

Abstract
Pompe disease is an autosomal recessive genetic disorder characterized by a deficiency of the enzyme responsible for degradation of lysosomal glycogen (acid α-glucosidase (GAA)). Cardiac dysfunction and respiratory muscle weakness are primary features of this disorder. To attenuate the progressive and rapid accumulation of glycogen resulting in cardiorespiratory dysfunction, adult Gaa–/– mice were administered a single systemic injection of rAAV2/9-DES-hGAA (AAV9-DES) or bimonthly injections of recombinant human GAA (enzyme replacement therapy (ERT)). Assessment of cardiac function and morphology was measured 1 and 3 months after initiation of treatment while whole-body plethysmography and diaphragmatic contractile function was evaluated at 3 months post-treatment in all groups. Gaa–/– animals receiving either AAV9-DES or ERT demonstrated a significant improvement in cardiac function and diaphragmatic contractile function as compared to control animals. AAV9-DES treatment resulted in a significant reduction in cardiac dimension (end diastolic left ventricular mass/gram wet weight; EDMc) at 3 months postinjection. Neither AAV nor ERT therapy altered minute ventilation during quiet breathing (eupnea). However, breathing frequency and expiratory time were significantly improved in AAV9-DES animals. These results indicate systemic delivery of either strategy improves cardiac function but AAV9-DES alone improves respiratory parameters at 3 months post-treatment in a murine model of Pompe disease.
Introduction
Pompe disease is an autosomal recessive disorder characterized by a deficiency of acid α-glucosidase (GAA), an enzyme responsible for the degradation of lysosomal glycogen. Mutations in the GAA gene cause a significant accumulation of lysosomal glycogen leading to swelling of the lysosome, displacement of myofibril contractile units, and impaired autophagy.1–6 Complete or near complete loss of GAA leads to the infantile-onset form of the disease resulting in cardiac or respiratory failure within the first year of life.7,8 Patients with the late-onset form of the disease, often diagnosed as adults, retain some residual GAA activity, and display a less severe yet progressive phenotype, where skeletal muscle weakness and respiratory complications occur later in life. The only Food and Drug Administration-approved treatment is bimonthly infusions of human recombinant GAA (enzyme replacement therapy (ERT)), which has been shown to reduce cardiac dilation, lessen respiratory insufficiency, and improve overall survival rate in early-onset patients.9 However, the majority of early and late-onset patients receiving ERT eventually require ventilatory assistance.9 Factors including inefficient mannose 6-phosphate receptor-mediated uptake of circulating enzyme, antibody-mediated clearance of hGAA, or inability to cross the blood–brain barrier could be limiting ERT therapeutic potential. Failure to clear neuronal glycogen may be key as gene therapy studies in a Gaa–/– knockout mouse model have demonstrated improved respiratory capacity following depletion of lysosomal glycogen in motoneurons.10–13 Another limitation of the efficacy of ERT is ~25% of patients with infantile onset of Pompe do not demonstrate any detectable GAA protein by molecular methods and are classified as cross-reactive immunologic material (CRIM)-negative.14 CRIM-negative patients receiving ERT treatment therefore produce high levels of anti-GAA circulating antibodies that may limit treatment. Several studies are currently underway investigating immunosuppression strategies in combination with ERT treatment; however, even with the use of immunosuppressants patient improvement is still limited.14
Successful gene therapy using recombinant adeno-associated virus (rAAV) vectors has been demonstrated in murine models of Pompe disease.15 We have shown that administration of rAAV-GAA greatly contributes to reducing lysosomal glycogen content and augmenting cardiac, respiratory, and motoneuron function in the GAA knockout mouse model (Gaa–/–).15 Initial studies were centered on rAAV1, which demonstrated high transduction in skeletal muscle through direct diaphragm application or direct intramuscular (quadriceps or gastrocnemius) injection, resulting in a significant increase in GAA activity and reduction of glycogen.16 However, improvement in functional cardiac and skeletal muscle indices of Gaa–/– animals had only been reported after intravenous administration of rAAV1 in neonates.17,18
In comparison to other rAAV serotypes, rAAV9 exhibits a more robust expression pattern and improved biodistribution when injected systemically.19 Moreover, rAAV9 has been shown to transduce the myocardium, skeletal muscle, as well as the central nervous system, at high levels in nonhuman primates.20 Adequate expression in these tissues is advantageous for Pompe disease as patients accumulate glycogen in virtually all cell types. This is in contrast to what has been observed with ERT as it does not efficiently cross the blood–brain barrier and therefore limits the therapeutic efficiency for correction of neural-related pathology.
Initial reports determining the efficacy of rAAV vectors utilized the chicken β-actin or CMV promoter to drive transgene expression. We recently tested a modified Desmin promoter (DES) to further increase tissue specific expression and potentially decrease the potential for promoter driven immunogenicity to the transgene. The DES promoter expression profile shows preferential expression in skeletal muscle and cardiac tissues, and is detectable in motor neurons.11,21 We have previously reported the biodistribution profiles following systemic administration at P0 in mice and similar patterns were observed in the myocardium and in phenotypically fast or slow-twitch skeletal muscle regardless of CMV or DES promoter.21
In this study, we evaluated the therapeutic potential of AAV9-DES following a single intravenous administration versus the bimonthly ERT administration in adult Gaa–/– animals. Outcome measurements consisted of respiratory and cardiac outcomes at 1 and 3 months after treatment initiation. Here, we demonstrate that an AAV-vector based approach is similar to ERT and augments respiratory and cardiac indices after a single administration with a marked improvement at 3 months of age.
