

Abstract
In humans, the vaginal microbiota is thought to be the first line of defense again pathogens including Chlamydia trachomatis. The guinea pig has been extensively used as a model to study chlamydial infection because it shares anatomical and physiological similarities with humans, such as a squamous vaginal epithelium as well as some of the long-term outcomes caused by chlamydial infection. In this study, we aimed to evaluate the guinea pig-C. caviae model of genital infection as a surrogate for studying the role of the vaginal microbiota in the early steps of C. trachomatis infection in humans. We used culture-independent molecular methods to characterize the relative and absolute abundance of bacterial phylotypes in the guinea pig vaginal microbiota in animals non-infected, mock-infected or infected by C. caviae. We showed that the guinea pig and human vaginal microbiotas are of different bacterial composition and abundance. Chlamydia caviae infection had a profound effect on the absolute abundance of bacterial phylotypes but not on the composition of the guinea pig vaginal microbiota. Our findings compromise the validity of the guinea pig-C. caviae model to study the role of the vaginal microbiota during the early steps of sexually transmitted infection.
Keywords: microbiome, model organism, Chlamydia trachomatis, Lactobacillus, vagina
The vaginal microbiota of the guinea pig differs from that of humans and cannot prevent chlamydial infections efficiently.
Graphical Abstract Figure.
The vaginal microbiota of the guinea pig differs from that of humans and cannot prevent chlamydial infections efficiently.
INTRODUCTION
The obligate intracellular bacterial pathogen, Chlamydia trachomatis, causes one of the most common human sexually transmitted infections with more than 1.4 million cases reported in the United States in 2012 (CDC 2014). In women, C. trachomatis can ascend from the lower to the upper genital tract, causing diseases with an inflammatory etiology such as pelvic inflammatory disease, which if left untreated can lead to infertility and ectopic pregnancy (Westrom 1975). The guinea pig is commonly used to model ocular and genital disease upon infection with C. caviae (Mount, Bigazzi and Barron 1972). Although C. caviae exhibits phenotypic differences with C. trachomatis, including resistance to sulfonamides (Gordon and Quan 1972), non-fusing inclusions (Rockey, Fischer and Hackstadt 1996) and a failure to accumulate glycogen (Fan and Jenkin 1970), the genome of C. caviae is similar to that of C. trachomatis in gene order and gene content (Read et al. 2003). The guinea pig-C. caviae model has been used to study sexual transmission from males to females (Rank et al. 2003) as well as the development of disease pathology associated with ascending infection in females (Rank, Batteiger and Soderberg 1990). This model is superior to the available mouse models in several aspects. First, similar to the human female genital tract and in contrast to the murine reproductive tract (Iguchi et al. 1983), the guinea pig vagina is lined with non-keratinized squamous epithelial cells (Durrani et al. 1985) that accumulate glycogen upon estradiol treatment (Rank et al. 1982). Second, the length of the 17-day estrous cycle of the guinea pig (Alkhalaf et al. 1992), although not identical, is closer to that of the human menstrual cycle than that of the mouse (4 days) (Parkes and Brambell 1928). Third, key features of the disease process, such as endometritis (Peipert et al. 1996), salpingitis (Mardh et al. 1977) and the lack of overt tubal disease (Wiesenfeld et al. 2012), are better reproduced in the guinea pig than in the mouse (Darville et al. 1997).
The vaginal microbiota is thought to be the first line of defense against sexually transmitted infections (Spear, St John and Zariffard 2007). Commensal lactic acid-producing Lactobacillus spp. that are present in the human vagina have long been considered the primary protective barrier, creating an acidic environment (pH < 4.5) that reduces colonization by pathogens (Stamey and Timothy 1975; Hanna et al. 1985; Boskey et al. 2001; Stefka et al. 2014). However, recent culture-independent studies have shown that only ∼73% of healthy asymptomatic women have a vaginal microbiota dominated by Lactobacillus spp. (Ravel et al. 2011), and a significant proportion of apparently healthy women have a vaginal microbiota that instead is comprised of a broad array of facultative and strictly anaerobic microorganisms (Ravel et al. 2011). Epidemiological studies have shown an association between a Lactobacillus-depleted vaginal microbiota and increased risks of acquisition and transmission of sexually transmitted infections, including HIV (Sha et al. 2005; Mirmonsef et al. 2012; Mitchell et al. 2013). While the guinea pig has been widely used in Chlamydia research, very little is known about the composition of the vaginal microbiota in this species. A previous study, done using culture-based methods, indicated that the guinea pig vaginal microbiota include species of Corynebacterium, Enterococcus, Propionibacterium and Streptococcus, as well as some Lactobacillus spp. (Hafner, Rush and Timms 1996). Culture-based methods however have significant well-known limitations (Bull and Hardman 1991; Hugenholtz, Goebel and Pace 1998), suggesting that in-depth characterization of the guinea pig vaginal microbiota should be investigated a new using high-throughput culture-independent 16S rRNA gene sequence analysis. Culture-independent analyses have shown that the composition of the human vaginal microbiota is dynamic over time and most influenced by menstruation and sexual activity (Gajer et al. 2012), although high estrogen/progesterone levels tend to stabilize it. While longitudinal studies in women are needed to answer questions about factors that influence the stability and resilience of vaginal microbiota, animal models are needed to examine the role of the vaginal microbiota in determining susceptibility to sexually transmitted diseases. In this study, we first investigated the composition of healthy guinea pig vaginal microbiota over two estrous cycles, and subsequently examined the impact of C. caviae infection on the vaginal microbiota over two estrous cycles using a culture-independent 16S rRNA gene sequence-based approach.
MATERIALS AND METHODS
Study design
A total of 15 female Hartley guinea pigs (Cavia porcellus) weighing 450–500 g were obtained from Charles River Laboratories, Wilmington, MA, and housed individually in cages with fiberglass filter tops. The estrous cycle for each animal was determined by observing the presence or absence of the vaginal membrane daily. In guinea pigs, the vaginal membrane spontaneously opens on the first day of estrus and after 2–3 days regenerates. Guinea pigs were observed through at least two complete estrous cycles to confirm that each was cycling normally before they were included in the experiment. Infection of animals was initiated on the first day of estrus (day 0), and samples from control animals were collected based on the first day of estrus. Three groups of five animals were either infected with C. caviae, non-infected or mock-infected. The infected group was inoculated intravaginally with 20 μl of C. caviae (104 IFU; approximately 200 ID50 units) in SPG (2:7:7 ratio: succinic acid: sodium dihydrogen phosphate: glycine) delivered with a pipette tip inserted 2–3 cm into the vagina. Mock-infected guinea pigs were inoculated intravaginally with 20 μl of SPG medium alone, while the non-infected group was untouched. For sampling, the guinea pigs were placed on their back and immobilized with a hand placed over their abdomen. A flocked pediatric Eswabs (Copan Diagnostics, Murrieta, CA, USA) was placed into the vaginal opening and inserted up to the cervix. Swabs were rotated 8–10 times, removed, placed in 1 ml of liquid Amies transport medium (Hindiyeh, Acevedo and Carroll 2001) and then stored at −80°C until processed. Samples were collected on days 2, 5, 8, 11, 14 and 16 post-infection in the first estrous cycle and at similar times during the second estrous cycle. In a separate set of five healthy and uninfected guinea pigs, measurements of vaginal pH were obtained with a MI-414P Tip 4 cm probe (Microelectrodes, Bedford, New Hampshire, USA). The protocol was approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences (AUP file # 3288).
