. Therefore, targeting p75NTR represents a potential therapeutic strategy for diseases associated with aberrant expression of proNGF.
Introduction
The retina, a typical neurovascular unit is composed of vascular cells (endothelial and pericyte) that interact with neurons and glia to maintain retinal homeostasis under variant conditions.1 Retinal ischemia gauged as an inadequacy, but not essentially a complete block of blood flow can result in failure to meet the high energy demand of retina cells. Central and branched retinal vein occlusion and diabetic retinopathy are the most well-defined retinal microangiopathy diseases in adults.2 Due to lack of effective treatments, retinal ischemia remains a common cause of visual impairment and blindness in the working age. Therefore, understanding of the molecular events that govern microvascular degeneration is critical for devising new treatments for such blinding diseases.
Neurons and glia are the main source of neurotrophins, essential factors for differentiation, development, and maturation of neuronal cells.3–6 Although typically studied in relation to their actions on neurons, the role of neurotrophins in maintaining the proper function of the vasculature has gained recent attention. The first discovered neurotrophin, nerve growth factor (NGF), is secreted as a proform NGF (proNGF) that is traditionally cleaved intracellular by furin and extracellular by the matrix metalloproteineases including MMP-7 and -9 into the mature ligand.7 NGF promotes cell survival by preferentially binding the prosurvival receptor tyrosine kinase A (TrkA), while proNGF has high affinity to bind and activate p75NTR, resulting in cell death.3,8 Accumulation of proNGF has been associated with neurodegenerative diseases such as Alzheimer’s,9 Down’s syndrome,10 and retinal neurodegenerative diseases.11,12 Our group was the first to demonstrate that diabetes-induced oxidative milieu impairs MMP-7 activity and maturation of NGF, resulting in accumulation of its precursor, proNGF, in clinical and experimental diabetes.13–15 Next, we attempted to mimic accumulation of proNGF via an artificial model of overexpressing cleavage-resistant proNGF in rat retina to examine molecular events triggered by proNGF apart from diabetic milieu.16–18 These studies elucidated that proNGF overexpression causes glial activation, retinal inflammation, and neurodegeneration.
Deleterious effects of the proNGF/p75NTR pathway on retinal neurodegeneration have been shown to occur via direct action on neurons16 or indirectly via paracrine stimulation of proinflammatory mediators in Müller cells.12,16,17 While the role of proNGF/p75NTR in mediating neuronal cell death is well-documented, the extent and mechanism by which proNGF/p75NTR contributes to retinal endothelial cell (EC) death in vitro and development of acellular capillaries in vivo remains unexplored. P75NTR lacks intrinsic kinase activity; instead, p75NTR undergoes two-step proteolytic cleavage by α- and γ-secretase to release the intracellular domain (p75ICD) that can recruit different protein adaptors to dictate specific signaling pathways.19,20 In neurons, a number of interacting proteins have been identified that can bind the p75ICD in the cytosol and activate proapoptotic signaling including neurotrophin receptor interacting factor (NRIF)21 and tumor necrosis factor receptor associated factor-6 (TRAF6).22 The significance of NRIF has been established based on phenotypic similarities between NRIF–/– and p75NTR–/– mice23 and on its essential role in p75NTR-mediated apoptosis in the developing mouse retina.24 Nevertheless, the impact of modulating expression p75NTR on EC death and the crosstalk between p75NTR and TrkA in EC remain largely unexplored.
The aim of this study was to elucidate the molecular mechanisms by which p75NTR contributes to proNGF-induced acellular capillary formation, a hallmark of retinal ischemia. A comprehensive approach using stable overexpression of cleavage-resistant proNGF, and pharmacological and molecular modulators of p75NTR expression and activity were used to probe the mechanisms in vivo and in vitro. The results demonstrate protective effect of modulating p75NTR expression at two levels: by inhibiting proapoptotic action of proNGF/p75NTR and by restoring endogenous survival pathways of NGF/TrkA.
Results
Silencing p75NTR prevents proNGF accumulation and restores NGF levels in rat retina
To examine the contribution of proNGF to retinal microvascular degeneration, we used our established protocol of overexpressing cleavage-resistant proNGF in rat retina.18 Before electroporation, rats received single intraocular injection of one of the following: pGFP plus scrambled shRNA (GFP+Scr), pGFP plus short hairpin RNA (shRNA) against p75NTR (GFP+shRNA); pGFP-proNGF123, a cleavage-resistant form of proNGF plus scrambled shRNA (proNGF+Scr); pGFP-proNGF123 plus shRNA against p75NTR (proNGF+shRNA). After 6-weeks, retinas were collected and examined to verify proNGF and p75NTR expression. As shown in Figure 1a, overexpression of proNGF induced expression of both total p75NTR and its intracellular domain (p75ICD) compared with GFP-control. A 2 × 2 analysis showed a significant interaction between proNGF overexpression and silencing p75NTR. Silencing p75NTR with shRNA prevented the increase in p75NTR and p75ICD expression in proNGF group but not in GFP controls (Figure 1b,c). Silencing p75NTR mitigated the increase in proNGF level in the proNGF-overexpressing group (proNGF+shRNA) comparable to GFP-controls (Figure 1d). We next examined impact of proNGF overexpression on NGF levels in rat retina. A 2 × 2 analysis showed a significant interaction between proNGF overexpression and silencing p75NTR. As shown in Figure 1e, proNGF overexpression resulted in significant decreases in NGF levels compared with GFP-control similar to our prior report.18 Silencing p75NTR with shRNA significantly increased NGF levels in proNGF overexpressing retina (Figure 1e). However, silencing p75NTR with shRNA significantly reduced NGF levels in GFP+p75shRNA compared with GFP+Scrambled controls.
Figure 1.