Results
Weight gain and mortality
Animal mass was recorded at 1 and 3 months post-treatment administration. At 1-month postadministration of AAV or ERT, both treatments resulted in minor improvement in weight gain. However, the increase in mass was significantly improved at 3 months with both treatments (P < 0.05). Although it has not been formally documented, the Gaa–/– mouse strain displays higher mortality than wild-type animals (unpublished data), yet Gaa–/– are still capable of reaching normal lifespan expectancies of the strain (~21–24 months). The survival of the initial group of Gaa–/– animals receiving ERT without diphenhydramine did not surpass a third ERT injection due to anaphylactic shock. This prompted a second ERT group that received diphenhydramine prior to ERT administration. No mortality was observed following diphenhydramine/ERT administration throughout the study protocol. In the present study, there was no incidence of mortality in Gaa–/–, wild-type, or AAV9-DES-treated animals (Figure 1a).
Figure 1.

Effects of AAV9-DES or continuous enzyme replacement therapy (ERT) on animal weight gain and cardiac characteristics at 1 and 3 months. (a) AAV9-DES or ERT did not significantly improve animals’ weights until 3 months after study initiation (P > 0.05). (b) Detection of ejection fraction (EF%) and (c) end diastolic cardiac mass/gram wet weight (EDM/c) at 4.7-T were not significantly altered compared to Gaa–/– following 1 month of therapy. (b) ERT and AAV9-DES resulted in a significant increase in EF% at 3 months. (c) EDM/c was significantly reduced at 3 months in AAV9-DES-treated animals. (d) Elongation of PR interval was significant following AAV9-DES at 3 months post-treatment. Values indicated are the mean ± SEM. *P ≤ 0.05. **P ≤ 0.01. ***P ≤ 0.001. ****P ≤ 0.0001. θ = wild-type at 6 months.
Impact of ERT or AAV9 therapy on cardiac function and morphology
The Pompe mouse model develops dilated cardiomyopathy, accompanied by alterations in electrophysiology as early as 6 months of age. Neither ERT- nor AAV9-treated animals demonstrated significant improvement in PR interval (i.e., elongation) at 1-month post-treatment (data not shown). In AAV9-DES-treated animals, elongation of the PR interval was observed at 3 months post-treatment suggesting a correction of cardiac conductance ((P < 0.05), Figure 1d).
To further evaluate the cardiac function, animals were subjected to high-field cardiac magnetic resonance imaging (4.7T) to assess left ventricular mass and ejection fraction. No differences were observed at 1 month following the initiation of treatment, but significance was detected by increased ejection fraction and a reduction in left ventricular mass after 3 months in ERT and AAV9-DES-treated groups compared to GAA-deficient mice (Figure 1b,c, respectively). There were no significant differences between the two treated groups.
Respiratory function following ERT or AAV9 therapy
Both the infantile and late-onset forms of Pompe disease cause respiratory insufficiency, resulting in a majority of patients requiring mechanical ventilation. Assessment of ventilatory function by whole body plethysmography was performed at 3 months following initiation of therapy. A period of normoxia followed by a brief hypercapnic challenge revealed significant variations in respiratory parameters in the AAV9-DES group when compared to Gaa–/– or ERT mice. At normoxic conditions, AAV9-DES animals demonstrated a significant increase in frequency (Figure 2a) and decrease in expiratory time (TE) and timing of the total respiratory cycle (TI/TTOT) (Figure 2e,f). No significant differences were detected in minute ventilation, tidal volume, or inspiratory time between all groups at this age (Figure 2b–d).
Figure 2.

Whole-body plethysmography during eupnea. AAV9-DES resulted in significant changes in (a) breathing frequency when compared to Gaa–/– or enzyme replacement therapy (ERT). No significant change was detected in (b) minute ventilation, (c) tidal volume, or (d) inspiratory time. AAV9-DES treatment significantly decreases (e) expiratory time and increased the (f) total respiratory cycle time. Values indicated are the mean ± SEM. *P ≤ 0.05. **P ≤ 0.01.
We have previously shown that Gaa–/– mice demonstrate a progressive decline in diaphragmatic contractile strength over time, and that administration of AAV1-CMV-GAA to the diaphragm can correct skeletal muscle dysfunction.16 Ex vivo isometric force-frequency measurements were performed on diaphragm muscle from treated and untreated groups. Contractile data demonstrated an overall reduction in specific force in Gaa–/– mice compared to wild-type controls; however, both ERT and AAV9 treatment resulted in a significant increase in force generation compared to Gaa–/– mice. There was no significant difference in specific force production between ERT and AAV9 treated groups, and both groups demonstrated a significant reduction in contractile force when compared to the wild-type group (Figure 3).
Figure 3.