Genomic DNA extraction, 16S rRNA gene sequence amplification, sequencing and analyses
Genomic analyses are described in Supplementary File 1 (Supporting Information). The sequencing data generated under this project have been deposited to the Sequence Read Archive under bioproject PRJNA270250.
Statistical modeling procedures
The packages MCMCglmm and rjags (Hadfield 2010; Plummer 2014) in R (version 12.1.3) were used to implement mixed effects Bayesian Markov Chain Monte Carlo (MCMC) models; this approach allows for the significance testing of differences between groups of animals, while accounting for correlations between samples collected from the same animals over time. Differences in bacterial phylotype relative and absolute abundances between experimental groups were evaluated using Bayesian negative binomial random intercept models (Sheldon 1969; Ntzoufras 2009). In these models, the response variable was 16S rRNA gene sequence read count. Both relative abundance and absolute abundance models included an offset term. In the case of relative abundance models, the offset was the log of the total 16S rRNA gene sequence read counts (calculated after excluding chlamydial reads). For the absolute abundance models, the offset was equal to the difference between the log of total 16S rRNA gene sequence read count and the log of the total 16S rRNA gene abundance. Only one random factor (Grune et al. 2004) was used in addition to multiple fixed factors including the day, cycle and experimental group.
We run two MCMC chains for all models. For each chain, 10 000 iterations with a thinning of 10 after a ‘burn-in’ of 10 000 were used. Models were checked for convergence and mixing by examining the Gelman–Rubin statistic (Gelman and Rubin 1992, 1996) among the two iterations, a potential scale reduction factor <1.1 was used for all parameters including fixed and random factors. The parameters considered were posterior means, and 95% credible intervals (CI). p-values associated with CIs were defined as the infimum value of q over q-CI that do not contain the reference mean value. To account for multiple testing, q-values were computed using false discovery rate proposed by Benjamini and Hochberg (1995) and Benjamini et al. (2001) as implemented in the p.adjust() routine of R (R Development Core Team 2013). In the pursuit of transparency, the R package knitr (Xie 2014) was used to make easy to follow scripts for Bayesian modeling and graphical output generation using the provided data files (Supplementary File 2, Supporting Information). In addition, input data files and R scripts are available at https://github.com/RavelLab/GuineaPigVMB.
RESULTS
Characterization of the microbial phylotypes as a function of depth of coverage
A total of 180 samples were obtained from 15 guinea pigs at 12 time points. Amplification and sequencing of the V1–V3 hypervariable regions of 16S rRNA genes resulted in a dataset consisting of 973 999 reads from 177 samples with an average length of 471 bp and ∼6300 reads per sample (range 1961–15 585). The depth of coverage was enough to detect taxa that comprise ∼0.02% of the population. Taxa found at less than this level are referred to as ‘rare’ taxa, but these reads are only rare in the context of sampling depth. Phylogenic classification of this dataset identified a total of 191 bacterial phylotypes. A total of 99.9% of the sequences were taxonomically assigned at the phylum level with >95% confidence, while 98.6% were at class level, 98.2% at order level, 97.4% at family level and 83.2% at genus level. Of the 191 bacterial taxa present in the guinea pig vaginal microbiota, 29 taxa accounted for ∼90% of the dataset; this subset of 29 bacterial taxa is shown in Figs 1 and S1a (Supporting Information). Rarefaction analysis, a common tool to estimate the ability of the sequencing depth to capture the diversity in each sample, was measured by Good's coverage estimator and (Esty 1986) was 92.4% (SD = 0.027), 91.3% (SD = 0.029) and 91.6% (SD = 0.030) in the non-infected, mock-infected and infected groups, respectively (Fig. S2, Supporting Information). The shape of the rarefaction curves indicated that sequence coverage was comprehensive and that additional sequences would have yielded little benefit (Gotelli and Colwell 2001).
Figure 1.

Relative abundances of bacterial taxa in the guinea pig vaginal microbiota of C. caviae-infected (a) and non-infected (b) animals sampled every 3 days over two estrous cycles. The top 29 most abundant phylotypes are shown. ‘Others’ represents the sum of the remaining phylotypes.
Composition of the guinea pig vaginal microbiota
The majority of bacterial taxonomic groups found in the non-infected guinea pig vaginal microbiota consisted of obligate and facultative anaerobic bacteria (Table S1a, Supporting Information), including members of the genera Corynebacterium, Anaerococcus, Peptoniphilus, Aerococcus, Facklamia and Allobaculum (Figs 1 and S1a, Supporting Information). Some of these genera, but not all, were previously cultivated from the guinea pig vaginal microbiota (Table S2, Allobaculum); these included Corynebacterium, Enterococcus, Streptococcus, Staphylococcus, Lactobacillus, Proteus, Propionibacterium, Bifidobacterium and Bacteroides (Hafner, Rush and Timms 1996). While Lactobacillus spp. were present in the guinea pig vaginal microbiota, they represented a small proportion of the total phylotypes (<0.48%) and in most instances were undetectable. Lactobacillus fermentum, often found in the gut microbiota, was detected with a mean relative abundance of 0.08% ± 0.1 SD, while L. crispatus and L. iners commonly found in the human vaginal microbiota were detected with mean relative abundance of 0.04% ± 0.06 SD and 0.01% ± 0.02 SD. Interestingly, Gardnerella vaginalis, BVAB1, BVAB3, Mobiluncus spp., Megasphaera spp. and Atopobium vaginae, which are all taxa associated with bacterial vaginosis in humans, were not detected in the guinea pig vaginal microbiota. However, BVAB2, which is a member of the Clostridiales and is also associated with bacterial vaginosis, was detected with a mean relative abundance of 0.11% ± 0.09 SD.
Effect of C. caviae infection on the composition of the guinea pig vaginal microbiota
Chlamydial 16S rRNA gene sequences were detected in samples from infected animals during the first 8 days of the first estrous cycle but their abundance varied widely (day 2 (7.49–37.33%), day 5 (1.52–76.48%) and day 8 (0.03–14.18%). However, after day 8 the number of C. caviae dramatically decreased (Fig. 1) and this persisted throughout the second menstrual cycle. Noteworthy, in guinea pig 251, chlamydial reads were not detected between day 8 of the first menstrual cycle and day 11 of the second menstrual cycle after which chlamydia relative abundance increased to 9.48% on day 14 (Fig. 1 and Table S1a, Supporting Information).
To better characterize the impact of chlamydial infection on the guinea pig vaginal microbiota, we compared diversity of bacterial communities in different sample types including the guinea pig vagina, guinea pig fecal samples and the human vagina. Because the range of Shannon diversity values depends on the number of phylotypes detected and that number is different in each sample type, the Shannon diversities indices were normalized by dividing each by ln(n), where n is the number of phylotypes in a given sample type. The resulting quantity is a measure of evenness of the community and its values range between 0 and 1(Sheldon 1969). Combining all animal and treatment groups, evenness measurements ranged between 0.291 and 0.918, with an average of 0.731 ± 0.01, indicating a high level of phylotype diversity, compared to the median evenness of the human vaginal microbiota (0.33 ± 0.16) (Fig. S3a, Supporting Information). In the non-infected and mock-infected guinea pig groups, the median evenness were 0.75 ± 0.11, 0.74 ± 0.07 and 0.75 ± 0.11, respectively (Fig. S3b, Supporting Information). Bayesian mixed effect modeling did not identify any significant difference between the evenness of non-infected and mock-infected samples. However, at peak infection (days 2, 5 and 8 of the first estrous cycle), the median evenness was 0.59 ± 0.15. The evenness was statistically significantly lower in the infected group compared to the mock and non-infected groups, 0.73 ± 0.08 (Fig. 2).
