

Abstract
It is a current regulatory requirement to demonstrate absence of detectable replication-competent lentivirus (RCL) in lentiviral vector products prior to use in clinical trials. Immune Design previously described an HIV-1-based integration-deficient lentiviral vector for use in cancer immunotherapy (VP02). VP02 is enveloped with E1001, a modified Sindbis virus glycoprotein which targets dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) expressed on dendritic cells in vivo. Vector enveloped with E1001 does not transduce T-cell lines used in standard HIV-1-based RCL assays, making current RCL testing formats unsuitable for testing VP02. We therefore developed a novel assay to test for RCL in clinical lots of VP02. This assay, which utilizes a murine leukemia positive control virus and a 293F cell line expressing the E1001 receptor DC-SIGN, meets a series of evaluation criteria defined in collaboration with US regulatory authorities and demonstrates the ability of the assay format to amplify and detect a hypothetical RCL derived from VP02 vector components. This assay was qualified and used to test six independent GMP production lots of VP02, in which no RCL was detected. We propose that the evaluation criteria used to rationally design this novel method should be considered when developing an RCL assay for any lentiviral vector.
Introduction
In order for lentiviral vector products to be used in human clinical trials, it is a regulatory requirement to demonstrate absence of detectable replication-competent lentivirus (RCL) in each batch of vector and end-of-production cells (EOPC).1 Third-generation lentiviral vectors are designed to minimize the risk of generating an RCL by (i) splitting vector genome components onto separate expression plasmids, (ii) minimizing homology between genetic segments to reduce the risk of reassembly by homologous recombination, (iii) optimizing codons for protein expression (to eliminate cis-acting viral replication functions, and (iv) employing a self-inactivating 3′-LTR, which ensures vector genomes are less likely to be mobilizable in target cells).2,3 Indeed, there has never been an RCL detected in a third-generation lentiviral vector to date. Nonetheless, regulatory bodies still require RCL testing for all lots of lentiviral vector, given that it is theoretically possible that during vector production a very rare series of nonhomologous recombination events could generate a functional viral genome, resulting in an RCL.1,4 Immune Design has developed an HIV-1-based lentiviral vector (VP02) designed to target the dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) receptor on human dendritic cells (DCs) in vivo.3 Following entry (transduction) into antigen-presenting cells, specific antigens encoded in the vector genome are expressed by cellular machinery and subsequently presented as peptides via the MHC class I pathway, which in animal models has resulted in the generation of robust CD8 T-cell responses and protective and therapeutic immunity.5 An NY-ESO-1-expressing VP02 vector is being developed as an active immunotherapy for use in the field of cancer and is currently under clinical investigation. We now report the design and qualification of an assay to detect the presence of RCL in preparations of VP02 vectors and associated EOPC.
VP02 has several unique features in comparison to other third-generation lentiviral vectors. First, unlike traditional lentiviral vectors that encode the pan-tropic vesicular stomatitis virus envelope glycoprotein (VSVG), VP02 utilizes a modified Sindbis virus envelope glycoprotein (E1001, Figure 1a) with post-translational carbohydrate modifications that enhance binding to human DCs via DC-SIGN. Modification of E1001 by high-mannose glycosylation is achieved by inclusion of the mannosidase-I inhibitor kifunensine during vector production.3,6,7 Second, the simian immunodeficiency virus (SIVmac) accessory protein Vpx is incorporated into vector particles and mediates VP02 transduction of human DCs by degrading the restriction factor sterile alpha motif (SAM) domain and HD domain-containing protein 1 present in the target cells.3,8,9 Third, VP02 is integration-deficient, because of a D64V Integrase mutation within the gag/pol gene and the deletion of the 3′-polypurine tract (3′-PPT) within the vector genome, a modification which promotes circularization of vector DNA in target cells following reverse transcription.3,10 Although an RCL might recruit functional sequences from nucleic acids derived from the production cell, it is assumed that any putative RCL generated during production of VP02 vector would most likely be derived from components of the VP02 vector production system, including these distinct elements.
Figure 1.

Schematic of the VP02 vector system and a generic replication-competent lentivirus (RCL) assay. (a) Structure of wild-type HIV-1 and the five components of the VP02 vector system. Regions of homology between vector components are marked by dotted lines. The vector genome encodes a modified ubiquitin promoter (UBp) upstream of an antigen (Ag), the woodchuck hepatitis post-transcriptional regulatory element (WPRE), and an extended deletion within the U3 and 3′-PPT regions (ΔU3). The gag/pol vector component has been codon-optimized to reduce homology to wild-type HIV-1; however, the sequence of the frame-shift region has been maintained to ensure proper translation of the Gag and Gag-Pol polypeptides. In addition, the pol gene encodes a D64V point mutation within the catalytic site of the Integrase protein to abrogate Integrase-dependent vector integration. VP02 contains two accessory proteins: Vpx from SIVmac and Rev from HIV-1. Shaded regions denote HIV-1 sequence conserved in the VP02 vector system. VP02 is pseudotyped with the heterologous envelope glycoprotein E1001. (b) Diagram of standard cell culture-based RCL assays. Vector product (test article) is used to transduce permissive amplification cells. Small amounts of replication-competent virus which may be present in the original test article are expected to replicate during subsequent cell passages (amplification phase). Following ~4 weeks in cell culture, cell supernatant is analyzed for the presence of virus using a sensitive detection method for components of the virus particle (p24 by ELISA) or RT enzymatic activity (F-PERT assay). F-PERT, fluorescent-product enhanced reverse transcriptase; SIV, simian immunodeficiency virus.