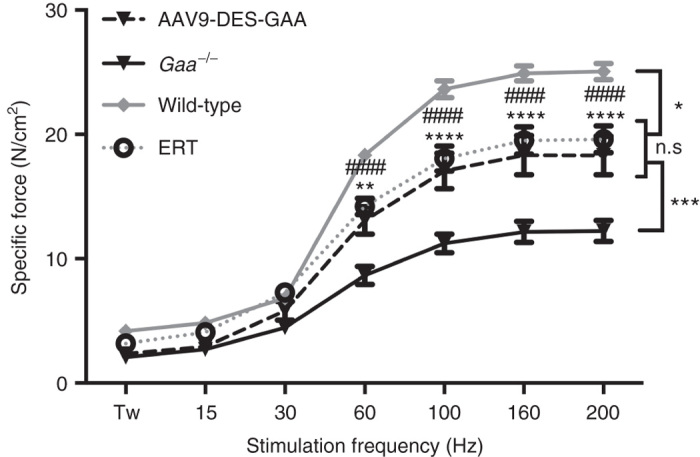
Diaphragmatic force-frequency response (in vitro) of costal diaphragm. AAV9-DES and enzyme replacement therapy (ERT) groups demonstrated a significant increase in force generating capacity of diaphragm myofibers ≥ 60 Hz when compared to Gaa–/–. AAV9-DES and ERT remained significantly lower than wild-type samples ≥ 60 Hz. Values indicated are the mean ± SEM. *WT = P ≤ 0.05 versus AAV9-DES or ERT. **AAV9-DES = P ≤ 0.01 versus Gaa–/–. ***AAV9-DES or ERT = P ≤ 0.001 versus Gaa–/–. ****AAV9-DES = P ≤ 0.0001 versus Gaa–/–. ####ERT = P ≤ 0.0001 versus Gaa–/–. n.s., not significant.
GAA immunogenicity of ERT or AAV9 therapy
Recently, modulation of immune responses following ERT therapy in Pompe patients has attracted much attention.14,22–24 Excessive antibody production against GAA may result in decreased circulating GAA following infusion and limiting receptor mediated enzyme uptake.
Serial serum collection was performed throughout the study to assess the long-term kinetics and degree of immune provocation of anti-GAA antibody production. Importantly, it was necessary to treat Gaa–/– animals receiving ERT with diphenhydramine prior to injection in order to dampen the immune response. Initially, we did not administer diphenhydramine and none of the ERT treated animals survived past the third dose of ERT (data not shown). Anti-GAA titer was significantly higher in the ERT group when compared to the AAV9-DES group, however; all treatments resulted in significant elevation of anti-GAA titer when compared to untreated Gaa–/– (Figure 4).
Figure 4.
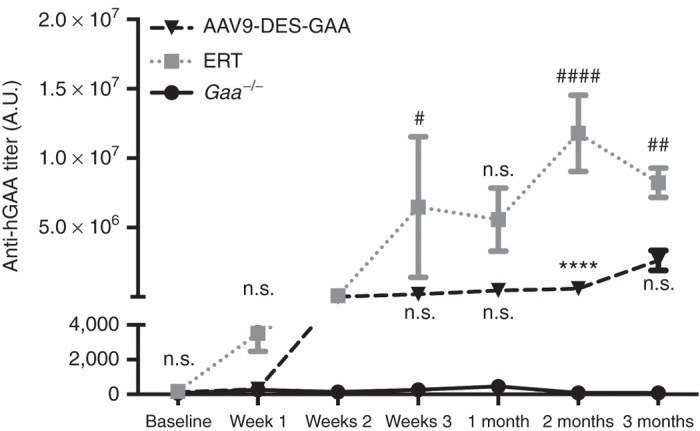
GAA antibody titers following systemic AAV9-DES or continuous ERT therapy. Continuous enzyme replacement therapy (ERT) administration resulted in significant elevation of GAA antibody titers at 3 weeks, 2 months, and 3 months when compared to naive Gaa–/–. ERT-generated GAA antibody titers were significantly increased at 2 months when compared to AAV9-DES treated Gaa–/– animals. Values indicated are the mean ± SEM. #ERT = P ≤ 0.05 versus Gaa–/–. ##ERT = P ≤ 0.01 versus Gaa–/–. ****AAV9-DES = P ≤ 0.0001 versus Gaa–/–. ####ERT = P ≤ 0.0001 versus Gaa–/–. n.s., not significant; GAA, acid α-glucosidase.
Cardiac and diaphragm transduction
Others and we have previously shown that systemically delivered AAV9 effectively transduces cardiac and skeletal muscle in the murine model of Pompe disease.11,21,25,26 We confirmed these observations in the current study: we examined AAV9-DES transduction efficiency in adult animals 3 months postinjection. High vector genome copy number was observed in the diaphragm and cardiac tissues from treated animals. AAV vector genomes were also detected in the spinal cord, suggesting either the travel of vector in a retrograde manner or travel across the blood–brain barrier (Figure 5).
Figure 5.