Silencing p75NTR prevents proNGF accumulation and restores NGF levels in rat retina. Rats received single intraocular injection of one of the following: pGFP plus scrambled shRNA (GFP+Scr), pGFP plus shRNA against p75NTR (GFP+shRNA); pGFP-proNGF123, a cleavage-resistant form of proNGF plus scrambled shRNA (proNGF+Scr); pGFP-proNGF123 plus shRNA against p75NTR (proNGF+shRNA). Retinas were lysed after 6 weeks. (a) Representative western blot image and (b,c) statistical analysis of total p75NTR (75 kDa) and its intracellular domain (ICD, 25 kDa) normalized to actin. A 2 × 2 analysis showed a significant interaction between proNGF overexpression and silencing p75NTR. Overexpression of proNGF induced expression of both total p75NTR and p75ICD compared with GFP. Silencing p75NTR with shRNA prevented the increase in p75NTR and p75ICD expression in proNGF group but not in GFP group. n = 4–5 per group, *P < 0.05 versus other groups. (d) Representative western blot image and statistical analysis of proNGF expression normalized to actin showing significant increases in proNGF in proNGF overexpressing group that were mitigated by silencing p75NTR compared with GFP controls. (e) Representative western blot images and statistical analysis of NGF expression normalized to actin. A 2 × 2 analysis showed a significant interaction between proNGF overexpression and silencing p75NTR. ProNGF overexpression resulted in significant decreases in NGF levels compared with GFP. Silencing p75NTR with shRNA significantly reduced NGF levels in GFP-controls and restored NGF levels in proNGF overexpressing retina. Results presented as mean ± SD. n = 4–5 per group. *P < 0.05 versus other groups.
Silencing p75NTR mitigates proNGF-induced retinal acellular capillaries formation
Prior work showed that proNGF overexpression can induce formation of acellular capillaries,18 a surrogate marker for retinal microangiopathy.25 To dissect the contribution of p75NTR in proNGF-mediated acellular capillary formation, p75NTR expression was silenced using single intravitreal delivery of lentiviral particles containing shRNA against p75NTR. Representative images of periodic acid–Schiff and hematoxylin stained, trypsin-digested, and flat-mounted retinas show acellular capillaries (arrows) identified as capillary-sized vessel tubes having no nuclei anywhere along their length (see typical example in Figure 2f). 2 × 2 way analysis showed interaction between proNGF overexpression and silencing p75NTR. Overexpression of GFP-proNGF induced significant (1.9-fold) increase in acellular capillaries compared with the control GFP-plasmid (Figure 2e). Silencing expression of p75NTR using shRNA mitigated proNGF-induced acellular-capillary formation but did not alter the number in GFP-plasmid control group.
Figure 2.

Silencing p75NTR mitigates acellular capillaries in proNGF-overexpression model. After 6 weeks of intraocular injection, retina vasculature was isolated using trypsin digest and stained with periodic acid–Schiff hematoxylin showing acellular capillaries. Representative images of trypsin digested retinas are shown for (a) GFP+Scr, (b) GFP+p75shRNA, (c) proNGF+Scr, and (d) proNGF+p75shRNA. Red arrows indicate acellular capillaries. (e) A 2 × 2 analysis showed a significant interaction between proNGF overexpression and silencing p75NTR. Overexpression of proNGF induced a significant increase of acellular capillaries compared with GFP-controls. These effects were mitigated by silencing of p75NTR. Results presented as mean ± SD. n = 6 per group. *P < 0.05 versus other groups. (f) A typical acellular capillary identified as capillary-sized vessel tubes having no nuclei anywhere along their length.
Silencing p75NTR mitigates proNGF-induced apoptotic markers in vivo
ProNGF-induced vascular cell death was also verified by performing TUNEL-staining on digested and periodic acid–Schiff and hematoxylin stained retina vasculature (trypsinized retinas). As shown in (Figure 3a–d), overexpression of GFP-proNGF induced numerous TUNEL-positive cells in retinal vasculature compared with GFP-control. This observation prompted us to investigate the impact of proNGF/p75NTR expression on activation of apoptotic signals in retina lysate. 2 × 2 analysis showed interaction of proNGF overexpression and silencing p75NTR expression. Overexpression of GFP-proNGF induced twofold increase in the phosphorylation of the stress kinase C-Jun N-terminal kinase (JNK; Figure 3e) and 1.6-fold in cleaved- PolyAdp-Ribose Polymerase (PARP; Figure 3f). Silencing p75NTR prevented the proNGF-induced numerous TUNEL-positive nuclei (Figure 3d) and suppressed activation of apoptotic signals suggesting that p75NTR is a critical mediator of proNGF-induced capillary degeneration.
Figure 3.

Silencing p75NTR suppresses proNGF-induced vascular cell death and apoptotic marker in rat retina. Representative images of trypsin-digested retinas stained with TUNEL (green) and counterstained with DAPI (blue) are shown for (a) GFP+Scr, (b) GFP+p75shRNA, (c) proNGF+Scr, and (d) proNGF+p75shRNA. TUNEL assay showed several positive cells in proNGF-overexpressing retinas, suggesting that capillary degradation occurs through apoptosis. (e) Representative western blots and statistical analysis of pJNK and JNK and (f) cleaved-PARP normalized to actin. 2 × 2 analysis showed interaction between proNGF expression and silencing p75NTR. Overexpression of proNGF induced significant activation of JNK and cleavage of PARP compared with GFP-controls. These effects were mitigated by p75shRNA but not scrambled. Results presented as mean ± SD. n = 4–6 per group. *P < 0.05 versus other groups.
Expression of proNGF is dependent on p75NTR in retinal endothelial cells
Overexpression of proNGF has been shown to induce triad of events in the retina including glial inflammation and neurodegeneration that can contribute to microvascular degeneration. To dissect the detrimental effects of proNGF in vasculature, we used proNGF overexpression model in human retinal EC. ECs were transduced with one of the followings: pGFP plus scrambled siRNA (GFP+Scr), pGFP plus siRNA against p75NTR (GFP+siRNA); pGFP-proNGF123, plus scrambled siRNA (proNGF+Scr); pGFP-proNGF123 plus siRNA against p75NTR (proNGF+siRNA) and cell lysate and supernatant were collected after 48 hours. 2 × 2 analysis showed interaction between proNGF overexpression and silencing p75NTR. Silencing p75NTR expression was first verified by in both GFP-controls and proNGF groups (Figure 4a,b). Silencing p75NTR downregulated proNGF protein expression back to GFP controls in EC (Figure 4a,c). We have verified that the proNGF detected in EC is expressed mainly by the cleavage-resistant plasmid. As shown in Supplementary Figure S1a, PCR analysis using mouse proNGF primers showed 4000-fold increase in the proNGF-GFP group that was significantly reduced by silencing p75NTR using siRNA. These results demonstrated that proNGF expression is p75NTR dependent in retinal EC. Moreover, EC cultures were treated with an inhibitor for Furin, the main endogenous protease responsible for intracellular cleavage of proNGF to NGF. As shown in Supplementary Figure S1b, inhibiting Furin did not result in further increase of proNGF levels in EC cultures transduced with proNGF alone or proNGF with p75siRNA. Interestingly, furin inhibitor significantly increased accumulation of proNGF only in GFP-controls, suggesting that the proNGF protein is mainly due to translation of the mouse proNGF plasmid and not from endogenous human proNGF.