Tests for RCL are typically carried out on each batch of lentiviral vector and the EOPC according to regulatory recommendations with a specified test sample (volume, percentage of batch, or number of cells).1 To test for a very rare, putative RCL, an assay typically starts with a biological amplification phase. First, permissive “amplification” cells are inoculated with a preparation of lentiviral vector (test article) or a positive control virus. These cells are then passaged sequentially and at the endpoint they are assayed for viral particles using a sensitive detection method (Figure 1b).11–13 This passaging regimen is designed to allow a single infection event because of a putative replication-competent virus to amplify to detectable levels above assay background (the “amplification phase”). Moreover, the serial passaging of the amplification phase also dilutes out assay signal contributed by input vector (test article) or contaminating nucleic acid sequences used to generate the vector, thus avoiding false-positive test results. For EOPC testing, EOPC are cocultured with amplification cells prior to the amplification phase. Virus amplification (either from the positive control virus or from a putative RCL) is detected using one of several methods, including a PCR-based fluorescent-product enhanced reverse transcriptase (F-PERT) assay or p24 ELISA, in the endpoint or “detection phase” of the RCL assay.11,13,14
Published reports have described an RCL assay format employing the C8166-45 T-cell line and this format has been used to meet RCL testing requirements for numerous manufacturing lots of VSVG-pseudotyped, HIV-1-based lentiviral vectors.11,12 However, exploratory studies we conducted demonstrated that C8166-45 cells do not express the DC-SIGN receptor targeted by the E1001 envelope, which prevents transduction by VP02 vectors. It therefore follows that the “standard” RCL assay is incompatible with testing VP02 vectors, because unmodified C8166-45 cells are not expected to amplify an E1001-enveloped RCL. Our aim was to design and qualify a novel assay to detect the presence of a putative RCL in preparations of E1001-enveloped vector.
Owing to the unique nature of the VP02 vector, in designing this novel RCL assay we evaluated five scientific approaches comprising different combinations of positive control virus and assay amplification cell type. The likelihood of each assay approach to be able to detect an RCL was assessed based on three evaluation criteria established in collaboration with US regulatory authorities. These criteria were designed to demonstrate the ability of the assay cell line to amplify both an E1001-enveloped RCL (should one exist) and the chosen positive control virus. This manuscript describes the evaluation process and ultimate scientific design of a novel method to detect the presence of RCL in E1001-enveloped lentiviral vector preparations. This assay has been qualified and used to test six independent, large-scale production lots of VP02 product and EOPC, during which no RCL was detected.
Results
RCL assay design approaches and evaluation criteria
Our goal was to adapt the standard RCL assay format (comprised of an amplification phase and a detection phase, see Figure 1b) to be compatible with testing VP02 vector and corresponding EOPC. To this end, it was necessary to identify both an appropriate positive control virus (that would best model a putative E1001-enveloped RCL) and an assay amplification cell line, permissive for VP02 transduction. Ideally, the positive control used in an RCL assay for VP02 would model aspects of a putative RCL that could in theory be derived from the VP02 production system, if technically feasible. In principle, engineering HIV-1 to express the E1001 envelope glycoprotein instead of gp160 would have yielded a positive control virus with a genomic structure most similar to a putative VP02-derived RCL, although the fitness of such a virus is not predictable. However, altering the tropism of a replication-competent human virus was deemed an unacceptable increase in risk to assay operators.15 Therefore, a primary objective of development was to identify a positive control virus that was as similar to a putative RCL as feasible, taking into consideration viral replication cycle, capsid and genome, mechanism of viral entry, and operator and environmental safety.
Specific design elements render VP02 to be integration deficient (Figure 1a). Based on the reported observation that integration is required for effective HIV-1 replication, it is highly likely that any “true” RCL derived from vector components will necessarily have acquired integration competence.16 To empirically test this theory, we investigated whether an RCL assay positive control virus could be engineered to be integration deficient, to match the expected replication kinetics of a putative integration-deficient RCL. To test this, two proviral HIV-1 clones were constructed: integration-competent HIV-1 positive control (HIVPC) and integration-deficient HIV-1 positive control (HIVPC-ID) (Figure 2a). HIVPC was based on HIV-1 strain NL4-3 (the strain from which HIV-1-based ID-VP02 vector elements are derived) and encodes the native HIV-1 envelope and multiple knock-out mutations of four accessory proteins (Vif, Vpr, Vpu, and Nef). HIVPC-ID encodes a D64V Integrase mutation and extended deletion of the 3′-PPT, mirroring the mutations that render VP02 integration defective (Figure 1a). Virus stocks were generated in HEK293T cells and assayed for replication in C8166-45 cells (see Supplementary Figure S1). Although integration-competent HIVPC amplified ~4 logs over the 9-day passage period, cells inoculated with HIVPC-ID virus were indistinguishable from mock-infected cells, even when input virus was increased by 10-fold. These results suggest that an integration-deficient RCL would likely not replicate, which underlines the utility of this safety feature engineered into the therapeutic VP02 vector. However, this design is not suitable for a positive control virus and thus all subsequent RCL assay development work encompassed only integration-competent positive control viruses.
Figure 2.