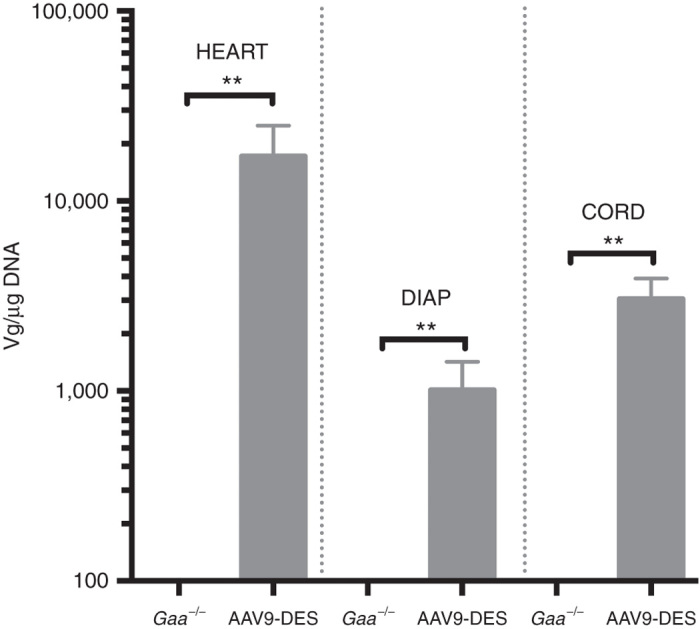
Vector genome quantification of AAV9 following systemic injection. Detection of vector copy number was significantly increased in cardiac, diaphragm, and spinal cord following AAV9 administration. Values indicated are the mean ± SEM. **AAV9-DES = P ≤ 0.01 versus Gaa–/–.
Enzymatic activity and lysosomal glycogen following therapy
The primary goal following administration of an AAV vector or repeated ERT is to increase GAA levels in tissues to reduce the lysosomal glycogen load. Tissues lysates from cardiac, diaphragm, and costal muscles were assayed for GAA activity. As seen in Figure 6, AAV9 resulted in a significant increase of GAA activity in cardiac, diaphragm, and costal muscle when compared to Gaa–/– or ERT. No detection of GAA activity was detected in the ERT group and this was attributed to the time of tissue harvest following the final administration of ERT (12 days).27
Figure 6.
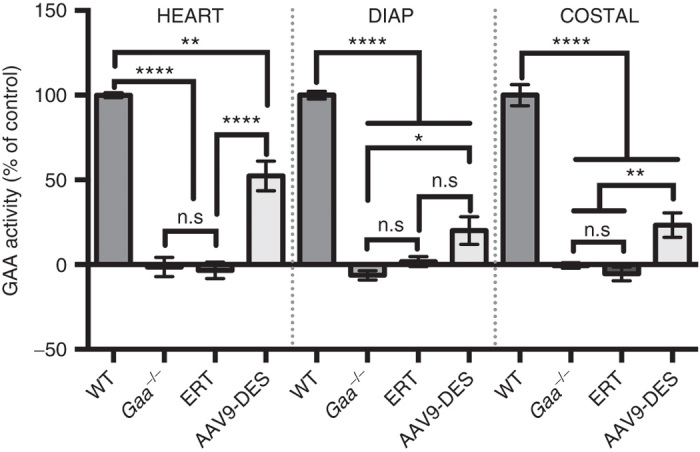
GAA enzymatic activity following systemic AAV9-DES or continuous enzyme replacement therapy (ERT) therapy. GAA activity in the heart, diaphragm, and costal muscle 3 months following AAV9-DES or ERT (2 weeks after final ERT administration). AAV9-DES administration resulted in significant detection of GAA in lysates of the heart, diaphragm, and costal muscle compared to Gaa–/–. Values indicated are the mean ± SEM. * = P ≤ 0.05. ** = P ≤ 0.01. **** = P ≤ 0.001. n.s., not significant; GAA, acid α-glucosidase.
To determine the extent of glycogen clearance in cardiac and respiratory muscles, the left ventricle and diaphragm were examined following Periodic Acid Schiff staining. When compared to Gaa–/–, AAV9 appeared to reduce glycogen deposition in both the heart (Figure 7i) and diaphragm (Figure 7j) compared to Gaa–/– and ERT, however this did not reach statistical significance.
Figure 7.

Glycogen accumulation in the myocardium and diaphragm. At 6 months of age, significant accumulation of glycogen is apparent in the Gaa–/– (b) myocardium and (f) diaphragm when compared to control (a) and (e), ERT (d) and (h), respectively. Reduction of glycogen in the myocardium and diaphragm of AAV9-DES (c and g, respectively) was not significantly elevated compared to WT. Quantification of glycogen content in the fraction of region of interest is presented for the myocardium (i) and diaphragm (j). Inset = 2× magnification. *WT = P ≤ 0.05 versus Gaa–/– or ERT. **WT = P ≤ 0.01 versus ERT. ERT, enzyme replacement therapy; WT, wild type.
Discussion
Without adequate levels of GAA in the lysosome, excessive glycogen accumulation occurs in virtually all tissues. Clinically, the only Food and Drug Administration-approved therapy to replace GAA is through ERT, which must be administered every other week for the entirety of the patient’s life. This strategy has been show to produce variable results in early and late-onset patients and has primarily been attributed to antibody-mediated clearance of circulating GAA and low cation-independent mannose-6-phosphate receptor density.28,29 Moreover, recent evidence has suggested alternative mechanisms such as impaired intracellular trafficking of GAA may limit the effectiveness of ERT.1 Here, we utilized an AAV9 vector to produce widespread cell autonomous production of hGAA to contrast with the existing ERT methodology in the murine model of Pompe disease. Major findings include improved diaphragmatic and cardiac contractile function in Gaa–/– animals receiving ERT or AAV9-DES and improvement in respiratory function was limited to AAV9-DES therapy.