Figure 2.

Species evenness on days 2, 5 and 8 combined, of guinea pig vaginal microbiota in non-infected and C. caviae-infected animals in cycle 1 and 2. The red lines indicate the 95% CI. The mean of the dataset is shown as a red dot. The mean evenness of the vaginal community in C. caviae-infected animals in cycle 1 and 2 during days 2, 5 and 8 combined are significantly different.
To estimate the effect of treatment and day in the estrous cycle on relative abundance of different taxa, 66 phylotypes present in at least 25% of all samples were selected for analysis. For each selected phylotype a zero-inflated negative binomial random effects model was fitted to the data with the count of 16S rRNA gene sequence reads for the phylotype as a response variable. In the models, the offset of log total 16S rRNA gene sequence read count was used to estimate the effect of treatment and day in the estrous cycle on the relative abundance of the given phylotype. Zero-inflated models are routinely used for modeling count data where the number of zeros cannot be explained by the base distribution (negative binomial in this case) (Lambert 1992). Animal-specific random intercept accounts for correlations between samples from the same animal.
Significant differences between the mean relative abundance of few selected phylotypes were detected at different time points during the two estrous cycles when comparing infected and mock or non-infected animals. However, there was no particular pattern over days in the estrous cycle and in the number of phylotypes for which a significant difference was detected in the mean relative abundance between infected and mock and non-infected animals (Fig. S4a, Supporting Information). The lack of major difference in the structure and composition of the guinea pig vaginal microbiota in these three groups was thus evaluated using total and taxa-specific absolute bacterial counts. Overall the differences in vaginal bacteria relative abundance between infected and non-infected animals were modest (Table S3, Supporting Information).
Changes in the absolute abundance of bacterial populations in non-infected, mock-infected and C. caviae-infected animals over two estrous cycles
The absolute abundance of bacterial populations in guinea pig vaginas was evaluated by quantifying 16S rRNA gene copy number per swab (cpn swab−1) using a pan-bacterial Taqman assay. The 16S rRNA gene copy number per swab ranged between 5.13 × 103 and 1.53 × 109 cpn swab−1 with an average of 5.51 × 108 cpn swab−1 (Fig. 3a). Changes in the non-chlamydial 16S rRNA gene copy number were not significantly different between the non-infected and mock-infected experimental groups at all time points, except on day 8 post-infection, where surprisingly, the mean 16S rRNA gene copy of mock-infected animals was higher than that of non-infected animals (Fig. 3a). In infected animals, at peak infection (first estrous cycle), the non-chlamydial mean total 16S rRNA gene copy number declined from a 8.93 × 107 cpn swab−1 on day 2 to a 1.95 × 105 cpn swab−1 on day 5, indicating ∼100-fold drop in the non-chlamydial 16S rRNA gene counts (Fig. 3a). In infected animals over both estrous cycles, the overall non-chlamydial 16S rRNA gene copy number averaged 2.53 × 107 cpn swab−1.
Figure 3.
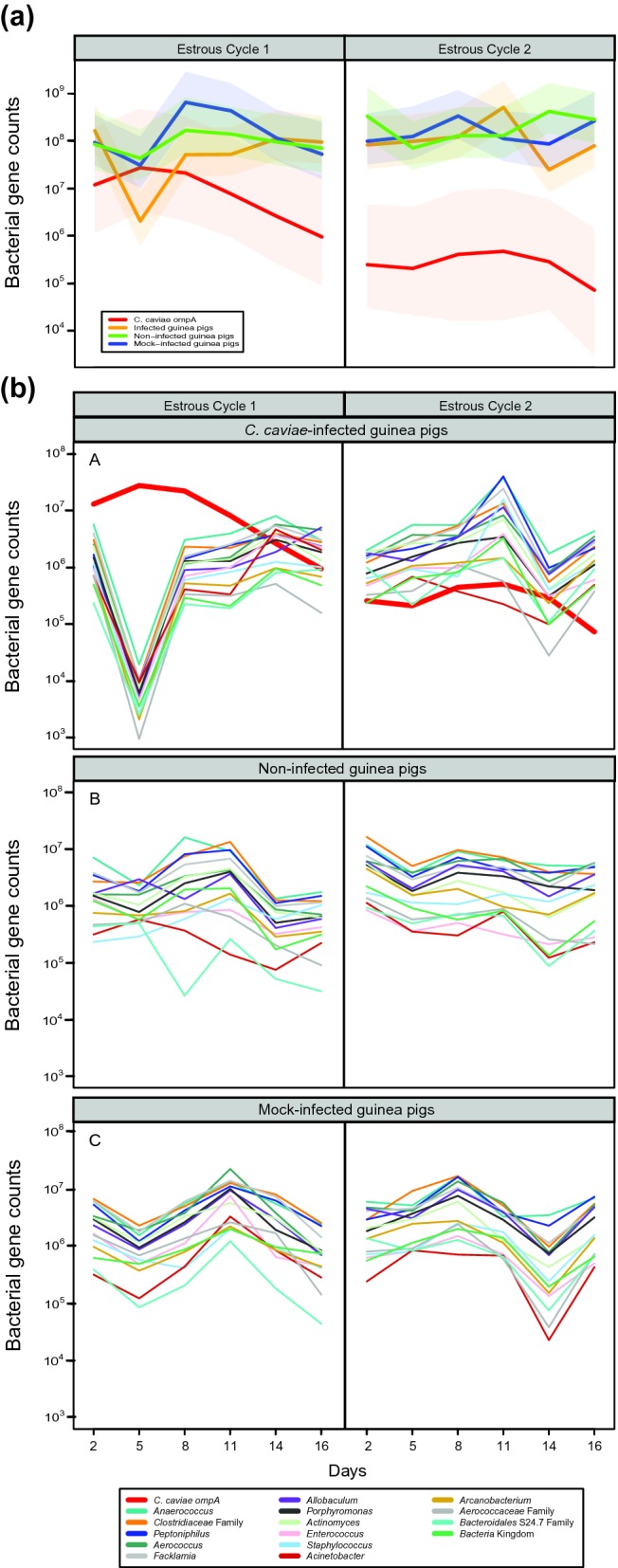
Changes in estimates of absolute bacterial abundance in the guinea pig vaginal microbiota over two estrous cycles. (a) Average bacterial 16S rRNA gene counts and C. caviae ompA counts in the vaginal microbiota of guinea pigs in each group. Values were normalized to gene counts per swab. ompA counts for non-infected animals were below the detection level and were excluded. The colored shaded areas indicate the standard errors. (b) Average bacterial count for the 15 most abundant taxa found in guinea pig vaginal microbiota of C. caviae-infected and non-infected animals over two estrous cycles. The count estimates were calculated by multiplying the relative abundance obtained by 16S rRNA gene sequencing by the total 16S rRNA gene counts in the corresponding sample. Values are normalized to gene counts per swab.