Figure 4.

Expression of proNGF is dependent on p75NTR in EC. Human retinal ECs were transduced with one of the followings: pGFP plus scrambled siRNA (GFP+Scr), pGFP plus siRNA against p75NTR (GFP+siRNA); pGFP-proNGF123, a mutant cleavage resistant form of proNGF, plus scrambled siRNA (proNGF+Scr); pGFP-proNGF123 plus siRNA against p75NTR (proNGF+siRNA), and cell lysate and supernatant were collected after 48 hours. (a,b) Representative western blots statistical analysis of total p75NTR expression normalized to actin. (a,c) Representative western blots and statistical analysis of proNGF expression normalized to actin. 2 × 2 analysis showed interaction between proNGF overexpression and silencing p75NTR. Silencing p75NTR significantly reduced its expression in both GFP-controls and proNGF-groups. Results showed that overexpression of proNGF induced p75NTR expression and that silencing p75NTR downregulated proNGF expression in EC. Results presented as mean ± SD. n = 5–6 per group. *P < 0.05 versus other groups. (d) Representative western blots and statistical analysis of NGF expression normalized to actin in EC. (e) Representative western blots and statistical analysis of TrkA activation normalized to total TrkA. 2 × 2 analysis demonstrated interaction between proNGF overexpression and silencing p75NTR. Overexpression of proNGF resulted in significant decreases in NGF levels and activation of its TrkA receptor compared with GFP-controls. Silencing p75NTR expression increased NGF levels and enhanced TrkA activation in proNGF overexpressing cells. Results are expressed as mean ± SD. n = 5 per group. *P < 0.05 versus other groups.
Overexpression of proNGF significantly reduces NGF level and activation of TrkA in EC
NGF is the mature ligand and well-known survival factor for both neurons and vasculature. In EC, overexpression of proNGF resulted in 75% reduction in NGF levels compared with GFP-controls (Figure 4d). The decision of survival or death may be dictated by the ratio of p75NTR and TrkA receptor.26 Overexpression of proNGF in EC resulted in 60% decrease in TrkA phosphorylation, compared with GFP-controls (Figure 4e). Silencing p75NTR increased NGF levels and TrkA activation back to normal levels in proNGF overexpressing cells but not in GFP-controls.
Overexpression of proNGF activates p75NTR-mediated apoptotic signal in EC
We next investigated the impact of proNGF/p75NTR expression on activation of apoptotic signals in EC. In comparison to GFP-controls, proNGF overexpression induced twofold phosphorylation of the stress kinase JNK (Figure 5a), 1.7-fold in activation of p38MAPK (Figure 5b) and 2.2-fold in cleaved-PARP (Figure 5c). 2 × 2 analysis showed interaction between proNGF overexpression and silencing p75NTR. Silencing p75NTR completely mitigated proNGF-mediated apoptotic signal in EC and have modest decreases in GFP-controls.
Figure 5.

Silencing p75NTR suppresses proNGF-induced apoptotic markers in EC. Representative western blots and statistical analysis of (a) pJNK, total JNK, (b) pP38MAPK, total p38MAPK, and (c) cleaved-PARP (C-PARP), actin in EC lysates. 2 × 2 way analysis showed interaction between proNGF overexpression and silencing p75NTR. Overexpression of proNGF induced EC apoptosis evident by activation of JNK, p38MAPK, and cleavage of PARP. These effects were blunted by silencing p75NTR. Results presented as mean ± SD. n = 4–6 per group. *P < 0.05 versus other groups.
Overexpression of proNGF does not exert proinflammatory effect in EC
We and others have shown that proNGF overexpression caused production and release of TNF-α in glial Müller cells.12,17 To explore whether proNGF can be proinflammatory in EC, we examined TNF-α expression as well as release of TNF-α and IL-1β in EC supernatant. Overexpression of proNGF in EC did not result in significant changes in release of proNGF (Figure 6a,b) TNF-α (Figure 6a,c) IL1β (data not shown) or TNF-α mRNA expression (Figure 6d). These results suggest that unlike the proinflammatory effect in glial Müller cells, proNGF does not induce a proinflammatory action in EC.
Figure 6.

Overexpression of proNGF does not exert proinflammatory effect in EC. (a–c) Representative western blots statistical analysis of proNGF and TNF-α of concentrated EC-supernatants normalized to BSA-IgG. (d) Statistical analysis of mRNA expression of TNF-α normlaized to 18S. 2 × 2 analysis showed no effect of proNGF overexpression or silencing p75NTR on altering levels of TNF-α expression in EC lysates or released proNGF or TNF-α into supernatant of EC cultures. Results presented as mean ± SD. n = 4 per group.
ProNGF induces p75NTR-dependent nuclear translocation of NRIF-p75ICD in EC
To elucidate how proNGF/p75NTR activates JNK apoptotic signal in EC, P75NTR, we switched to using human mutant (hm-proNGF)12 instead of proNGF-plasmid electroporation. We first verified the apoptotic effects of hm-proNGF on human EC. As shown in Supplementary Figure S2, hm-proNGF induced cell death assessed by a death and life assay. Interestingly, 10, 20, and 50 ng/ml but not 100 ng/ml of hm-proNGF induced significant EC death compared with control. Therefore, we selected 50 ng/ml to probe the apoptotic signal in EC. As shown in Figure 7a, hm-proNGF (50 ng/ml) induced 2.5-fold in caspase-activity compared with controls that was blunted by a small peptide and a selective antagonist (A) against p75NTR receptor.17,27 To explore the mechanism by which proNGF/p75NTR mediates JNK-apoptotic signals in EC, we investigated whether increases in p75ICD in the cytosol can recruit the DNA binding protein, neurotrophin interacting factor (NRIF), resulting in NRIF-p75ICD nuclear translocation. Immunocytochemistry clearly showed that hm-proNGF (50 ng/ml) induced redistribution of NRIF (green) from the cytoplasm and perinuclear to be in the nuclei (blue) of EC after 48 hours (Figure 7b). Treatment with hm-proNGF also induced translocation of p75NTR (red), NRIF (green) to the nuclie (blue) (Figure 7c). Nuclear translocation of NRIF was blocked by the specific p75NTR antagonist (A, 20 μmol/l). Of note, the antibody used for immunocytochemistry can recognize both total p75NTR and p75ICD. To further characterize whether the total p75NTR receptor or the p75ICD is translocated to the nuclei, we attempted to inhibit γ-secretase-mediated cleavage of p75NTR using compound E or DAPT. However, results were not conclusive due to the harsh effects of vehicle used (dimethylsulfoxide and ethanol) for extended time (48 hours of exposure). Next, we examined NRIF and p75ICD protein interaction using immunoprecipitation. As shown in Figure 7d, proNGF markedly increased protein interactions between p75ICD (detected ~17kDa) and NRIF (detected ~90 kDa) compared with controls. Interestingly, the NRIF band had additional band at 100 kDa that was detected in all groups. The interaction between NRIF and p75ICD was abolished by a pharmacological antagonist of p75NTR. These results demonstrate that proNGF-induced NRIF nuclear translocation plays an essential role in executing p75NTR-mediated EC death.