Significant advances have been made since the advent of AAV-based gene therapy for cardiac and skeletal muscle disease. Moreover, vector-mediated transgene delivery has been effective in resultant transgene expression in rodent, feline, canine, porcine, and nonhuman primate models. Specific to Pompe, numerous studies have independently shown that AAV-GAA vectors can efficiently transduce, clear glycogen, and improve skeletal muscle function at various stages of disease in the murine model.15,30 Work from our laboratory has shown that both AAV1 and AAV9 vectors enhance cardiac function and morphology when administered to neonate animals.17,18,25
Gene therapy as an alternative strategy may hold several advantages over ERT for Pompe disease. For example, one inherent difference of ERT compared to AAV-directed therapies is the trafficking of GAA to the lysosome. ERT therapy relies on engagement of the cation-independent mannose 6-phosphate receptor and may be limited due to receptor density and a dampened efficiency in lysosomal targeting from the extracellular pathway.1,31–35 This is in contrast to AAV therapy where transduction of target tissues elicits endogenous production of GAA that is long-lasting and effective in clearance of glycogen.15
We tested and compared an AAV9 vector driven by a tissue-restricted, nonviral promoter (DES) against the current approved therapy for replacement of GAA in Pompe disease (ERT; 20 mg/kg bimonthly). Significant correction of cardiorespiratory measures was not detected at 1 month post-therapy but did emerge at 3 months after initiation of either AAV9 or ERT therapy. Although glycogen levels are immediately reduced in the cell, this may suggest a mechanism where the tissue requires an extended period of time to stabilize cellular function. One of the more interesting findings is related to the disparate findings between the ERT and AAV9 groups by plethysmography. In contrast to ex vivo assessment of diaphragm contractile function, plethysmography allows the assessment of an intact neural-respiratory system in unanesthetized animals. Significant changes in frequency (f), expiratory time (TE), and total respiratory cycle duration (TTOT), were observed in the AAV9-DES group and suggest partial correction of lower motoneurons and improved respiratory timing that ultimately contributes to enhanced respiratory function. We have previously shown that Gaa–/– animals demonstrate decreased hypoglossal and phrenic motoneuron function as a result of disease manifestation10,13,16,36–38 and that transduction limited to the cervical spinal cord improves respiratory function.12 Therefore, these results suggest an additional advantage of AAV9 therapy opposed to ERT due to the transduction properties of motoneurons by AAV9.39–42
Adequate substrate (i.e., lysosomal glycogen) reduction is the goal of therapy for Pompe disease. In this report, we observed persistent levels of GAA in the AAV group alone. We did not observe GAA levels in tissue lysates analyzed at 12 days postinjection. This is in contrast to previous reports where detection of GAA in tissues was observed 12 (ref. 43) and 190 days post-adenovirus vector–mediated liver GAA secretion despite minimal detection of plasma GAA.44 This may be the result of stabilization of GAA once localized to the lysosome resulting in a longer half-life.44 Further studies will elucidate the mechanism by which stability of the enzyme may be enhanced by alternative mechanisms45 to allow persistence of GAA past the reported half-life.
Compared to ERT, a single administration of AAV9-DES significantly reduced modulation of anti-GAA antibody formation over the course of the study. Moreover, animals receiving ERT required preadministration of diphenhydramine HCL to survive the study. Antibody formation following ERT is common and may be dependent on CRIM status and play a role in clinical outcomes.14,29 High antibody titers have been linked to reduced gross motor function and increased cardiac dimension in patients. Furthermore, initiation of ERT therapy may play a role in antibody formation irrespective of CRIM-status.46 This has brought a new emergence of strategies to induce tolerance to GAA. In animal models, liver specific Gaa expressing Pompe models,2 vector-mediated liver restricted GAA expression,47–50 or coadministration of AAV vector and anti-CD4 monoclonal antibody51 has shown an enhanced therapeutic benefit attributed to a dampened immune responses.
Gene therapy for cardiac and skeletal myopathies is an emerging field with a host of ongoing clinical trials and more currently in the stage of enrollment. Identification of serotype tropism and enhancement of transduction efficiencies is key in conferring a single AAV-based strategy to address myopathies that affect both the myocardium and skeletal muscle. Factors that may limit transduction of specific cell types have not been closely studied and may elucidate whether disease severity and antibody formation limits treatment capacity. Clinical trials of AAV-based gene therapy have been well tolerated and shown to provide benefit in the presenting disease phenotype.
Materials and Methods
Packaging and purification of rAAV9 vectors
Recombinant single-stranded AAV9 vector was produced using the triple plasmid transfection method, purified and titered as previously described at the Powell Gene Therapy Center Vector Core at the University of Florida.52 The DES promoter21 and p43.2-GAA plasmid53 have been described previously. The AAV9 packaging plasmid pRep2/Cap9 was a kind gift from Dr. James Wilson (University of Pennsylvania, Philadelphia, PA).
Animals
All animals were group housed with littermates under controlled conditions of temperature and light/dark cycles (12 hours/12 hours). Food and water were provided ad libitum. Three-month-old male and female Gaa–/– mice (Taconic, Germantown, NY) originally developed by Raben et al.3 were outbred to a 129SVE background. All animal studies were approved in accordance with the guidelines set forth by the University of Florida Institutional Animal Care and Use Committee.