After day 8, 16S rRNA gene sequence reads assigned to Chlamydia constituted less than 1% of phylotypes in three out of five animals. Because the pan-bacterial TaqMan assay (Liu et al. 2012) underrepresented the overall abundance of C. caviae, a specific Taqman qPCR assay using ompA of C. caviae was designed to accurately quantify C. caviae genome copy numbers. Because C. caviae carries a single ompA, ompA copy number is equivalent to the number of C. caviae genomes. This assay showed that the C. caviae absolute abundance between days 2 and 14 of the first estrous cycle ranged from 9.68 × 103 to 5.73 × 107 genomes per swab, peaking on day 5 (Fig. 3b). After day 14 of the first estrous cycle, the abundance of C. caviae generally started to decrease. This continued throughout the second estrous cycle but was not the case in all animals (Figs 3b and S5, Supporting Information). Results from non-infected and mock-infected animals were at or below background fluorescence, which after normalization was 3.8 × 103 C. caviae genomes per swab. In the infected animals, the absolute abundance of C. caviae was modeled using a negative binomial mixed effect model with the mean absolute abundance of C. caviae established by ompA qPCR (Fig. 3b). The mean abundance of C. caviae at each day of the first estrous cycle was significantly higher than the mean abundance of C. caviae on the last day of the second estrous cycle. In addition, the mean abundance of C. caviae at days 2, 5, 8, 11 of the first cycle were significantly higher from the mean abundance of C. caviae at day 16 of the first cycle and every day of the second cycle (Fig. 3b).
To identify time in the estrous cycle (days) where the mean absolute abundance of infected samples was significantly different from the mean absolute abundance of non-infected or mock-infected samples, Bayesian zero-inflated negative binomial mixed effects models were applied with the 16S rRNA gene read count as the outcome variable and the difference of the log of total 16S rRNA gene sequence read count (calculated after excluding chlamydial reads) and the log of the total 16S rRNA gene count as the offset (Table S1b, Supporting Information). In contrast to the results obtained for relative abundance, the number of phylotypes for which the mean absolute abundance in infected animals was significantly lower from the mean absolute abundance in non-infected and mock-infected animals showed a clear pattern of dependence on the day in the estrous cycles with a maximum of 34 phylotypes (out of 66 phylotypes included in the analysis) affected on day 5 of the first estrous cycle (Tables S4a and S4b and Fig. S4b, Supporting Information).
Comparison of the guinea pig and the human vaginal microbiota
The most common bacteria found in the ‘healthy’ human vaginal microbiota are Lactobacillus spp. (L. crispatus, L. iners, L. jensenii and L. gasseri). Lactobacillus spp. produce copious amount of lactic acid and thereby create an acidic environment (pH < 4.5) (Ravel et al. 2011) that is thought to be protective against opportunistic pathogens. In the guinea pig vaginal microbiome, the relative abundance of Lactobacillus spp. was less than 0.48% and the absolute abundance ranged from 0 to 1.58 × 106 cpn swabs−1 (Tables S1a and S1b, Supporting Information). Importantly, lactobacilli were only detected in 35% of the samples. In a separate set of five uninfected guinea pigs, vaginal pH measured over a complete estrous cycle indicated that on average the vaginal environment is slightly basic ranging between 6.83 and 7.21 (Fig. S6, Supporting Information). This is consistent with the relative paucity of lactic acid-producing bacteria in the guinea pig vaginal microbiota.
To further examine the differences between the human and guinea pig microbiota, we compared the composition of the vaginal microbiota of 177 guinea pig samples to 338 samples from reproductive age women with and without chlamydial infection (unpublished data) (Fig. 4). A total of 19 of the 42 bacterial species with average relative abundance >0.01% detected in the human vaginal microbiota was detected in the guinea pig vaginal microbiota but their relative abundances were dramatically different (Table S5, Supporting Information). Some of the phylotypes common to both the human and guinea pig vaginal microbiota included Aerococcus spp., Actinomyces spp., members of the Order Clostridiales, L. crispatus, L. iners, Bifidobacterium spp. and Prevotella spp. (Table S5, Supporting Information). Using Ward linkage hierarchical clustering, the human vaginal microbiota grouped into four of the previously described community state types (Ravel et al. 2011), while most of the guinea pig vaginal microbiota clustered into a novel community state type (CST GPII). Evenness was significantly higher in the guinea pig vaginal microbiota compared to that of human (Figs S3a and S3b, Supporting Information), indicating higher diversity of the guinea pig vaginal microbial communities.
Figure 4.

Heatmaps showing the relative abundances vaginal bacterial taxa relative abundance in 177 guinea pig and 338 human samples. Bacterial composition and Ward linkage hierarchical clustering of (a) infected and (b) non-infected samples is shown. The top 35 most abundant bacterial taxa are shown.
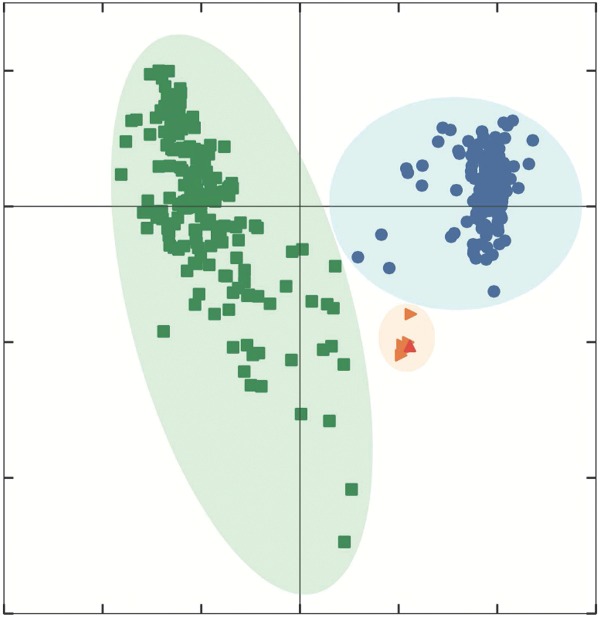
To further visualize the differences in the structure of the guinea pig and human vaginal microbiota, principal coordinate analysis (PCoA) was performed using weighted Unifrac distances (Lozupone et al. 2011). No clustering by infection status was observed, both in human and the guinea pig (Fig. 5a). On the other hand, two distinct clusters were identified that separated human vaginal samples from other sample types including guinea pig vaginal samples (Fig. 5b). However, C. trachomatis infected human vaginal microbiota had a statistically significant overabundance of CST IV-B and CST III, both of which were already represented in non-infected samples. These results indicate that while there are overlapping bacterial phylotypes between the human and guinea pig vaginal microbiota, these microbiota have distinct community composition and structure.
Figure 5.

PCoA of guinea pig vaginal and gut microbiota and human vaginal microbiota using weighted Unifrac distances. Samples are color coded by infection status (a) and collection site (b).