Figure 7.

ProNGF causes protein interaction and nuclear translocation of p75NTR and neurotrophin interacting factor (NRIF). Human retinal endothelial cells were treated with human mutant (hm-proNGF, 50 ng/ml) in the presence or absence of a selective p75NTR antagonist (A, 20 μmol/l) for 48 hours. (a) Cell death was assessed by measuring activity of caspase enzyme and the results were presented as mean ± SD. n = 3–5 per group. *P < 0.05 versus other groups. (b) Representative images of immunolocalization showing that hm-proNGF induced nuclear translocation of NRIF (green) within the nucleus (blue) that was blocked by p75NTR antagonist (A) (proNGF+A). (c) Enlarged micrographs of EC treated with hm-proNGF showing colocalization of p75NTR (red), NRIF (green) within the nuclei (blue). Similar results were observed in two independent experiments. (d) Representative blots and statistical analysis showing that hmproNGF induced protein association between NRIF (90 kDa) and p75ICD (17 kDa) in EC lysate immunoprecipitated with anti-p75NTR and immunoblotted by anti-NRIF. Additional band for NRIF was detected at 100 kDa. Results presented as mean ± SD. n = 4 per group. *P < 0.05 versus all groups.
Discussion
The main findings of this study are: (i) Similar to neurodegenerative disease, overexpression of proNGF shift the balance to less neurotrophic support evident by less NGF and impaired TrkA activation. Silencing p75NTR expression restored balance by decreasing proNGF/p75NTR levels and increasing NGF/TrkA in proNGF-overexpression models in vitro and in vivo (Figures 1 and 4). (ii) ProNGF induced NRIF nuclear translocation and EC apoptosis in a p75NTR-dependant manner (Figures 5 and 7). (iii) Modulating p75NTR expression prevented proNGF-induced development of acellular capillaries and hence retinal ischemia in vivo (Figures 2 and 3). We believe that this is the first report dissecting the contribution of upregulated proNGF/p75NTR and impaired NGF/TrkA in mediating EC death. A scheme of the proposed molecular mechanism and findings is depicted in (Figure 8).
Figure 8.
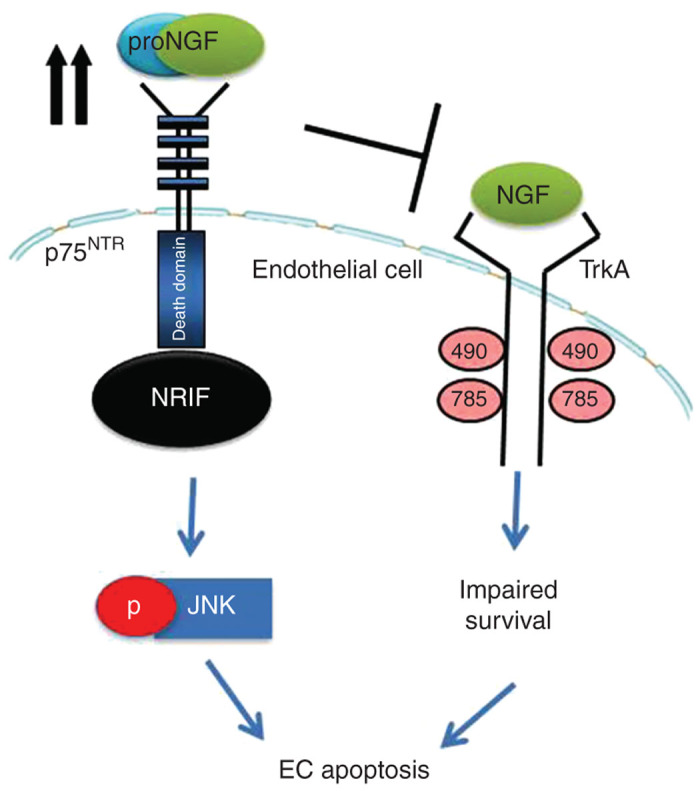
Schematic diagram depicting the proposed role of proNGF/p75NTR in mediating EC death and development of acellular capillaries. Overexpression of proNGF impairs endogenous NGF/TrkA survival signaling pathway and activates p75NTR-mediated apoptotic signal. ProNGF triggers interaction of intracellular domain of p75NTR (p75ICD) and the transcriptional factor neurotrophin interacting factor (NRIF) to activate JNK apoptotic signal. Targeting p75NTR prevents the apoptotic signal and stimulates endogenous cell survival.
Neurotrophins are traditionally perceived as essential growth factors for differentiation, development, maturation, and regulation of cell death of retinal neurons.3–6 However, over the past two decades, a growing body of evidence identified the actions of neurotrophins on other cell types in the retina.1 Our group has been leading studies that demonstrated the multiple and complex ways by which diabetes alters homeostasis of NGF maturation resulting in upregulation of proNGF/p75NTR receptor and diminishing NGF/TrkA receptor activation.13,15,17 Here, we examined the impact of accumulated proNGF/p75NTR on EC apoptosis and acellular capillary formation using cleavage-resistant proNGF-overexpression model. Our data showed that silencing p75NTR expression prevented proNGF-induced acellular capillary formation in vivo (Figure 2) as well as EC death in vitro (Figures 5 and 7). These effects were associated with restoring the balance of proNGF and NGF ratio back to the control levels, which are essential for proper retinal function (Figures 1 and 4). We next examined whether proNGF activates apoptosis via direct activation of the apoptotic pathway in EC and/or via release of proinflammatory mediators that can contribute to EC death. We and others have demonstrated proinflammatory action of proNGF in Müller cells via a p75NTR-dependent pathway.12,16–18 Interestingly, overexpression of proNGF in EC did not induce significant increases in expression of TNF-α or release of proNGF, TNF-α, and IL-1β compared with controls (Figure 6), suggesting that EC apoptosis was mediated through direct activation of proNGF/p75NTR signaling at least within in vitro setting. In support of this concept, prior studies have demonstrated a direct apoptotic effect of proNGF and p75NTR in brain EC28,29 and in vascular cells of the ischemic heart.30 However, the contribution of paracrine action of retinal neuro- and glial inflammation cannot be ruled out in the in vivo setting.