In vivo delivery of drug
Mice were anesthetized using 2% isoflurane (1L O2). Under sterile conditions, a 0.5 cm incision was made to expose the jugular vein. Viral vector was administered at day 0 of the study. ERT was administered at day 0 and every 2 weeks following the initiation of the study. Recombinant AAV9 (1 × 1011 vg) was diluted in lactated ringers solution to a quantity sufficient volume of 150 µl for intravenous delivery. Administration of ERT (20 mg/kg) was preceded by an intraperitoneal injection of diphenhydramine (5 mg/kg) 20 minutes prior to intravenous ERT (every other week) over the course of 3 months.
Biochemical assessment
At 3 months postinjection, tissue homogenates were assayed for GAA enzyme activity as described previously.53 Briefly, tissue lysates were assayed for GAA activity by measuring the cleavage of 4-methylumbelliferyl-α-D-glucoside (Sigma M9766, Sigma-Aldrich, St. Louis, MO) after 1 hour incubation at 37 °C.
Histological analysis
Cardiac and diaphragm samples were fixed immediately in 3% glutaraldehyde in 0.2 mol/l Na Cacodylate buffer (pH 7.3) before embedding in epon as described previously.11 Glycogen deposition was quantified in the left ventricle (n = 3/group) and diaphragm (n = 3/group) with Cellsens software (Olympus, Pittsburgh, PA). Images were obtained with a 40× objective. A random region of interest (ROI) was placed on each image and glycogen was analyzed by performing threshold detection followed by count and measure on ROI. This resulted in a percent area occupied on each ROI.
Vector pharmacology
Real-time DNA polymerase chain recation detection was performed as previously described.16 An n = 6 for each group was analyzed and data are reported as AAV vector genome copies per μg DNA ± SE.
Cardiac measurements
Assessment of ejection fraction and cardiac mass.
Cardiac magnetic resonance imaging was performed on a 4.7 T Bruker Avance spectrometer (Bruker BioSpin Corporation, Billerica, MA) at the University of Florida AMRIS facility. The animals were anesthetized using 1.5% isoflurane and 1 L/minute oxygen. The animals were placed prone on a home-built quadrature transmit-and-receive surface coil, with the heart placed at the center of the coil. Images were acquired using IntraGate and were retrospectively reconstructed. The heart was visualized by acquiring single short-axis slices along the length of the left ventricle. Images were processed using CAAS MRV for mouse (Pie Medical Imaging, Maastricht, The Netherlands). Contours were drawn for the epicardium and the endocardium for each slice along the length of the left ventricle at both end diastole and end systole. The results were exported and analyzed and ejection fraction % and end diastolic myocardial mass was calculated following normalization to animal weight. Data were collected at 1 and 3 months after initiation of therapy.
Electrocardiography.
Five lead electrocardiogram tracings were acquired as described previously with modifications.20,25 Once animals reached an anesthetic plane, an electrode was placed in the left lower leg, tail, right scapula region, and right and left forelimb. Following steady acquisition of electrocardiogram tracings, data was recorded and averaged for 3 minutes using AD Instrument Chart software. Data were collected at 1 and 3 months after initiation of therapy.
Ventilation.
These studies were undertaken to determine the impact of systemic therapy on ventilation. Ventilation was quantified using whole-body plethysmography in unrestrained, unanesthetized mice as previously described.13,36 Mice were placed inside a 3.5” × 5.75” Plexiglas chamber which was calibrated with known airflow and pressure signals before data collection. Data were collected in 10-second intervals and respiratory volumes including tidal volume and minute ventilation were calculated as described previously.36 During both a 30 minutes acclimation period and subsequent 30–60 minutes baseline period, mice were exposed to normoxic air (21% O2, 79% N2). At the conclusion of the baseline period, the mice were exposed to a brief respiratory challenge which consisted of a 10-minute hypercapnic exposure (7% CO2, balance O2). Experiments were conducted using naïve and Gaa−/− mice that had received systemic AAV or repeated ERT injection. Data were collected at 3 months after initiation of therapy.
Assessment of diaphragm contractile function.
Isometric force-frequency measurements of costal diaphragm muscle bundles were performed as previously described.54 After determination of Lo, peak isometric tetanic force was measured at 10, 20, 40, 80, 100, 150, and 200 Hz. Single 500-ms trains were used, with a 2-minute recovery period between trains. Data were collected at 3 months after initiation of therapy.
Detection of GAA antibodies.
Briefly, plates were coated with 5 µg/ml of rhGAA in PBS and incubated at 4 °C overnight. Plates were washed three times with phosphate-buffered saline-tween 20 (PBS-T) (0.05%). Blocking was performed with 10% FBS in PBS at room temperature for 2 hours. Following three washes in PBS-T, serum samples were diluted in blocking buffer and incubated in the plate at 4 °C overnight. Plates were washed three times in PBS-T and then incubated with anti-mouse IgG-HRP conjugate (Amersham NA931, GE Healthcare, Piscataway, NJ) diluted 1:10,000 in blocking buffer for 2 hours at room temperature. Following three washes in PBS-T, 3,3’,5,5’-tetramethylbenzidine (Invitrogen 00-2023, Grand Island, NY) was added to each well for 3 minutes. Reaction was stopped following addition of 0.5M H2SO4 and absorbance was read at 450 nm. Samples were run in duplicate and reported as titer.
Statistical analysis
Statistical studies were performed using GraphPad Prism Software. Descriptive statistics were used for each group, and one-way or two-way analysis of variance, followed by Tukey’s post hoc comparison if necessary was used among groups. All results are presented as mean ± SE. P < 0.05 was considered significant.