Comparison of the guinea pig gut and vaginal microbiota
Some abundant bacterial phylotypes found in the guinea pig vaginal microbiota are commonly found in the gut microbiota such as Enterococcus spp. To evaluate the possibility that bacteria from guinea pig feces might have colonized the guinea pig vagina (cage effect), the composition and structure of the microbiota in four guinea pig fecal samples and one cage bedding sample were characterized in a separate set of animals maintained in similar conditions and environment. A total of 52 bacterial phylotypes were identified in these samples. The most abundant phylotypes in the guinea pig gut microbiota were members of the Orders Clostridiales and Bacteroidales (median relative abundance 23.17 and 5.76%, respectively), members of the Families Ruminococcaceae and Lachnospiraceae (median relative abundance 3.22 and 11.74%, respectively), Allobaculum spp. (median relative abundance 5.71%), Ruminococcus spp. (median relative abundance 3.22%) and Bifidobacterium spp. (median relative abundance 11.6%), and in aggregate these comprised between 49.55 and 85.23% of each sample (Fig. S1b and Table S1c, Supporting Information). Interestingly, 41 of these 52 bacterial taxa were also detected in the guinea pig vaginal microbiota (Table S1a, Supporting Information); however, their abundance and the overall structure of the microbiota were significantly different (Fig. 5b). Overall, this analysis indicates that, (1) despite the relatively low number of classified phylotypes, the guinea pig gut microbiota is highly diverse with an average evenness of 0.88 ± 0.1 (Figs S3a and S3b, Supporting Information); (2) of the 52 bacterial taxa detected in the guinea pig gut microbiota, only 11 were found with average relative abundance >1% (Fig. S1b and Table S1c, Supporting Information); (3) of the 41 bacterial taxa detected in both the guinea pig gut and vaginal microbiota, only Bacteroidales S24–7 and Allobaculum spp. are present at >1% in the guinea pig vaginal microbiota (Figs 1 and S1a, Supporting Information). Differences in guinea pig gut and vaginal bacterial community structure were further demonstrated by PCoA analysis. The two types of samples formed two separate clusters (Fig. 5). The possibility of transfer from feces to the vaginal microbiota cannot, however, be excluded.
DISCUSSION
In the present study, we used culture-independent next-generation 16S rRNA gene sequencing to characterize the relative abundance of bacterial taxa in the vaginal microbiota in non-infected and C. caviae-infected guinea pigs. Broad range 16S rRNA gene quantitative PCR was used to estimate the absolute abundance of these bacterial taxa. The guinea pig vaginal microbiota consists of obligate and facultative anaerobic bacteria, including members of the genera Corynebacterium, Anaerococcus, Peptoniphilus, Aerococcus, Facklamia and Allobaculum. Interestingly, the vaginal microbiota bacterial composition and relative abundance in C. caviae-infected guinea pigs were not noticeably different from that of the non-infected or mock-infected guinea pig vaginal microbiota. However, during the first 5 days post-infection, while the C. caviae genome copies were increasing, the remaining bacterial taxa decreased in numbers. These changes were reversed as the infection cleared and C. caviae genome copies decreased. In the second estrous cycle, the vaginal microbiota of infected animals was indistinguishable from that of non-infected or mock-infected animals, both in terms of composition and relative abundance, as well as absolute bacterial counts. Lactobacillus spp. were observed in only 35% of the samples analyzed, and when present they constituted a small fraction of the microbiota (<0.48%). Interestingly, bacterial species such as G. vaginalis and A. vaginae, which are often found in women's vagina when Lactibacillus spp. are present in low abundance, were not observed in the guinea pig vaginal microbiota. These organisms have previously been found in the vaginal microbiota of non-human primates (Spear et al. 2010; Rivera et al. 2011), which also do not contain high numbers of Lactobacillus spp. The features and structure of the guinea pig vaginal microbiota highlight the differences between this animal model and humans, and questions the model's validity, particularly to study the early steps of chlamydial infection where a role for the vaginal microbiota is likely.
Traditional cultivation methods, in which bacterial populations are isolated by cultivation on solid culture medium have long been used to characterize the microbial communities associated with many environments, including human and animal body sites (see Syed, Svanberg and Svanberg 1981; Onderdonk et al. 1986; Noguchi, Tsukumi and Urano 2003; Noguchi et al. 2004). Such studies have provided significant insight into host-associated microbial ecology. However, culture-based approaches are biased and do not provide a complete picture of bacterial diversity and community structure. Every facet of the enrichment and isolation process including the nutrient composition of the culture medium, temperature and atmospheric gases, exposure to ambient light, competition between organisms and the human bias associated with the selection of colonies for further study constrains the growth of many bacterial taxa from the original samples. Thus, error and subjectivity is systematically introduced on the evaluated composition of the natural population present in the sample. The cumulative effect is a significant reduction in the breadth of recovered bacterial diversity (Staley and Konopka 1985). These limitations are evident when comparing the culture-based characterization of the guinea pig vaginal microbiota by Hafner, Rush and Timms (1996) with the present data generated using culture-independent 16S rRNA gene fragment sequencing. While most cultured taxa are present in the molecular dataset, including Corynebacterium, Enterococcus, Propionibacterium, Streptococcus, Staphylococcus, Proteus, Bifidobacterium as well as Lactobacillus spp., bacterial 16S rRNA gene counts for these bacterial taxa were not consistent with the CFU-based counts reported by Hafner, Rush and Timms (1996) (Table S2, Supporting Information). The present molecular survey of the guinea pig vaginal microbiota is likely to be more comprehensive, in that it captured bacterial taxa that were not cultured by Hafner, Rush and Timms (1996), including many difficult-to-grow anaerobic bacteria.
16S rRNA gene sequencing-based taxonomic surveys provide information on the relative abundance of bacterial taxa composing a microbiota. Comparison of the relative abundance of taxa in the three groups showed there were few differences, suggesting that C. caviae infection had little effect on the structure of the guinea pig vaginal microbiota. Fig. 1 shows that in infected animals chlamydial 16S rRNA gene relative abundance was maximal on days 2 and 5 and drastically decreased by day 11. Previously published data suggested that in guinea pigs, an active cervical infection persists for approximately 20 days post-infection (Golden et al. 2000). Chlamydia caviae-specific ompA qPCR detected chlamydial DNA in the guinea pig vagina throughout the first estrous cycle and into the second estrous cycle, i.e. ∼48 days post-infection (Fig. 3), considerably beyond the 20 days post-infection previously reported at the cervix (Rank, Sanders and Kidd 1993). However, qPCR measures the presence of DNA, inclusive potentially of dead chlamydiae. In contrast, previous studies measured infectious EBs (IFUs). This raises the possibility that qPCR might yield an overestimate of the total number of viable chlamydiae present in a sample. We did observe resolution of the infection in the animals, which is consistent with the self-resolving property of the C. caviae infection in the guinea pig (Rank, Sanders and Kidd 1993).
Measurements of the overall bacterial burden (bacteria absolute abundance) by qPCR revealed a clear inverse relationship between bacterial abundance and the abundance of C. caviae (Fig. 3). After resolution of the infection in the second estrous cycle, no differences were found between the three groups, indicating that while C. caviae DNA was detected in the second estrous cycle, it did not affect the structure of the vaginal microbiota. Peak C. caviae infection (days 2 to 11 of the first estrous cycle) negatively impacted all bacterial species somewhat equally. It is likely that this decrease in bacterial burden was the result of the strong immune response to C. caviae infection (Rank and Whittum-Hudson 2010), as well as the increased shedding of the squamous epithelium (Ojcius et al. 1998; Quayle 2002;Hafner, Beagley and Timms 2008). Both have been shown to be maximal early in infection (Ojcius et al. 1998; Quayle 2002; Rank and Whittum-Hudson 2010). While a reduction of bacterial burden during chlamydial infection has not been previously reported, similar findings have been observed with other pathogens such as Salmonella enterica, Streptococcus pneumoniae, rotavirus, astrovirus, norovirus and adenovirus (Barman et al. 2008; Yasuda et al. 2010; Ma et al. 2011). Ma et al. (2011) noted a reduction in fecal total bacterial abundance, as well as community diversity during viral diarrhea using denaturing gradient gel electrophoresis of 16S rRNA gene amplicons and taxon-specific qPCR. Similar findings were observed upon S. enterica serovar Typhimurium infection on the intestinal microbiota using molecular quantification of 16S rRNA gene copies (Barman et al. 2008). However, none of these studies analyzed samples collected longitudinally post-infection, so the long-term effects on the microbiota are unknown. Further work is needed to better characterize the role of the host factors such as the immune response, in reducing bacterial burden in the guinea pig vaginal microbiota post-chlamydial infection.