The apoptotic p75NTR signal requires phosphorylation of JNK and stress kinase p38MAPK.8,31–33 Here, we demonstrated that proNGF overexpression induced activation of JNK and p38MAPK as well as its downstream apoptotic markers caspase activation and cleaved-PARP in whole retina and EC (Figures 3 and 5). These effects were mitigated by silencing p75NTR expression using shRNA in vivo and siRNA in vitro. While prior studies documented the clear apoptotic effect of proNGF or p75NTR in neurons,23,31,33–36 this is the first report that demonstrated activation of p75NTR-dependent JNK activation in EC in response to proNGF. The use of human mutant (hm-proNGF) resulted also in EC death assessed by death/life assay and caspase activity. We used 50 ng/ml as this level induced maximum cell death (Supplementary Figure S2). A high dose of proNGF (100 ng/ml) had less cytotoxic effect possibly due to activation of other receptors including TrkA resulting in activation of survival signal.
P75NTR, like Notch receptors, undergoes γ-secretase-intramembranal proteolysis to liberate the intracellular domain (ICD), which can recruit several apoptotic intracellular binding proteins such as NRIF (reviewed in refs. 20,37). NRIF has been described as an essential mediator of apoptotic signals by p75NTR in neurons.21,38 To explore whether proNGF-mediated JNK activation and EC apoptosis involve interaction of p75NTR and NRIF, we switched to using human mutant (hm-proNGF) instead of proNGF-plasmid electroporation. Treatment of EC with hm-proNGF caused nuclear translocation of DNA binding NRIF as evident by localization of NRIF (green) and p75NTR (red) in nuclei of proNGF-treated EC, but not in controls or proNGF+A (Figure 7). Whether cleavage of p75NTR and release of p75ICD is a necessary step before nuclear translocation remains to be explored. One of the barriers to answer this question is the reliability of selective inhibitors of γ-secretase enzyme and their limited solubility in aqueous vehicles that affected cell viability. Several proteins have been shown to accumulate into the nucleolus during cell growth or death. Of note, the recruitment of NRIF and p75NTR in the nucleolus may represent an advanced DNA fragmentation and a stress response triggered by proNGF-apoptotic signal in endothelial cells. Furthermore, immunoprecipitation showed strong interaction between NRIF and p75ICD in response to proNGF, an effect that was abolished by selective antagonist of p75NTR, inhibitor A (Figure 7). Interestingly, the NRIF band was detected higher at 100 kDa instead of 90 kDa, which suggests that NRIF may undergo protein modification such as ubiquitination, as proposed by prior studies.38,39 Our findings suggest that NRIF/p75ICD translocate as a complex to mediate the cell death process in EC. Similarly, previous reports showed nuclear translocation of NRIF in neuronal cells.23,26,40 The significance of nuclear translocation p75NTR to directly activate JNK or other pathways warrant investigation.
While proNGF can promote neuronal cell death by binding p75NTR, NGF mediates survival by binding TrkA.15 Similar to prior findings,18 overexpression of cleavage-resistant proNGF significantly reduced mature NGF levels in vivo and in vitro in EC (Figures 1 and 4). Interestingly, these effects were associated with significant impairment of TrkA receptor phosphorylation (Figure 4). Supplementation of exogenous NGF has been shown to be neuro- and vascular protective in experimental models.41–43 However, the distribution and actual level of NGF in the retina after topical administration remains controversial. In this study, we took a different approach by suppressing the proNGF/p75NTR signal which stimulated endogenous NGF/TrkA expression that together improved cell survival and preserved the retinal vasculature. Silencing p75NTR expression restored NGF levels and TrkA receptor phosphorylation in proNGF-overexpressing retinas and in EC (Figure 4). We also examined whether observed increases in NGF were due to endogenous production by EC as a protective mechanism or due to improved processing and cleavage of proNGF-plasmid expression. Hence, we treated EC with an inhibitor for the intracellular protease “furin” and results showed that inhibiting furin resulted in accumulation of proNGF levels only in GFP-controls but not in GFP-proNGF expressing cells (Supplementary Figure S1b). These results confirmed that the detected proNGF protein is primarily coming from the exogenous non-cleavable proNGF-plasmid. Overexpression of proNGF reduced NGF levels in vivo and in EC probably due to feedback inhibition at the transcription level (see Supplementary Figure S1a). These results came similar to our prior findings using proNGF overexpression model.18 In support, our previous studies using diabetic model showed that deletion of p75NTR resulted in restoring the ratio between proNGF and NGF that was associated with preserving retinal barrier function.17 Studies are warranted to further elucidate the molecular events involved in how silencing p75NTR restored NGF levels.
Accumulation of proNGF has been documented in neurodegenerative diseases in the brain and retina including Alzheimer’s,9 Down’s syndrome,10 as well as models of optic neuropathy, light damage and diabetic retinopathy.11,12,15,16 In this study, we demonstrated unexplored role of p75NTR in proNGF-mediated EC cell apoptosis and subsequent acellular capillary formation, beyond its known role in neurodegeneration. These findings support the notion that targeting p75NTR expression represents a viable therapeutic option in diseases of the neurovascular unit. Modulating expression of p75NTR can protect against proNGF mediated-EC death at two levels by inhibiting apoptotic effect of proNGF and by stimulating NGF expression and activating the TrkA survival pathway to mitigate retinal cell death and acellular capillary formation.
Materials and Methods
Animals
All animal procedures were performed in accordance with Association for Research in Vision and Ophthalmology statement for use of animals in ophthalmic and vision research, and Charlie Norwood VA Medical Center Animal Care and Use Committee (ACORP#13–01–055).