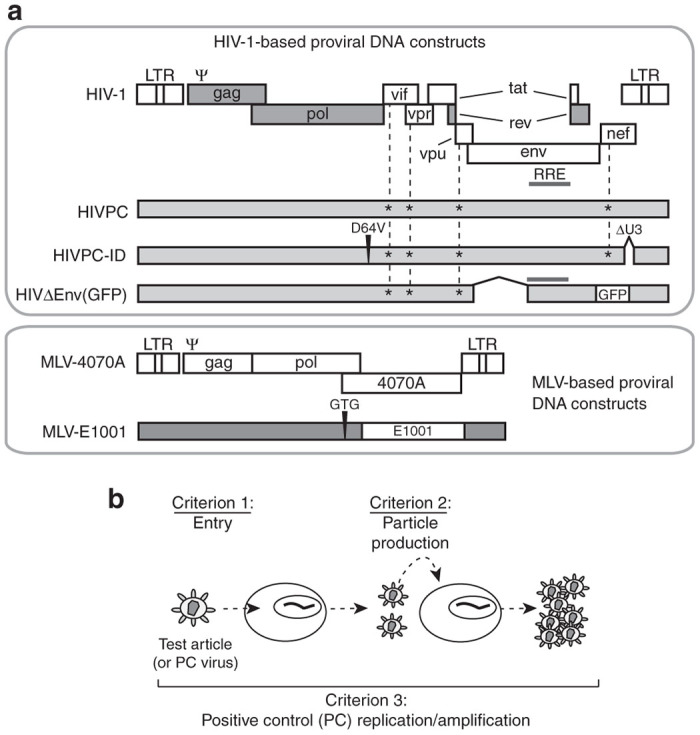
Positive control proviral DNA constructs used and replication-competent lentivirus (RCL) assay evaluation criteria. (a) Proviral DNA constructs generated in this study. HIVPC encodes knock-out mutations (*) of Vif, Vpr, Vpu, and Nef. HIVPC-ID additionally encodes a D64V Integrase mutation and an extended deletion within the U3 and 3′-PPT regions (ΔU3), rendering it integration defective. HIVΔEnv(GFP) lacks an encoded envelope gene and encodes GFP in the nef transcription unit. MLV-E1001 was generated by mutating the initiator methionine of the 4070A ORF in MLV-4070A, and replacing downstream sequences with the E1001 ORF. (b) RCL assay evaluation criteria. Criterion 1: Ability of E1001-enveloped vector particles to enter the chosen amplification cells. Criterion 2: Release of transduction-competent, E1001-enveloped vector particles from amplification cells. Criterion 3: Replication and amplification of the chosen positive control (PC) virus in the assay cell line from low MOI. GFP, green fluorescence protein; HIVPC, HIV-1 positive control; HIVPC-ID, integration-deficient HIV-1 positive control; MOI, multiplicity of infection.
To meet sensitivity, specificity, and reproducibility requirements of an RCL assay suitable for testing VP02, we pursued five different RCL assay design approaches (designated Approaches A–E, summarized in Table 1), comprising different combinations of positive control virus and assay amplification cell type. Potential positive control viruses were selected based on their similarity to a putative E1001-enveloped RCL in one or more respects, noting whether critical positive control elements are derived from HIV-1, Sindbis virus, or murine leukemia virus (MLV).
Table 1. RCL assay design approaches.
| Approach |
Positive control characteristics |
Amplification cell line characteristics |
||||||
|---|---|---|---|---|---|---|---|---|
| Positive control | Envelope | Capsid, genome and replication | Cell type | DC-SIGN | CD4 | CXCR4 | E1001 | |
| A | HIVPCa | gp120 | HIV-1 | C8166-45 | Introduced | Native | Native | — |
| B | HIV-1b | gp120 | HIV-1 | 293F | Introduced | Introduced | Native | — |
| C | HIVΔEnv(GFP)c | E1001 | HIV-1 | 293F | Introduced | — | Native | Introduced |
| D | MLV-E1001d | E1001 | MLV | 293F | Introduced | — | Native | — |
| E | MLV-4070Ae | 4070A | MLV | 293F | Introduced | — | Native | — |
| Putative E1001-enveloped RCL | E1001 | HIV-1 | ||||||
HIV-1 strain NL4-3 encoding knock-out mutations of Vif, Vpr, Vpu, and Nef. NL4-3 is the laboratory strain of HIV-1 that is most similar to the HIV-1-based vector components of VP02.
Wild-type HIV-1 strain NL4-3.
HIVPC modified to remove the envelope gene and to encode GFP in the nef transcription unit; conditionally replication-competent when an envelope glycoprotein is provided in trans.
MLV modified to encode E1001 envelope glycoprotein instead of the MLV envelope.
Hybrid moloney/amphotropic MLV encoding amphotropic 4070A envelope glycoprotein.
MLV, murine leukemia virus; RCL, replication-competent lentivirus; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin; HIVPC, HIV-1 positive control; CXCR4, C-X-C chemokine receptor type 4; GFP, green fluorescence protein.
The suitability of each RCL assay design approach was assessed based on three evaluation criteria designed in collaboration with US regulatory authorities (Figure 2b). Criterion 1 is to demonstrate the ability of E1001-enveloped vector particles to enter the chosen amplification cells, as determined by transduction and expression of a green fluorescence protein (GFP) transgene. Criterion 2 is to demonstrate the release of transduction-competent, E1001-enveloped vector particles from amplification cells. Both of these evaluation criteria must be met to demonstrate that a putative RCL present in a test article would be capable of entering (criterion 1) and being produced in (criterion 2) the chosen assay cell line. Criterion 3 is to demonstrate replication and amplification of the chosen positive control virus in the assay cell line from low multiplicity of infection (MOI), to demonstrate high assay sensitivity. An RCL assay design approach had to satisfy all three of these evaluation criteria to be deemed suitable for release testing of a sample of VP02 lentiviral vector and EOPC.