The purpose of a model organism is to address questions that are difficult to investigate in humans because of ethical or logistical issues. In this study, we aimed to evaluate the guinea pig-C. caviae model of genital infection as a surrogate for studying the role of the vaginal microbiota in the early steps of infection in human C. trachomatis genital infection. Our results revealed major differences in bacterial composition, diversity and overall community structure between the guinea pig and human vaginal microbiota. Importantly, a Lactobacillus-dominated microbiota, the hallmark of a healthy human vagina (Gajer et al. 2012), is not found in the guinea pig. Several phylotypes were present in both the vaginal microbiota of human and guinea pigs; however, those were present at very low relative abundance in the guinea pig compared to human. Such phylotypes included those found in women who normally have non-Lactobacillus-dominated microbiota, higher bacterial diversity and pH, and intermediate Nugent scores (Ravel et al. 2011). Further, the guinea pig vaginal microbiota does not resemble that of women with a bacterial vaginosis-like state (Fig. 5). Interestingly, the lack of Lactobacillus spp. in the guinea pig results in a vaginal milieu with a neutral pH, drastically different from the acidic (pH < 4) of the human vagina resulting from the production of lactic acid by Lactobacillus spp. These findings are consistent with surveys of the vaginal microbiota in other animals, including non-human primates (Patton et al. 1996a,b, 1997, 1998, 1999, 2001, 2003, 2004; Schlievert et al. 2008; Spear et al. 2010, 2012; Rivera et al. 2011), mice (Noguchi, Tsukumi and Urano 2003; Joo et al. 2012), rats (Larsen, Markovetz and Galask 1976; Noguchi, Tsukumi and Urano 2003; Dewi et al. 2013) and rabbits (Jacques et al. 1986; Noguchi, Tsukumi and Urano 2003). The human microbiota appears unique in the animal kingdom, and its role remains to be elucidated, while several hypotheses have been put forward that would explain why women harbor these unique Lactobacillus-dominated vaginal microbiota (Stumpf et al. 2013). A recent paper by Gong et al. (2014) demonstrated that lactic acid produced by vaginal Lactobacillus spp. exhibits anti-chlamydial properties in vitro, suggesting it plays a critical role in protecting women against infection. Ultimately, our findings compromise the validity of the guinea pig-C. caviae model, not as a model to study Chlamydia pathogenesis, but as a model to study the role of the vaginal microbiota during the early steps of sexually transmitted infections or in priming the host response.
Interestingly, when animals are studied in captivity, the possibility of a cage effect cannot be excluded. In non-human primates such as the baboon, comparison of captive and wild animals did not reveal major differences, and wild baboons maintain a non-Lactobacillus vaginal microbiota (Rivera et al. 2011). Similarly, the unique community structure of the guinea pig vaginal microbiota was not explained by the presence of bacterial taxa found in the guinea pig gut or cage bedding. We hypothesized that some of these taxa might transfer from the cage environment, which contains feces, to the guinea pig vagina. Analysis of the gut (stool) and cage-bedding microbiota revealed similar bacterial community compositions that were distinct from that of the guinea pig vagina. These communities shared some phylotypes, but all were present in very low abundance in the guinea pig vagina. Thus, it is unlikely that the guinea pig vaginal microbiota is shaped by exposure to stool or cage-bedding microbiota; however, that possibility cannot be excluded until wild guinea pigs are studied. Furthermore, outside of the 14–19 day estrous period, the guinea pig vagina is ‘closed’, protected by a membrane (Stockard and Papanicolaou 1919), making microbial transfer from the cage environment to the vagina highly unlikely. Interestingly, while the animals in this study were kept in separate cages, their vaginal microbiota were highly similar and stable over the time of the study, suggesting that functional and genetic characteristics of the communities are conserved and perhaps driven by local genital physiological and immunological factors that shape the composition of the guinea pig vaginal microbiota.
In conclusion, the guinea pig, an animal model used to study chlamydial infection, does not harbor a vaginal microbiota similar to that of humans, it has a neutral pH and is not dominated by Lactobacillus spp. Unlike mice, which have a keratinized vaginal epithelium, the guinea pig vaginal epithelium is squamous and similar to that of humans (Iguchi et al. 1983), suggesting that a humanized guinea pig vagina could be colonized by ‘human’ Lactobacillus spp. Importantly, while we believe that guinea pigs are not recommended to study the role of the vaginal microbiota in the early steps of chlamydial infection, the guinea pig-C. caviae model does remain a valid system to study chlamydial genital pathogenesis and disease or even sexual transmission.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSPD online.
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
FUNDING
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number U19AI084044.
Conflict of interest. None declared.
REFERENCES
- Alkhalaf M, Propper AY, Chaminadas G, et al. Ultrastructural changes in guinea pig endometrial cells during the estrous cycle. J Morphol. 1992;214:83–96. doi: 10.1002/jmor.1052140106. [DOI] [PubMed] [Google Scholar]
- Barman M, Unold D, Shifley K, et al. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–15. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg ME. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- Boskey ER, Cone RA, Whaley KJ, et al. Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum Reprod. 2001;16:1809–13. doi: 10.1093/humrep/16.9.1809. [DOI] [PubMed] [Google Scholar]
- Bull A, Hardman D. Microbial diversity. Curr Opin Biotech. 1991;2:421–8. [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Sexually Transmitted Disease Surveillance 2013. Atlanta: Department of Health and Human Services; 2014. [Google Scholar]
- Darville T, Andrews CW, Jr, Laffoon KR, et al. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun. 1997;65:3065–73. doi: 10.1128/iai.65.8.3065-3073.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewi AH, Ana ID, Wolke J, et al. Behavior of plaster of Paris-calcium carbonate composite as bone substitute. A study in rats. J Biomed Mater Res A. 2013;101:2143–50. doi: 10.1002/jbm.a.34513. [DOI] [PubMed] [Google Scholar]
- Durrani MJ, Kusai A, Ho NFH, et al. Topical vaginal drug delivery in the guinea pig. I. Effect of estrous cycle on the vaginal membrane permeability of vidarabine. Int J Pharm. 1985;24:209–18. [Google Scholar]
- Esty WW. The efficiency of Good's nonparametric coverage estimator. Ann Stat. 1986;14:1257–60. [Google Scholar]
- Fan VS, Jenkin HM. Glycogen metabolism in Chlamydia-infected HeLa-cells. J Bacteriol. 1970;104:608–9. doi: 10.1128/jb.104.1.608-609.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajer P, Brotman RM, Bai G, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132–52. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Rubin DB. Inference from iterative simulation using multiple sequences. Statist. Sci. 1992;7:457–72. [Google Scholar]