Overexpression of the cleavage-resistant proNGF in rat retinas
Four sets of, a total of 48 animals, Male Sprague-Dawley rats (~250 g body weight) were purchased from Harlan Laboratories (Indianapolis, IN) and randomly assigned to four groups. Stable overexpression of proNGF was achieved via intravitreal injection of proNGF123 plasmid and electroporation as described previously by our group.18 Animals received either lentiviral particles containing scrambled or shRNA against p75NTR (5,000 infectious units/eye) were obtained from Santa Cruz BioTechnology (Dallas, TX). Animal groups included intravitreal injection of pGFP plasmid (5 μg/eye) plus lentiviral particles containing scrambled shRNA (5,000 infectious units/eye); pGFP plasmid plus shRNA against p75NTR; pGFP-proNGF123, a cleavage resistant proNGF construct, plus scrambled shRNA; or pGFP-proNGF123 plus shRNA against p75NTR. Rats were sacrificed after 6 weeks, and eyes were enucleated for further investigations.
Isolation of retinal vasculature and determination of acellular capillaries
The retinal vasculature was isolated as described previously.44 Freshly enucleated eyes were fixed with 2% paraformaldehyde overnight. Retinal cups were dissected, washed in phosphate-buffered saline, then incubated with 3% Difco-trypsin 250 (BD Biosciences, San Jose, CA) in 25 mmol/l Tris buffer, pH 8, at 37 °C for 2 hours. Vitreous and nonvascular cells were gently removed from the vasculature, which was soaked in several washes of 0.5% Triton X-100 to remove the neuronal retina. Trypsin-digested retinas were stained with periodic acid–Schiff and hematoxylin. Numbers of acellular capillaries were quantified in six different areas of the mid-retina under the microscope (×20) in a masked manner by two different researchers. Acellular capillaries were identified as capillary-sized blood vessel tubes with no nuclei along their length. TUNEL assay was performed using ApopTaG in whole flat-mounted retinas that went through tryptic digest as described previously by our group.44
Cell culture
All human retinal endothelial cell (EC) studies were in accordance with Association for Research in Vision and Ophthalmology and Charlie Norwood Veterans Affairs Medical Center, research and ethics committee. EC and supplies were purchased from Cell Systems Corporations (Kirkland, WA) and VEC Technology (Rensselaer, NY) as described previously.45
Overexpression of proNGF and manipulation of p75NTR in EC culture
Plasmids used to overexpress cleavage-resistant proNGF (0.5 μg/ml) and silencing of p75NTR expression using small interfering RNA siRNA (0.6 μmol/l) were performed as described by our group.31,45 Both the SMARTpool of siRNA specific for p75NTR gene (also known as Ngfr) and its scrambled form were obtained commercially from Dharmacon (Lafayette, CO). The groups included: Control, pGFP alone plus scrambled siRNA (GFP); control-treated, pGFP plus siRNA against p75NTR (GFP-siRNA); proNGF, pGFP-proNGF123 plus scrambled siRNA (proNGF); and proNGF-treated, pGFP-proNGF123 plus siRNA against p75NTR (pro-siRNA). EC transduction was performed using Amaxa nucleofector primary endothelial cells kit (Lonza, Germany). EC cultures were transduced for 6 hours, then left to recover in complete medium for 24 hours before performing experiments. Transfection efficiency was 60–70% for both methods as indicated by number of cells expressing green fluorescent protein (GFP) or FITC-labeled siRNA. Silencing of p75NTR expression was verified by western blot analysis.
Pharmacological treatments
For cell viability experiments, human mutant proNGF (Alomone Labs, Jerusalem, Israel) was used to induce apoptosis in EC. A specific pharmacological antagonist of p75NTR (A) was provided by Dr Uri Saragovi (McGill University, Montreal, QC, Canada) and was used at 20 μmol/l as previously described17 for 48 hours. To examine whether inhibition of proteolytic activity would modify proNGF levels, a furin enzyme inhibitor Decanoyl-Arginyl-Valyl-Lysyl-Arginyl-Chloromethyl Ketone (Millipore, MA) was used at concentration 25 μmol/l, which did not affect cell viability up to 12 hours.46
Western blot assay and immunoprecipitation
Retinas and EC samples (30 μg protein) were homogenized in RIPA buffer and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).13 Antibodies were purchased as follows: anti-proNGF and anti-NGF (Alomone Labs, Jerusalem, Israel), JNK, p-JNK, p-p38, p38MAPK (Santa Cruz Biotechnology), and cleaved-PARP (Cell Signaling, Danvers, MA). Antibodies for p75NTR, p75ICD, and NRIF were kindly provided by Dr Bruce Carter (Department of Biochemistry, Vanderbilt University). Membranes were reprobed with β-actin (Sigma-Aldrich, St Louis, MO) to confirm equal loading. The primary antibody was detected using horseradish peroxidase-conjugated sheep antirabbit antibody (GE Healthcare, Piscataway, NJ) and enhanced chemiluminescence (Pierce, Thermo Fischer Scientific, Waltham, MA). For immunoprecipitation, EC lysates (150 g) were immunoprecipitated with anti-p75 antibody kindly provided by Dr Moses Chao (Skirball Institute of Biomolecular Medicine, New York University). The proteins were separated by SDS-PAGE, transferred to nitrocellulose membrane, and probed with antibody directed against NRIF. Band intensity was quantified using densitometry software (alphEaseFC) and normalized to p75ICD.
Caspase activity assay
This assay was performed as described previously.47 Briefly, 40 μl of 2× reaction buffer followed by 4 μl of 1 mol/l zDEVD-AFC (Enzo Life, Farmingdale, NY) were added to an equal volume of each sample. Cell lysates (10 μg) were incubated at 37 °C for 30 minutes, and caspase activity determined by measuring relative fluorescence units (RFU) at 505 nm following excitation at 400 nm using a Microplate reader BioTek (BioTek Instrument, Winooski, VT).
Death and life cell viability assay
This assay was performed following manufacturer’s protocol (Thermo Fischer Scientific) as described previously by our group.45 ECs were viewed under fluorescent microscope (Axiovert-200; Zeiss, Jena, Germany) with filter 488 nm, green for viable cells, and 568 nm, red nonviable. Death/ life ratio was counted in a masked manner.