Evaluation of RCL design Approach A: HIVPC virus and C8166-DC-SIGN cells
The principle of the first assay design approach (Approach A, Table 1) was to utilize an attenuated HIV-1-based positive control virus (HIVPC, Figure 2a), which would closely model the genome and capsid of the VP02 vector, but for safety reasons, not the envelope glycoprotein (or by definition, the mechanism of cell entry). Utilizing this positive control virus required the identification of a cell line permissive for both HIV-1 replication and transduction by vector enveloped with E1001. This approach was in principle the most similar to RCL assay methods commonly used for VSVG-pseudotyped, HIV-1-based lentiviral vectors.11,12,17 Therefore, we chose to modify the standard RCL assay cell line (C8166-45) to introduce the cellular receptor for E1001 envelope, DC-SIGN (C8166-DCSIGN, data not shown). E1001-enveloped vector effectively transduced C8166-DCSIGN cells (evaluation criterion 1) and this transduction was significantly enhanced when vector was produced in the presence of kifunensine (Figure 3a), similar to results previously observed in other cell lines.3 In comparison, unmodified C8166-45 cells were not transduced, as predicted based on their lack of DC-SIGN expression. C8166-DCSIGN cells were also permissive for HIVPC replication, satisfying evaluation criterion 3 (data not shown).
Figure 3.

Evaluation of a replication-competent lentivirus assay format utilizing C8166-DCSIGN cells and an attenuated HIV-1 positive control (Approach A). (a) Transduction efficiency of E1001-enveloped HIV-1 vector encoding GFP and produced in the presence or absence of kifunensine was assessed in C8166-45 parental cells and those engineered to express DC-SIGN (C8166-DCSIGN). Cells were incubated with vector and then assayed for GFP fluorescence at 3 days post-transduction. (b) Analysis of total vector particle release from 293F, C8166-45, and C8166-DCSIGN cells. Replication defective HIVΔEnv(GFP) was delivered to target cells in combination with one of the following: E1001 envelope (E); E1001 envelope + 1 μg/ml kifunensine (E + kifu); or no envelope (–). At 5 days post-transduction, cell supernatants were analyzed by F-PERT assay to detect RT activity (arbitrary units, ArbU). (c) Cell supernatants from b were titered onto fresh 293F-DCSIGN or C8166-DCSIGN cells and analyzed for GFP fluorescence. Vector produced by the standard transient transfection process in HEK293T cells was included as a positive control for transduction. F-PERT, fluorescent-product enhanced reverse transcriptase; GFP, green fluorescence protein.
The engineered C8166-DCSIGN cell line was next evaluated for the ability to produce transduction-competent E1001 vector particles (evaluation criterion 2). This criterion is important because it demonstrates whether or not the cell line in question could release replicated RCL to propagate and amplify a putative E1001-enveloped RCL, should it exist in a preparation of vector or EOPC. To determine whether transduction-competent particles could be released from C8166-DCSIGN cells, we needed a method of producing E1001-enveloped vector in this cell type. C8166-DCSIGN cells were refractory to all transient transfection methods attempted, such that no plasmid-derived protein expression was observed; therefore, vector components were delivered to these cells by vector transductions. In brief, a replication-defective HIV-1 provirus was constructed lacking an encoded envelope gene and encoding GFP in the nef transcription unit (HIV∆Env(GFP), Figure 2a). This provirus encodes all the components sufficient to assemble vector particles that may be pseudotyped with an envelope provided in trans. HIV∆Env(GFP) vector pseudotyped with VSVG was incubated with target cells in combination with an MLV vector engineered to deliver the E1001 open reading frame (ORF) (to trans-complement HIV∆Env(GFP)). Cells were then incubated with or without kifunensine. At 5 days postdelivery of vector components, cell supernatant was harvested and assayed for release of total particles (F-PERT assay) or transduction-competent particles (transduction assay). 293F cells, a derivative of HEK293 cells, were tested in parallel as a positive control for production of transduction-competent vector.
Reverse transcriptase (RT)-containing particles were detectable in supernatants from all cell types, demonstrating that vector particles were assembled and released (Figure 3b). However, only supernatants from 293F cells produced GFP fluorescence when titrated on target C8166-DCSIGN cells or 293F cells expressing DC-SIGN (293F-DCSIGN) (Figure 3c). These results demonstrated that only 293F cells produced transduction-competent vector. E1001 expression in C8166-45-based cells appeared to have a negative impact on HIV-1 particle release; however, these differences in total particle release between 293F cells and C8166-derived cell lines cannot account for the failure of transduction-competent particles to be detected in C8166-45 or C8166-DCSIGN cell supernatants. Western blot showed that E1001 envelope was highly expressed in C8166-DCSIGN cells in these experiments, demonstrating that defective particle release was not because of the lack of available envelope glycoprotein (data not shown). Overall, these data demonstrate that C8166-based cells produced noninfectious vector particles, despite abundant expression of E1001 envelope glycoprotein by the cell. For this reason, RCL assay design Approach A failed to meet evaluation criterion 2 (Figure 2b).
Evaluation of RCL design Approach B: wild-type HIVPC virus and 293F-DCSIGN-CD4 cells
In contrast to C8166-45 cells, the 293F cells tested were capable of producing transduction-competent VP02 vector (evaluation criterion 2, Figure 3c). Therefore, subsequent RCL assay design approaches focused on utilizing HEK293 derivatives for the assay amplification cell line. In assay design Approach B (Table 1), the viability of using this cell type in combination with HIV-1, the standard RCL assay positive control virus, was examined. To enable transduction of E1001-enveloped vector (test article) and infection of gp120-enveloped virus (HIV-1-based vector or HIVPC), 293F cells were engineered to stably express DC-SIGN and CD4, in addition to low-level endogenous expression of C-X-C chemokine receptor type 4 (CXCR4) (293F-DCSIGN-CD4) (Figure 4a).18 The capacity of the engineered receptors to support cell entry was tested by incubating 293F-DCSIGN-CD4 cells with gp120- or E1001-enveloped vector encoding GFP and assaying the target cells for GFP fluorescence (Figure 4b). Transductions were performed in the absence or presence of nevirapine, a reverse-transcriptase inhibitor, to measure assay background. 293F-DCSIGN-CD4 cells were transduced by gp120-enveloped vector at levels similar to C8166-45 cells, demonstrating that the CD4 and CXCR4 proteins expressed in the new cell line were functional. In parallel, 293F-DCSIGN-CD4 cells were also transduced by E1001-enveloped vector, demonstrating that DC-SIGN expressed in this cell line was similarly functional (evaluation criterion 1).