Acknowledgments
The authors gratefully acknowledge the University of Florida Powell Gene Therapy Center Vector Core Laboratory for production and titering of rAAV vectors and the University of Florida Toxicology Core for vector genome analysis. This work was supported by grants from the National Institutes of Health, NIAMS K01AR066077 (D.J.F.), NHLBI PO1 HL59412-06 (B.J.B.), 2R01HD052682-06A1 (D.D.F., B.J.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Arthritis and Musculoskeletal and Skin Diseases or National Heart, Lung, and Blood Institute of the National Institutes of Health. D.J.F., B.J.B., D.D.F., C.S.M., The Johns Hopkins University and the University of Florida could be entitled to patent royalties for inventions described in this manuscript.
The authors declare no conflict of interest.
References
- Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–708. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- Raben N, Danon M, Gilbert AL, Dwivedi S, Collins B, Thurberg BL. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Raben N, Nagaraju K, Lee E, Kessler P, Byrne B, Lee L. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 1998;273:19086–19092. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- Raben N, Takikita S, Pittis MG, Bembi B, Marie SK, Roberts A. Deconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sand. Autophagy. 2007;3:546–552. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]
- Raben N, Wong A, Ralston E, Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet. 2012;160C:13–21. doi: 10.1002/ajmg.c.31317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Galperin M, Melvin G, Horowits R, Raben N, Plotz P. Impaired organization and function of myofilaments in single muscle fibers from a mouse model of Pompe disease. J Appl Physiol (1985) 2010;108:1383–1388. doi: 10.1152/japplphysiol.01253.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D, Infantile-Onset Pompe Disease Natural History Study Group A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BJ, Kishnani PS, Case LE, Merlini L, Müller-Felber W, Prasad S. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103:1–11. doi: 10.1016/j.ymgme.2011.02.004. [DOI] [PubMed] [Google Scholar]
- ElMallah MK, Falk DJ, Lane MA, Conlon TJ, Lee KZ, Shafi NI. Retrograde gene delivery to hypoglossal motoneurons using adeno-associated virus serotype 9. Hum Gene Ther Methods. 2012;23:148–156. doi: 10.1089/hgtb.2012.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk DJ, Mah CS, Soustek MS, Lee KZ, Elmallah MK, Cloutier DA. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther. 2013;21:1661–1667. doi: 10.1038/mt.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu K, Falk DJ, Reier PJ, Byrne BJ, Fuller DD. Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol Ther. 2012;20:21–27. doi: 10.1038/mt.2011.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmallah MK, Falk DJ, Nayak S, Federico RA, Sandhu MS, Poirier A. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol Ther. 2014;22:702–712. doi: 10.1038/mt.2013.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder ME, Nayak S, Collins SW, Lawson LA, Kelley JS, Herzog RW. B-Cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2013;163:847–54.e1. doi: 10.1016/j.jpeds.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BJ, Falk DJ, Pacak CA, Nayak S, Herzog RW, Elder ME. Pompe disease gene therapy. Hum Mol Genet. 2011;20 R1:R61–R68. doi: 10.1093/hmg/ddr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah CS, Falk DJ, Germain SA, Kelley JS, Lewis MA, Cloutier DA. Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol Ther. 2010;18:502–510. doi: 10.1038/mt.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah C, Cresawn KO, Fraites TJ, Jr, Pacak CA, Lewis MA, Zolotukhin I. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther. 2005;12:1405–1409. doi: 10.1038/sj.gt.3302550. [DOI] [PubMed] [Google Scholar]
- Mah C, Pacak CA, Cresawn KO, Deruisseau LR, Germain S, Lewis MA. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol Ther. 2007;15:501–507. doi: 10.1038/sj.mt.6300100. [DOI] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- Pacak CA, Sakai Y, Thattaliyath BD, Mah CS, Byrne BJ. Tissue specific promoters improve specificity of AAV9 mediated transgene expression following intra-vascular gene delivery in neonatal mice. Genet Vaccines Ther. 2008;6:13. doi: 10.1186/1479-0556-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Doerfler PA, Porvasnik SL, Cloutier DD, Khanna R, Valenzano KJ. Immune responses and hypercoagulation in ERT for Pompe disease are mutation and rhGAA dose dependent. PLoS One. 2014;9:e98336. doi: 10.1371/journal.pone.0098336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, Plug I, van der Ploeg AT, Reuser AJ. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis. 2014;38:305–314. doi: 10.1007/s10545-014-9707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banugaria SG, Patel TT, Mackey J, Das S, Amalfitano A, Rosenberg AS. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012;105:677–680. doi: 10.1016/j.ymgme.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- Sun B, Young SP, Li P, Di C, Brown T, Salva MZ. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol Ther. 2008;16:1366–1371. doi: 10.1038/mt.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuser AJ, Kroos MA, Ponne NJ, Wolterman RA, Loonen MC, Busch HF. Uptake and stability of human and bovine acid alpha-glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients. Exp Cell Res. 1984;155:178–189. doi: 10.1016/0014-4827(84)90779-1. [DOI] [PubMed] [Google Scholar]