- Gelman A, Rubin DB. Markov chain Monte Carlo methods in biostatistics. Stat Methods Med Res. 1996;5:339–55. doi: 10.1177/096228029600500402. [DOI] [PubMed] [Google Scholar]
- Golden MR, Schillinger JA, Markowitz L, et al. Duration of untreated genital infections with chlamydia trachomatis: a review of the literature. Sex Transm Dis. 2000;27:329–37. doi: 10.1097/00007435-200007000-00006. [DOI] [PubMed] [Google Scholar]
- Gong Z, Luna Y, Yu P, et al. Lactobacilli inactivate Chlamydia trachomatis through lactic acid but not H2O2. PLoS One. 2014;9:e107758. doi: 10.1371/journal.pone.0107758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon FB, Quan AL. Susceptibility of Chlamydia to antibacterial drugs: test in cell cultures. Antimicrob Agents Ch. 1972;2:242–4. doi: 10.1128/aac.2.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotelli NJ, Colwell RK. Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett. 2001;4:379–91. [Google Scholar]
- Grune B, Fallon M, Howard C, et al. Report and recommendations of the international workshop ‘Retrieval approaches for information on alternative methods to animal experiments’. ALTEX. 2004;21:115–27. [PubMed] [Google Scholar]
- Hadfield JD. MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J Stat Softw. 2010;33:1–22. [Google Scholar]
- Hafner L, Beagley K, Timms P. Chlamydia trachomatis infection: host immune responses and potential vaccines. Mucosal Immunol. 2008;1:116–30. doi: 10.1038/mi.2007.19. [DOI] [PubMed] [Google Scholar]
- Hafner LM, Rush CM, Timms P. The vaginal microbiota of guinea pigs. Microb Ecol Health D. 1996;9:123–7. [Google Scholar]
- Hanna NF, Taylor-Robinson D, Kalodiki-Karamanoli M, et al. The relation between vaginal pH and the microbiological status in vaginitis. Brit J Obstet Gynaec. 1985;92:1267–71. doi: 10.1111/j.1471-0528.1985.tb04874.x. [DOI] [PubMed] [Google Scholar]
- Hindiyeh M, Acevedo V, Carroll KC. Comparison of three transport systems (Starplex StarSwab II, the new Copan Vi-Pak Amies Agar Gel collection and transport swabs, and BBL Port-A-Cul) for maintenance of anaerobic and fastidious aerobic organisms. J Clin Microbiol. 2001;39:377–80. doi: 10.1128/JCM.39.1.377-380.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–74. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi T, Uchima FD, Ostrander PL, et al. Growth of normal mouse vaginal epithelial cells in and on collagen gels. P Natl Acad Sci USA. 1983;80:3743–7. doi: 10.1073/pnas.80.12.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques M, Olson ME, Crichlow AM, et al. The normal microflora of the female rabbit's genital tract. Can J Vet Res. 1986;50:272–4. [PMC free article] [PubMed] [Google Scholar]
- Joo HM, Kim KA, Myoung KS, et al. Lactobacillus helveticus HY7801 ameliorates vulvovaginal candidiasis in mice by inhibiting fungal growth and NF-kappaB activation. Int Immunopharmacol. 2012;14:39–46. doi: 10.1016/j.intimp.2012.05.023. [DOI] [PubMed] [Google Scholar]
- Lambert D. Zero-inflated poisson regression, with an application to defects in manufacturing. Technometrics. 1992;34:1–14. [Google Scholar]
- Larsen B, Markovetz AJ, Galask RP. The bacterial flora of the female rat genital tract. Proc Soc Exp Biol Med. 1976;151:571–4. doi: 10.3181/00379727-151-39261. [DOI] [PubMed] [Google Scholar]
- Liu CM, Aziz M, Kachur S, et al. BactQuant: an enhanced broad-coverage bacterial quantitative real-time PCR assay. BMC Microbiol. 2012;12:56. doi: 10.1186/1471-2180-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Lladser ME, Knights D, et al. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5:169–72. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Wu X, Nawaz M, et al. Molecular characterization of fecal microbiota in patients with viral diarrhea. Curr Microbiol. 2011;63:259–66. doi: 10.1007/s00284-011-9972-7. [DOI] [PubMed] [Google Scholar]
- Mardh PA, Ripa T, Svensson L, et al. Chlamydia trachomatis infection in patients with acute salpingitis. New Engl J Med. 1977;296:1377–9. doi: 10.1056/NEJM197706162962403. [DOI] [PubMed] [Google Scholar]
- Mirmonsef P, Krass L, Landay A, et al. The role of bacterial vaginosis and trichomonas in HIV transmission across the female genital tract. Curr HIV Res. 2012;10:202–10. doi: 10.2174/157016212800618165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, Balkus JE, Fredricks D, et al. Interaction between lactobacilli, bacterial vaginosis-associated bacteria, and HIV Type 1 RNA and DNA Genital shedding in U.S., Kenyan women. AIDS Res Hum Retrov. 2013;29:13–9. doi: 10.1089/aid.2012.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount DT, Bigazzi PE, Barron AL. Infection of genital tract and transmission of ocular infection to newborns by the agent of guinea pig inclusion conjunctivitis. Infect Immun. 1972;5:921–6. doi: 10.1128/iai.5.6.921-926.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi K, Tsukumi K, Udono T, et al. Normal vaginal flora in chimpanzees (Pan troglodytes): qualitative and quantitative study. Comp Med. 2004;54:705–12. [PubMed] [Google Scholar]
- Noguchi K, Tsukumi K, Urano T. Qualitative and quantitative differences in normal vaginal flora of conventionally reared mice, rats, hamsters, rabbits, and dogs. Comp Med. 2003;53:404–12. [PubMed] [Google Scholar]
- Ntzoufras I. Bayesian Modeling Using WinBUGS. Hoboken, New Jersey: John Wiley & Sons; 2009. [Google Scholar]
- Ojcius DM, Souque P, Perfettini JL, et al. Apoptosis of epithelial cells and macrophages due to infection with the obligate intracellular pathogen Chlamydia psittaci. J Immunol. 1998;161:4220–6. [PubMed] [Google Scholar]
- Onderdonk AB, Zamarchi GR, Walsh JA, et al. Methods for quantitative and qualitative evaluation of vaginal microflora during menstruation. Appl Environ Microb. 1986;51:333–9. doi: 10.1128/aem.51.2.333-339.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes AS, Brambell FW. The causation of the anoestrous period. J Physiol. 1928;64:388–92. doi: 10.1113/jphysiol.1928.sp002448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DL, Cosgrove-Sweeney YT, Antonio MA, et al. Lactobacillus crispatus capsules: single-use safety study in the Macaca nemestrina model. Sex Transm Dis. 2003;30:568–70. doi: 10.1097/00007435-200307000-00007. [DOI] [PubMed] [Google Scholar]
- Patton DL, Cosgrove-Sweeney YT, Rabe LK, et al. The pig-tailed macaque rectal model: microflora and chlamydial infection. Sex Transm Dis. 2001;28:363–6. doi: 10.1097/00007435-200107000-00001. [DOI] [PubMed] [Google Scholar]
- Patton DL, Kidder GG, Sweeney YC, et al. Effects of nonoxynol-9 on vaginal microflora and chlamydial infection in a monkey model. Sex Transm Dis. 1996a;23:461–4. doi: 10.1097/00007435-199611000-00004. [DOI] [PubMed] [Google Scholar]
- Patton DL, Kidder GG, Sweeney YC, et al. Effects of multiple applications of benzalkonium chloride and nonoxynol 9 on the vaginal epithelium in the pigtailed macaque (Macaca nemestrina) Am J Obstet Gynecol. 1999;180:1080–7. doi: 10.1016/s0002-9378(99)70598-3. [DOI] [PubMed] [Google Scholar]