Figure 4.

Evaluation of a replication-competent lentivirus assay format utilizing a wild-type HIV-1 positive control virus and 293F-DCSIGN-CD4 cells (Approach B). (a) 293F, 293F-DCSIGN, 293F-DCSIGN-CD4, and C8166-45 cells were analyzed by flow cytometry for expression of DC-SIGN, the HIV-1 receptor CD4, and coreceptor CXCR4. Open histograms indicate unstained controls. (b) Transduction by gp120- or E1001-enveloped vector was assessed in the following cell lines: C8166-45, 293F-DCSIGN, and 293F-DCSIGN-CD4. Cells were incubated with vector encoding GFP in the presence or absence of the reverse-transcriptase inhibitor nevirapine (Nev). At 48 hours post-transduction, cells were analyzed for GFP fluorescence. (c) E1001-enveloped vector production was assessed in 293F, 293F-DCSIGN, and 293F-DCSIGN-CD4 cells. Using calcium phosphate, each cell type was transfected with vector component plasmids to generate E1001-enveloped HIV-1 vector encoding GFP. At 48 hours post-transfection, harvested supernatant was incubated with fresh cognate cells in the presence or absence of nevirapine; GFP fluorescence was analyzed after 72 hours. (d) C8166-45, 293F-DCSIGN, or 293F-DCSIGN-CD4 cells were inoculated with wild-type HIV-1 (strain NL4-3) at the MOI noted. Subsequent release of viral particles into supernatant fluid was measured using the F-PERT assay over four passages. As a negative control, spent media from mock-infected parental C8166-45 or 293F cells was assayed in parallel at each passage. F-PERT, fluorescent-product enhanced reverse transcriptase; GFP, green fluorescence protein.
We next investigated whether the 293F-DCSIGN-CD4 cell line was capable of producing transduction-competent, E1001-enveloped vector (evaluation criterion 2). 293F, 293F-DCSIGN, or 293F-DCSIGN-CD4 cells were transfected with component plasmids to generate GFP-encoding, E1001-enveloped vector; supernatants were then assayed for transduction on the various cell types. All three cell types tested produced viable vector particles (Figure 4c). These results demonstrate that the 293F-DCSIGN-CD4 cell line (as well as Approach B) satisfies evaluation criterion 2.
The 293F-DCSIGN-CD4 cell line was next evaluated for the ability to support replication of an HIV-1-based positive control virus. Initial experiments utilizing the multiply-attenuated HIVPC virus (Figure 2a) failed to demonstrate any viral replication (data not shown). Previous work has shown that the accessory proteins Nef and Vpu (knocked out in HIVPC) act to downregulate CD4 in cells infected with wild-type HIV-1, thereby preventing sequestration of gp120 envelope by CD4 within internal cellular compartments (reviewed in ref. 19). We hypothesized that Nef and Vpu may be required to facilitate egress of progeny gp120-enveloped virions from the engineered 293F-DCSIGN-CD4 cell line. Thus, a wild-type HIV-1 provirus (strain NL4-3) was used to investigate this hypothesis. C8166-45, 293F-DCSIGN, and 293F-DCSIGN-CD4 cells were inoculated with wild-type HIV-1 at low MOI, and shedding of RT-containing viral particles into the media was assayed for by F-PERT analysis at subsequent passage points. Input virus at an MOI of 0.2 infected 293F-DCSIGN-CD4 cells, resulting in low level, steady-state de novo viral particle production (Figure 4d). However, there was no amplification of virus in this cell line during the first four cell passages, demonstrating failure of the virus to replicate. Infection of 293F-DCSIGN-CD4 cells initiated at an MOI of 0.02 also produced a similar steady-state pattern (data not shown), indicating that cells infected at the higher dose had not become maximally infected within the first few days of the time course. In contrast, viral replication was clearly demonstrated in C8166-45 cells, which were inoculated at an MOI of 0.002. Although 293F-DCSIGN-CD4 cells expressed functional CD4 and CXCR4, this cell line did not support replication of an HIV-1-based positive control virus, and therefore Approach B failed to meet RCL assay evaluation criterion 3.
Evaluation of RCL design Approach C: HIVΔEnv(GFP) positive control vector and 293F-DCSIGN-E1001 cells
Because in Approach B it appeared that 293F-based cells could not be easily made permissive to an HIV-1 enveloped positive control through expression of CD4, the concept of engineering an E1001-enveloped positive control was examined in a third line of investigation—Approach C (Table 1). As discussed previously, it was not desirable to generate a replication-competent HIV-1-based virus encoding the E1001 envelope, for reasons of operator safety. As an alternative, Approach C considered the use of a conditionally replication-competent HIVPC vector (i.e., not a fully functional virus) named HIVΔEnv(GFP), which lacks an encoded envelope gene and encodes GFP in the nef transcription unit (Figure 2a). The 293F-DCSIGN cell line was modified to stably express the envelope glycoprotein E1001 (293F-DCSIGN-E1001), thereby constitutively providing an envelope in trans to the vector positive control (Figure 5a). In this system, kifunensine should in principle be included in the medium during cell passaging, to enhance E1001 binding to DC-SIGN on target cells and thereby promote vector amplification. It was hypothesized that this conditional vector-replicating system could mimic virus replication without increasing the general safety risk posed by engineering a chimeric HIV-1 virus with altered tropism.