- Chien YH, Hwu WL, Lee NC. Pompe disease: early diagnosis and early treatment make a difference. Pediatr Neonatol. 2013;54:219–227. doi: 10.1016/j.pedneo.2013.03.009. [DOI] [PubMed] [Google Scholar]
- Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol. 2013;260:951–959. doi: 10.1007/s00415-012-6636-x. [DOI] [PubMed] [Google Scholar]
- Fuller DD, ElMallah MK, Smith BK, Corti M, Lawson LA, Falk DJ. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol. 2013;189:241–249. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Ploeg AT, Kroos MA, Willemsen R, Brons NH, Reuser AJ. Intravenous administration of phosphorylated acid alpha-glucosidase leads to uptake of enzyme in heart and skeletal muscle of mice. J Clin Invest. 1991;87:513–518. doi: 10.1172/JCI115025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Kikuchi T, Hagiwara Y, Mizutani M, Chen YT, Van Hove JL. Recombinant human acid alpha-glucosidase corrects acid alpha-glucosidase-deficient human fibroblasts, quail fibroblasts, and quail myoblasts. Pediatr Res. 1998;43:374–380. doi: 10.1203/00006450-199803000-00011. [DOI] [PubMed] [Google Scholar]
- Maga JA, Zhou J, Kambampati R, Peng S, Wang X, Bohnsack RN. Glycosylation-independent lysosomal targeting of acid α-glucosidase enhances muscle glycogen clearance in pompe mice. J Biol Chem. 2013;288:1428–1438. doi: 10.1074/jbc.M112.438663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberl DD, Austin S, Case LE, Smith EC, Buckley AF, Young SP. Adjunctive albuterol enhances the response to enzyme replacement therapy in late-onset Pompe disease. FASEB J. 2014;28:2171–2176. doi: 10.1096/fj.13-241893. [DOI] [PubMed] [Google Scholar]
- Li S, Sun B, Nilsson MI, Bird A, Tarnopolsky MA, Thurberg BL. Adjunctive β2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J. 2013;27:34–44. doi: 10.1096/fj.12-207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH, Jr, Mah C. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk DJ, Mah CS, Soustek MS, Lee KZ, Elmallah MK, Cloutier DA. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther. 2013;21:1661–1667. doi: 10.1038/mt.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KZ, Qiu K, Sandhu MS, Elmallah MK, Falk DJ, Lane MA. Hypoglossal neuropathology and respiratory activity in pompe mice. Front Physiol. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockstael O, Foust KD, Kaspar B, Tenenbaum L. Recombinant AAV delivery to the central nervous system. Methods Mol Biol. 2011;807:159–177. doi: 10.1007/978-1-61779-370-7_7. [DOI] [PubMed] [Google Scholar]
- Hester ME, Foust KD, Kaspar RW, Kaspar BK. AAV as a gene transfer vector for the treatment of neurological disorders: novel treatment thoughts for ALS. Curr Gene Ther. 2009;9:428–433. doi: 10.2174/156652309789753383. [DOI] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan AK, Duque S, Foust KD, Morales PR, Braun L, Schmelzer L. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther. 2011;19:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalfitano A, McVie-Wylie AJ, Hu H, Dawson TL, Raben N, Plotz P. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci USA. 1999;96:8861–8866. doi: 10.1073/pnas.96.16.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding EY, Hodges BL, Hu H, McVie-Wylie AJ, Serra D, Migone FK. Long-term efficacy after [E1-, polymerase-] adenovirus-mediated transfer of human acid-alpha-glucosidase gene into glycogen storage disease type II knockout mice. Hum Gene Ther. 2001;12:955–965. doi: 10.1089/104303401750195917. [DOI] [PubMed] [Google Scholar]
- Khanna R, Flanagan JJ, Feng J, Soska R, Frascella M, Pellegrino LJ. The pharmacological chaperone AT2220 increases recombinant human acid α-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS One. 2012;7:e40776. doi: 10.1371/journal.pone.0040776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C van Gelder MK, Ozkan L, Plug I, Reuser A, van der Ploeg A.2013Antibody formation to enzyme therapy in classic infantile Pompe disease: implications of patient age BMC Musculoskelet Disord 14 (suppl 2)1823302451 [Google Scholar]
- Franco LM, Sun B, Yang X, Bird A, Zhang H, Schneider A. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12:876–884. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Zhang P, Sun B, Osada T, Rodriguiz R, Yang XY, Luo X. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther. 2012;23:460–472. doi: 10.1089/hum.2011.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Kulis MD, Young SP, Hobeika AC, Li S, Bird A. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther. 2010;18:353–360. doi: 10.1038/mt.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SO, Li S, Brooks ED, Masat E, Leborgne C, Banugaria S. Enhanced efficacy from gene therapy in pompe disease using coreceptor blockade. Hum Gene Ther. 2015;26:26–35. doi: 10.1089/hum.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ., Jr Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- Fraites TJ, Jr, Schleissing MR, Shanely RA, Walter GA, Cloutier DA, Zolotukhin I. Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol Ther. 2002;5 5 Pt 1:571–578. doi: 10.1006/mthe.2002.0580. [DOI] [PubMed] [Google Scholar]
- Mah C, Pacak CA, Cresawn KO, Deruisseau LR, Germain S, Lewis MA. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol Ther. 2007;15:501–507. doi: 10.1038/sj.mt.6300100. [DOI] [PubMed] [Google Scholar]