Quantitative real-time PCR
The One-Step qRT-PCR kit (Thermo Fischer Scientific) was used to amplify 10 ng retinal mRNA and quantification was performed as described previously.31,45 PCR primers designed to amplify human TNF-α were purchased from Integrated DNA Technologies (Coralville, IA). Quantitative PCR was performed using a Realplex Master cycler (Eppendorf, Germany) using primers (F: 5′CCAGGGACCTCTCTCTAATCA3′; R 5′TCAGCTTGAGGGTTTGCTAC3′). Primers for mouse proNGF are F: 5′TTTGATCGGCGTACAGGCAG3′ and R: 5′ AGTGGAGTCTCGCTGCCTTAAACA3′. Expression of human TNF-α or mouse proNGF was normalized to the 18S level and expressed as relative expression to control.
Immunofluorescence
Immunofluorescence was performed as described previously.48 EC were fixed with 2% PFA and incubated with blocking buffer (phosphate-buffered saline containing 5% goat serum, 0.02% Tween 20) followed by primary antibody (1:200) against antirabbit NRIF (provided by Dr Bruce Carter, Vanderbilt University) and antimouse p75NTR (Abcam, Cambridge, MA) followed by Alexa 488-conjugated goat antirabbit or goat antimouse IgG (1:200, Thermo Fischer Scientific) in blocking buffer. Specimens were covered with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Micrographs of EC were taken using a fluorescent microscope (Axiovert-200) at ×20 and ×40.
Data analysis
Results are expressed as mean ± SD, and the data were evaluated for normality and appropriate transformations were used when necessary. Data was processed for statistical analysis with one-way ANOVA to examine the effect of human-proNGF and p75NTR antagonist in endothelial cells. Two-way ANOVA was used to examine the effect of proNGF gene overexpression (GFP versus proNGF) and silencing p75NTR (scrambled versus shRNA) and their interaction on cell death, mRNA, and protein expression. Two-way ANOVA followed by Bonferroni posttests were used to adjust for the multiple comparisons used to assess significant effects using Prism (Graphpad version 6). Significance, GraphPad software, La Jolla, CA was defined as P < 0.05.
Acknowledgments
This work was supported by grant from EY-0022408, JDRF (2-2008-149), and Vision Discovery Institute to A.B.E.-R. AHA-postdoctoral fellowship to B.A.M. Authors are grateful for Moses Chao and Bruce Carter for providing p75NTR, NRIF antibodies.
A.Y.S. and A.B.E.-R. conceived the hypothesis. A.Y.S., B.A.M., S.M., and A.B.E.-R. performed experiments and data analysis. A.Y.S. and A.B.E.-R. wrote the manuscript. B.A.M. and A.B.E.-R. edited the manuscript. A.Y.S., B.A.M., S.M., and A.B.E.-R. approved final version.
The authors declared no conflict of interest.
References
- Mysona BA, Shanab AY, Elshaer SL, El-Remessy AB. Nerve growth factor in diabetic retinopathy: beyond neurons. Expert Rev Ophthalmol. 2014;9:99–107. doi: 10.1586/17469899.2014.903157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS. Neurotrophins in the developing and regenerating visual system. Histol Histopathol. 1998;13:437–459. doi: 10.14670/HH-13.437. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinle JJ, Granger HJ. Nerve growth factor regulates human choroidal, but not retinal, endothelial cell migration and proliferation. Auton Neurosci. 2003;108:57–62. doi: 10.1016/S1566-0702(03)00151-6. [DOI] [PubMed] [Google Scholar]
- Jadhao CS, Bhatwadekar AD, Jiang Y, Boulton ME, Steinle JJ, Grant MB. Nerve growth factor promotes endothelial progenitor cell-mediated angiogenic responses. Invest Ophthalmol Vis Sci. 2012;53:2030–2037. doi: 10.1167/iovs.11-8430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstead BL. Commentary: regulating proNGF action: multiple targets for therapeutic intervention. Neurotox Res. 2009;16:255–260. doi: 10.1007/s12640-009-9054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Allard S, Leon WC, Pakavathkumar P, Bruno MA, Ribeiro-da-Silva A, Cuello AC. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J Neurosci. 2012;32:2002–2012. doi: 10.1523/JNEUROSCI.1144-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iulita MF, Do Carmo S, Ower AK, Fortress AM, Aguilar LF, Hanna M. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain. 2014;137 Pt 3:860–872. doi: 10.1093/brain/awt372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AM, López-Sánchez N, Martín-Oliva D, de la Villa P, Cuadros MA, Frade JM. Sortilin participates in light-dependent photoreceptor degeneration in vivo. PLoS One. 2012;7:e36243. doi: 10.1371/journal.pone.0036243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR, Di Polo A. ProNGF induces TNFalpha-dependent death of retinal ganglion cells through a p75NTR non-cell-autonomous signaling pathway. Proc Natl Acad Sci USA. 2010;107:3817–3822. doi: 10.1073/pnas.0909276107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB. Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes. 2008;57:889–898. doi: 10.2337/db07-1669. [DOI] [PubMed] [Google Scholar]
- Al-Gayyar MM, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Thioredoxin interacting protein is a novel mediator of retinal inflammation and neurotoxicity. Br J Pharmacol. 2011;164:170–180. doi: 10.1111/j.1476-5381.2011.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali TK, Al-Gayyar MM, Matragoon S, Pillai BA, Abdelsaid MA, Nussbaum JJ. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia. 2011;54:657–668. doi: 10.1007/s00125-010-1935-1. [DOI] [PubMed] [Google Scholar]
- Al-Gayyar MM, Mysona BA, Matragoon S, Abdelsaid MA, El-Azab MF, Shanab AY. Diabetes and overexpression of proNGF cause retinal neurodegeneration via activation of RhoA pathway. PLoS One. 2013;8:e54692. doi: 10.1371/journal.pone.0054692. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mysona BA, Al-Gayyar MM, Matragoon S, Abdelsaid MA, El-Azab MF, Saragovi HU. Modulation of p75(NTR) prevents diabetes- and proNGF-induced retinal inflammation and blood-retina barrier breakdown in mice and rats. Diabetologia. 2013;56:2329–2339. doi: 10.1007/s00125-013-2998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matragoon S, Al-Gayyar MM, Mysona BA, Abdelsaid MA, Pillai BA, Neet KE. Electroporation-mediated gene delivery of cleavage-resistant pro-nerve growth factor causes retinal neuro- and vascular degeneration. Mol Vis. 2012;18:2993–3003. [PMC free article] [PubMed] [Google Scholar]