Figure 5.

Evaluation of a replication-competent lentivirus (RCL) assay format utilizing a conditionally replication-competent HIVΔEnv(GFP) positive control vector and 293F-DCSIGN-E1001 cells (Approach C). (a) Schematic of RCL assay design Approach C, in which the positive control is a vector lacking an encoded envelope gene; instead, the E1001 envelope glycoprotein is provided in trans by the assay cell line (293F-DCSIGN-E1001). (b) Amplification of the HIVΔEnv(GFP) positive control vector was evaluated in 293F-DCSIGN cells alone (DCS) or stably expressing the E1001 envelope glycoprotein (DCS/E1001). Cells were transduced with HIVΔEnv(GFP) positive control vector enveloped with E1001. Low input (lo) corresponds to 100-fold less vector, as compared to high (hi). At each passage, cells were analyzed for GFP expression and culture supernatants were transferred onto fresh cells. Where noted, kifunensine was present in the culture media at 1 µg/ml for the duration of the experiment. Untreated 293F-DCSIGN-E1001 cells served as a negative control (mock). GFP, green fluorescence protein.
To test the conditional vector-replicating system, 293F-DCSIGN or 293F-DCSIGN-E1001 cells were transduced with increasing doses of HIVΔEnv(GFP) positive control vector enveloped with E1001, then washed to remove input vector. Duplicate cultures were then incubated +/– kifunensine. At subsequent cell passages, culture supernatants were transferred onto fresh cells (without cell carryover) and the cells from that culture step were analyzed for GFP expression to detect transduction. In this method, only de novo transduction events are measured at each passage. As expected, the HIVΔEnv(GFP) positive control vector could not be serially transferred through control 293F-DCSIGN cells from passage to passage, at either the high or low input vector doses tested (Figure 5b). In contrast, 293F-DCSIGN-E1001 cells allowed entry and exit of apparently functional E1001-enveloped HIVΔEnv(GFP) particles at every subsequent passage when initiating the “infection” at high doses. When starting with low amounts of input vector, this effect gradually decreased over subsequent passages, indicating lack of fitness of this “infection” paradigm. Culturing the vector-producing cells in the presence of kifunensine (to increase affinity of released E1001-enveloped particles for DC-SIGN on target cells) increased the number of GFP-positive cells obtained from the low dose input vector ~10-fold, but did not result in net vector amplification. Similar results were observed when cells (not supernatant) were transferred at each passage (data not shown), indicating that efficiency of vector transfer could not be enhanced through cell-mediated mechanisms. In conclusion, the HIVΔEnv(GFP) vector positive control could conditionally replicate in 293F-DCSIGN-E1001 amplification cells from relatively high titer inputs (satisfying evaluation criteria 1 and 2), but no significant biological amplification was observed when using limiting input quantities (MOI < 0.5), demonstrating failure of Approach C to meet evaluation criterion 3.
Evaluation of RCL design Approaches D and E: MLV-based positive control virus and 293F-DCSIGN cells
The goal of Approaches D and E (Table 1) was to identify a positive control virus that replicates in the 293F-DCSIGN amplification cell line and is genetically similar to HIV-1 (i.e., a retrovirus). The 293F-DCSIGN cell line was chosen based on its ability to be transduced by E1001-enveloped vector (Figure 4b) and to produce transduction-competent vector enveloped with E1001 (Figure 4c), thereby satisfying the first two assay evaluation criteria. Furthermore, 293F-DCSIGN cells are genetically distinct from the HEK293T vector production cell line, making it possible to rule out false-positive signal should such an event occur during testing EOPC in the final RCL assay.13 In identifying a potential positive control virus, nonhuman tropic lentiviruses were precluded from consideration, as replication of these viruses requires expression of a species-specific Cyclin-T1 and cellular receptor, neither of which is present in either 293F-DCSIGN cells or the HEK293T vector (and RCL) production cells. As the next closest genetic model to an HIV-1-based RCL, we considered members of the broader retrovirus family. The gammaretrovirus MLV encoding the amphotropic 4070A envelope glycoprotein (MLV-4070A, Figure 2a) was our primary candidate, as it is permissive in HEK293-derived cells and is the standard positive control virus for replication-competent retrovirus assays.4 Gammaretroviruses are simple retroviruses and lack the auxiliary and accessory genes encoded by HIV-1; furthermore, they are independent of Cyclin-T1 restriction. However, the structural proteins are similar to those of HIV-1 and, as in the case of all retroviruses, viral replication proceeds through reverse transcription of the viral genomic RNA into DNA, followed by integration into the host genome. Similarity of the MLV-4070A and HIV-1 lifecycles makes MLV-4070A a reasonable model for the internal components of a putative VP02-based RCL. Furthermore, as MLV-4070A does not cause disease in humans, it was considered safe for altering tropism.
In Approach D, we replaced the native amphotropic MLV envelope coding sequence (4070A) with E1001, to generate a chimeric virus: MLV-E1001 (Figure 2a). In this provirus, expression of E1001 is driven from the native MLV env transcription/splicing unit, to mimic temporal expression of the 4070A glycoprotein that occurs during wild-type MLV-4070A infection in permissive cells. Unlike the positive control viruses evaluated in previous approaches, MLV-E1001 should better model the mechanism of entry of a putative RCL derived during vector production. Finally, in Approach E we evaluated using unmodified MLV-4070A virus as the RCL assay positive control.