- Patton DL, Sweeney YC, Bohannon NJ, et al. Effects of doxycycline and antiinflammatory agents on experimentally induced chlamydial upper genital tract infection in female macaques. J Infect Dis. 1997;175:648–54. doi: 10.1093/infdis/175.3.648. [DOI] [PubMed] [Google Scholar]
- Patton DL, Sweeney YC, Rabe LK, et al. The vaginal microflora of pig-tailed macaques and the effects of chlorhexidine and benzalkonium on this ecosystem. Sex Transm Dis. 1996a;23:489–93. doi: 10.1097/00007435-199611000-00009. [DOI] [PubMed] [Google Scholar]
- Patton DL, Sweeney YC, Tsai CC, et al. Macaca fascicularis vs. Macaca nemestrina as a model for topical microbicide safety studies. J Med Primatol. 2004;33:105–8. doi: 10.1111/j.1600-0684.2004.00059.x. [DOI] [PubMed] [Google Scholar]
- Patton DL, Sweeney YT, McKay TL, et al. 0.25% chlorhexidine gluconate gel. A protective topical microbicide. Sex Transm Dis. 1998;25:421–4. doi: 10.1097/00007435-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Peipert JF, Boardman L, Hogan JW, et al. Laboratory evaluation of acute upper genital tract infection. Obstet Gynecol. 1996;87(5 Pt 1):730–6. doi: 10.1016/0029-7844(96)00040-3. [DOI] [PubMed] [Google Scholar]
- Plummer M. rjags: Bayesian Graphical Models Using MCMC. R Package Version 3.13. 2014 http://mcmc-jags.sourceforge.net. [Google Scholar]
- Quayle AJ. The innate and early immune response to pathogen challenge in the female genital tract and the pivotal role of epithelial cells. J Reprod Immunol. 2002;57:61–79. doi: 10.1016/s0165-0378(02)00019-0. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. http://www.R-project.org. [Google Scholar]
- Rank RG, Batteiger BE, Soderberg LS. Immunization against chlamydial genital infection in guinea pigs with UV-inactivated and viable chlamydiae administered by different routes. Infect Immun. 1990;58:2599–605. doi: 10.1128/iai.58.8.2599-2605.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Bowlin AK, Reed RL, et al. Characterization of chlamydial genital infection resulting from sexual transmission from male to female guinea pigs and determination of infectious dose. Infect Immun. 2003;71:6148–54. doi: 10.1128/IAI.71.11.6148-6154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Sanders MM, Kidd AT. Influence of the estrous cycle on the development of upper genital tract pathology as a result of chlamydial infection in the guinea pig model of pelvic inflammatory disease. Am J Pathol. 1993;142:1291–6. [PMC free article] [PubMed] [Google Scholar]
- Rank RG, White HJ, Hough AJ, Jr, et al. Effect of estradiol on chlamydial genital infection of female guinea pigs. Infect Immun. 1982;38:699–705. doi: 10.1128/iai.38.2.699-705.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Whittum-Hudson JA. Protective immunity to chlamydial genital infection: evidence from animal studies. J Infect Dis. 2010;201(Suppl 2):S168–77. doi: 10.1086/652399. [DOI] [PubMed] [Google Scholar]
- Ravel J, Gajer P, Abdo Z, et al. Vaginal microbiome of reproductive-age women. P Natl Acad Sci USA. 2011;108(Suppl 1):4680–7. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Myers GS, Brunham RC, et al. Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003;31:2134–47. doi: 10.1093/nar/gkg321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera AJ, Frank JA, Stumpf R, et al. Differences between the normal vaginal bacterial community of baboons and that of humans. Am J Primatol. 2011;73:119–26. doi: 10.1002/ajp.20851. [DOI] [PubMed] [Google Scholar]
- Rockey DD, Fischer ER, Hackstadt T. Temporal analysis of the developing Chlamydia psittaci inclusion by use of fluorescence and electron microscopy. Infect Immun. 1996;64:4269–78. doi: 10.1128/iai.64.10.4269-4278.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlievert PM, Strandberg KL, Brosnahan AJ, et al. Glycerol monolaurate does not alter rhesus macaque (Macaca mulatta) vaginal lactobacilli and is safe for chronic use. Antimicrob Agents Ch. 2008;52:4448–54. doi: 10.1128/AAC.00989-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha BE, Zariffard MR, Wang QJ, et al. Female genital-tract HIV load correlates inversely with Lactobacillus species but positively with bacterial vaginosis and Mycoplasma hominis. J Infect Dis. 2005;191:25–32. doi: 10.1086/426394. [DOI] [PubMed] [Google Scholar]
- Sheldon AL. Equitability indices: dependence on the species count. Ecol Soc Am. 1969;50:466–67. [Google Scholar]
- Spear GT, Gilbert D, Sikaroodi M, et al. Identification of rhesus macaque genital microbiota by 16S pyrosequencing shows similarities to human bacterial vaginosis: implications for use as an animal model for HIV vaginal infection. AIDS Res Hum Retrov. 2010;26:193–200. doi: 10.1089/aid.2009.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear GT, Kersh E, Guenthner P, et al. Longitudinal assessment of pigtailed macaque lower genital tract microbiota by pyrosequencing reveals dissimilarity to the genital microbiota of healthy humans. AIDS Res Hum Retrov. 2012;28:1244–9. doi: 10.1089/aid.2011.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear GT, St John E, Zariffard MR. Bacterial vaginosis and human immunodeficiency virus infection. AIDS Res Ther. 2007;4:25. doi: 10.1186/1742-6405-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–46. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- Stamey TA, Timothy MM. Studies of introital colonization in women with recurrent urinary infections. I. The role of vaginal pH. J Urol. 1975;114:261–3. doi: 10.1016/s0022-5347(17)67003-4. [DOI] [PubMed] [Google Scholar]
- Stefka AT, Feehley T, Tripathi P, et al. Commensal bacteria protect against food allergen sensitization. P Natl Acad Sci USA. 2014;111:13145–50. doi: 10.1073/pnas.1412008111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockard CR, Papanicolaou GN. The vaginal closure membrane, copulation, the vaginal plug in the guinea-pig, with further considerations of the oestrous rhythm. Biol Bull. 1919;37:222–45. [Google Scholar]
- Stumpf RM, Wilson BA, Rivera A, et al. The primate vaginal microbiome: comparative context and implications for human health and disease. Am J Phys Anthropol. 2013;152(Suppl 57):119–34. doi: 10.1002/ajpa.22395. [DOI] [PubMed] [Google Scholar]
- Syed SA, Svanberg M, Svanberg G. The predominant cultivable dental plaque flora of beagle dogs with periodontitis. J Clin Periodontol. 1981;8:45–56. doi: 10.1111/j.1600-051x.1981.tb02023.x. [DOI] [PubMed] [Google Scholar]
- Westrom L. Effect of acute pelvic inflammatory disease on fertility. Am J Obstet Gynecol. 1975;121:707–13. doi: 10.1016/0002-9378(75)90477-9. [DOI] [PubMed] [Google Scholar]
- Wiesenfeld HC, Hillier SL, Meyn LA, et al. Subclinical pelvic inflammatory disease and infertility. Obstet Gynecol. 2012;120:37–43. doi: 10.1097/AOG.0b013e31825a6bc9. [DOI] [PubMed] [Google Scholar]
- Xie Y. Dynamic Documents With R and Knitr. Boca Raton: Chapman & Hall/CRC Press; 2014. [Google Scholar]
- Yasuda Y, Matsumura Y, Kasahara K, et al. Microbial exposure early in life regulates airway inflammation in mice after infection with Streptococcus pneumoniae with enhancement of local resistance. Am J Physiol-Lung C. 2010;298:L67–78. doi: 10.1152/ajplung.00193.2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.