- Zampieri N, Xu CF, Neubert TA, Chao MV. Cleavage of p75 neurotrophin receptor by alpha-secretase and gamma-secretase requires specific receptor domains. J Biol Chem. 2005;280:14563–14571. doi: 10.1074/jbc.M412957200. [DOI] [PubMed] [Google Scholar]
- Fortini ME. Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol. 2002;3:673–684. doi: 10.1038/nrm910. [DOI] [PubMed] [Google Scholar]
- Linggi MS, Burke TL, Williams BB, Harrington A, Kraemer R, Hempstead BL. Neurotrophin receptor interacting factor (NRIF) is an essential mediator of apoptotic signaling by the p75 neurotrophin receptor. J Biol Chem. 2005;280:13801–13808. doi: 10.1074/jbc.M410435200. [DOI] [PubMed] [Google Scholar]
- Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL. NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron. 2000;27:279–288. doi: 10.1016/s0896-6273(00)00036-2. [DOI] [PubMed] [Google Scholar]
- Kenchappa RS, Zampieri N, Chao MV, Barker PA, Teng HK, Hempstead BL. Ligand-dependent cleavage of the P75 neurotrophin receptor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neurons. Neuron. 2006;50:219–232. doi: 10.1016/j.neuron.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA. The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J. 1999;18:6050–6061. doi: 10.1093/emboj/18.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Gong B, Hatala DA, Kern TS. Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest Ophthalmol Vis Sci. 2007;48:361–367. doi: 10.1167/iovs.06-0510. [DOI] [PubMed] [Google Scholar]
- Skeldal S, Matusica D, Nykjaer A, Coulson EJ. Proteolytic processing of the p75 neurotrophin receptor: a prerequisite for signalling?: neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR) Bioessays. 2011;33:614–625. doi: 10.1002/bies.201100036. [DOI] [PubMed] [Google Scholar]
- LeSauteur L, Wei L, Gibbs BF, Saragovi HU. Small peptide mimics of nerve growth factor bind TrkA receptors and affect biological responses. J Biol Chem. 1995;270:6564–6569. doi: 10.1074/jbc.270.12.6564. [DOI] [PubMed] [Google Scholar]
- Kim H, Li Q, Hempstead BL, Madri JA. Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem. 2004;279:33538–33546. doi: 10.1074/jbc.M404115200. [DOI] [PubMed] [Google Scholar]
- Caporali A, Pani E, Horrevoets AJ, Kraenkel N, Oikawa A, Sala-Newby GB. Neurotrophin p75 receptor (p75NTR) promotes endothelial cell apoptosis and inhibits angiogenesis: implications for diabetes-induced impaired neovascularization in ischemic limb muscles. Circ Res. 2008;103:e15–e26. doi: 10.1161/CIRCRESAHA.108.177386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siao CJ, Lorentz CU, Kermani P, Marinic T, Carter J, McGrath K. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J Exp Med. 2012;209:2291–2305. doi: 10.1084/jem.20111749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Gayyar MM, Matragoon S, Pillai BA, Ali TK, Abdelsaid MA, El-Remessy AB. Epicatechin blocks pro-nerve growth factor (proNGF)-mediated retinal neurodegeneration via inhibition of p75 neurotrophin receptor expression in a rat model of diabetes [corrected] Diabetologia. 2011;54:669–680. doi: 10.1007/s00125-010-1994-3. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EB, Said F. Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J Neurosci. 2003;23:11373–11381. doi: 10.1523/JNEUROSCI.23-36-11373.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA. Death of injured neurons caused by the precursor of nerve growth factor. Proc Natl Acad Sci USA. 2004;101:5703–5704. doi: 10.1073/pnas.0401374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng KK, Felice S, Kim T, Hempstead BL. Understanding proneurotrophin actions: recent advances and challenges. Dev Neurobiol. 2010;70:350–359. doi: 10.1002/dneu.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geetha T, Kenchappa RS, Wooten MW, Carter BD. TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 2005;24:3859–3868. doi: 10.1038/sj.emboj.7600845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav TS, Wooten MW, Wooten MC. Mining the TRAF6/p62 interactome for a selective ubiquitination motif. BMC Proc. 2011;5 (suppl. 2):S4. doi: 10.1186/1753-6561-5-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volosin M, Trotter C, Cragnolini A, Kenchappa RS, Light M, Hempstead BL. Induction of proneurotrophins and activation of p75NTR-mediated apoptosis via neurotrophin receptor-interacting factor in hippocampal neurons after seizures. J Neurosci. 2008;28:9870–9879. doi: 10.1523/JNEUROSCI.2841-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1:527–534. [PMC free article] [PubMed] [Google Scholar]
- Colafrancesco V, Coassin M, Rossi S, Aloe L. Effect of eye NGF administration on two animal models of retinal ganglion cells degeneration. Ann Ist Super Sanita. 2011;47:284–289. doi: 10.4415/ANN_11_03_08. [DOI] [PubMed] [Google Scholar]
- Mantelli F, Lambiase A, Colafrancesco V, Rocco ML, Macchi I, Aloe L. NGF and VEGF effects on retinal ganglion cell fate: new evidence from an animal model of diabetes. Eur J Ophthalmol. 2014;24:247–253. doi: 10.5301/ejo.5000359. [DOI] [PubMed] [Google Scholar]
- Al-Gayyar MM, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Neurovascular protective effect of FeTPPs in N-methyl-D-aspartate model: similarities to diabetes. Am J Pathol. 2010;177:1187–1197. doi: 10.2353/ajpath.2010.091289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed IN, Hafez SS, Fairaq A, Ergul A, Imig JD, El-Remessy AB. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia. 2014;57:413–423. doi: 10.1007/s00125-013-3101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi M, Lu D, Zhang YQ, Sawada Y, Sasaki T, Kayo T. Upregulatory expression of furin and transforming growth factor-beta by fluid shear stress in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2001;21:785–790. doi: 10.1161/01.atv.21.5.785. [DOI] [PubMed] [Google Scholar]
- el-Remessy AB, Bartoli M, Platt DH, Fulton D, Caldwell RB. Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI 3-kinase tyrosine nitration. J Cell Sci. 2005;118 Pt 1:243–252. doi: 10.1242/jcs.01612. [DOI] [PubMed] [Google Scholar]
- Shanab AY, Nakazawa T, Ryu M, Tanaka Y, Himori N, Taguchi K. Metabolic stress response implicated in diabetic retinopathy: the role of calpain, and the therapeutic impact of calpain inhibitor. Neurobiol Dis. 2012;48:556–567. doi: 10.1016/j.nbd.2012.07.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.