Replication of MLV-E1001 and MLV-4070A was evaluated in 293F-DCSIGN cells, using virus stocks generated by transfection of HEK293T cells with proviral DNA (Figure 6). Target cells were inoculated with virus at MOI ranging from 0.05 to 5, and kifunensine was added to cultures that received MLV-E1001. At each passage postinoculation, culture supernatant from infected cells was analyzed by F-PERT to detect de novo virus production and cells were passaged into fresh culture medium. In this experimental design, the viral particles quantified may result from either de novo infection events or from stable integration events that occurred in earlier cell passages (MLV viral proteins are not toxic and HEK293 cell cultures can remain chronically infected). Cultures infected with the parental MLV-4070A virus produced RT-containing viral particles in a dose-dependent manner that amplified over multiple cell passages, demonstrating viral replication (satisfying evaluation criterion 3). In contrast, MLV-E1001 infection resulted in steady-state release of RT-containing particles for the duration of the 16-day culture period, reflecting the initial inoculum dose. Therefore, while the seed stock of MLV-E1001 virus was capable of entering 293F-DCSIGN cells and producing de novo viral particles in “first-round” cells, these particles were not capable of cell-spreading infection, demonstrating the failure of progeny MLV-E1001 virus to amplify. In summary, of the two MLV-based positive control viruses tested, only MLV-4070A (Approach E) met evaluation criterion 3. MLV-4070A was therefore selected as the RCL assay positive control virus.
Figure 6.
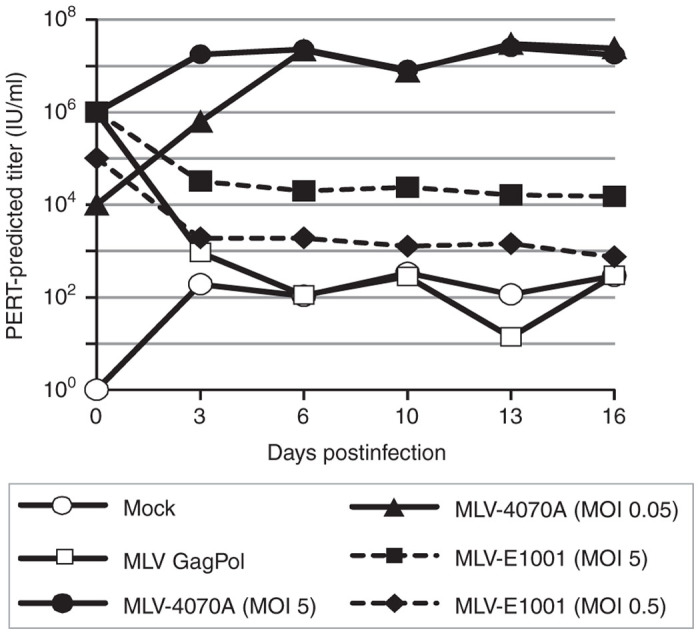
Evaluation of a replication-competent lentivirus assay format utilizing 293F-DCSIGN cells and an MLV-based positive control (Approaches D and E). 293F-DCSIGN cells were inoculated with MLV-4070A or MLV-E1001 at the indicated MOIs and amplification of virus was evaluated ~16 days in culture. At each passage, cell supernatant was analyzed by F-PERT assay and cells were passaged 1 : 6 into fresh medium. Kifunensine was present at 1μg/ml in cultures inoculated with MLV-E1001. Unenveloped MLV Gag-Pol particles and media alone were included as negative controls for viral replication. F-PERT, fluorescent-product enhanced reverse transcriptase; MLV, murine leukemia virus; MOI, multiplicity of infection.
Investigation of the effects of kifunensine in the final RCL assay format
Having identified a viable combination of positive control virus (MLV-4070A) and assay cell line (293F-DCSIGN), one outstanding question remained: should the small molecule kifunensine should be included in the final RCL assay design? Kifunensine is used in the VP02 manufacturing process, as it increases the infectivity of vector produced from the HEK293T production cell line by promoting high-mannose glycoforms of E1001 that enhance receptor targeting.3 It was assumed that kifunensine would similarly enhance the infectivity of a putative RCL enveloped with E1001, arguing in favor of including kifunensine in the culture media during the assay amplification phase. However, kifunensine would be expected to modify the carbohydrate structure of not only the E1001 envelope, but also the cellular proteins during the amplification phase, including DC-SIGN. Experiments in a model cell line demonstrated that kifunensine does modify the DC-SIGN receptor, which can negatively impact subsequent transduction by E1001-enveloped vector (data not shown). Presumably, this effect is because of high-mannose modification of DC-SIGN altering the binding affinity for the E1001 envelope glycoprotein. An experiment was therefore designed to evaluate the impact of kifunensine in the final RCL assay format. The 293F-DCSIGN cells were continuously passaged for 25 days in the absence or presence of kifunensine (modeling the extended RCL assay cell culture amplification phase), and these cells were subsequently transduced with E1001-enveloped HIV-1 GFP-vector stocks that had been produced from HEK293T cells in the absence or presence of kifunensine, respectively. Transduction of target cells was evaluated by measuring GFP fluorescence. Results from this experiment demonstrated that there was no net benefit (no increased transduction) from adding kifunensine to the assay system (see Supplementary Figure S2). Therefore, we omitted kifunensine from the amplification phase of the final RCL assay format. However, it should be noted that in all instances VP02 test article will be produced in the presence of kifunensine, thus ensuring optimal cell entry of a E1001-enveloped, putative RCL in the first step of the RCL assay.
Design and performance of an RCL assay specific for lentiviral vector based on the VP02 platform
After defining components of the RCL assay (Figure 7a), a viral stock of MLV-4070A was generated and experiments were performed at 48-well plate scale to determine the minimal infectious dose (MID) on the 293F-DCSIGN assay amplification cell line. We then performed mixing experiments at the MID to investigate potential inhibition of MLV-4070A positive control virus from either VP02 vector or EOPC, thereby mimicking amplification of a VP02-derived RCL present in a test article. Results from these studies demonstrated that VP02 vector should be assayed at a final dilution of no less than 1-in-4 at the final RCL assay scale to avoid any potential inhibitory effects on viral replication of the positive control (data not shown). EOPC is generally assayed for RCL by coculturing at a 1 : 1 ratio with the RCL assay cell line (293F-DCSIGN cells in this instance), after which the culture supernatant fluid is harvested and used to inoculate a fresh monolayer of assay amplification cells; subsequent passages are performed as for vector test article. When VP02 EOPC were cocultured with the 293F-DCSIGN assay cell line in the presence of 1 × MID of MLV-4070A virus, no inhibition of viral replication was observed, demonstrating that EOPC coculture had no effect on the sensitivity of the RCL assay for VP02 (data not shown).
Figure 7.
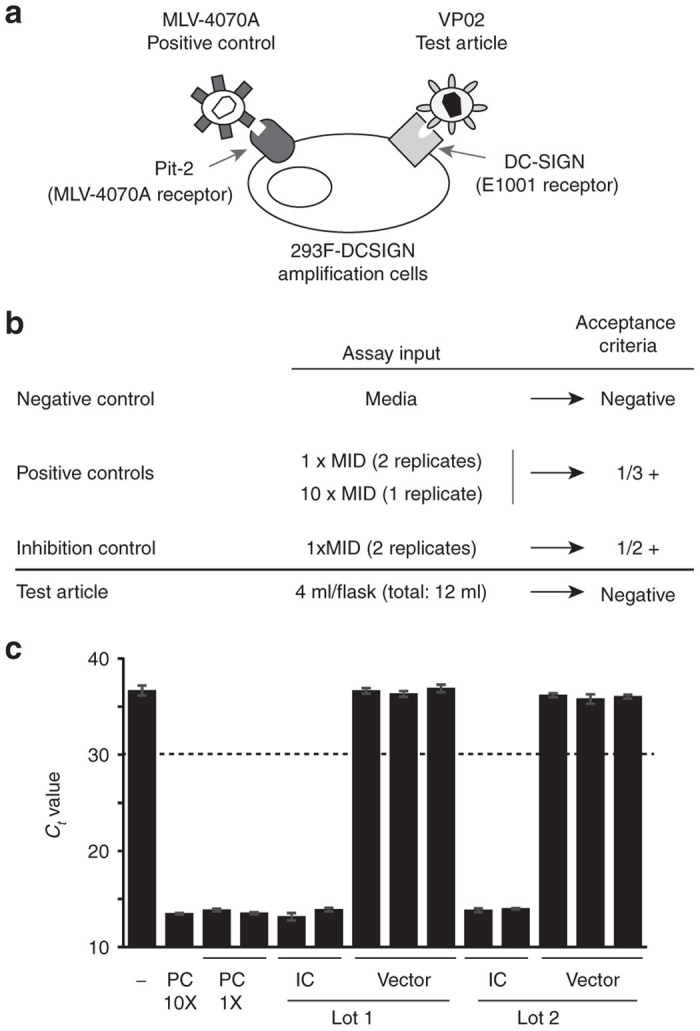
Replication-competent lentivirus (RCL) testing of large-scale vector production lots. (a) Schematic of the final design for an RCL assay specific for VP02 vector and EOPC. The assay positive control virus consists of MLV-4070A and the amplification cell line is 293F modified to express DC-SIGN (293F-DCSIGN), the receptor for the E1001 envelope glycoprotein present on the surface of VP02. (b) Controls and acceptance criteria for the VP02-specific RCL assay. The positive controls consist of MLV-4070A in cell culture media, whereas the inhibition control consists of vector test article spiked with MLV-4070A. At least one of the positive controls and one of the inhibition controls must be positive for the assay to be valid. (c) Test results for two of six independent production lots of VP02 clinical drug substance. A 12-ml sample of each bulk drug substance was split equally into three T-flasks. Two additional flasks contained an identical amount of vector, in addition to the MLV-4070A positive control spike (positive controls, PC; inhibition controls, IC). F-PERT analysis was performed on cell supernatants after six passages. Each bar represents a separate flask; error bars represent standard deviation from the mean for F-PERT replicates. Dashed line represents cut-off for assay positivity; ≥ 2 F-PERT replicates must return a Ct value of ≤30 in order for a flask to be deemed positive. F-PERT, fluorescent-product enhanced reverse transcriptase; MLV, murine leukemia virus.
The endpoint readout of our RCL assay is F-PERT analysis. Titration of recombinant MLV-4070A and HIV-1 RT enzymes demonstrated parallel dilutional linearity when analyzed by F-PERT, indicating comparable sensitivity of detection for these two enzymes (from positive control virus or a putative RCL, respectively) in the F-PERT assay (data not shown). The threshold for the initial definition of a positive RT signal in a cell culture flask was based on F-PERT signal observed in negative samples, such as extended cultures of noninfected cells.
Using our novel assay, two large-scale production lots of VP02 lentiviral vector and associated EOPC were tested for RCL. The current US FDA recommendation is to test at least 5% of the total supernatant of a clinical vector lot or 300 ml (the minimum theoretical crude vector volume required to detect RCL at a concentration of 0.01 IU/ml with 95% confidence interval), whichever is lesser.4 As elements present in the cell culture medium at vector harvest may be inhibitory to the RCL assay, we chose to perform testing on a downstream sample of bulk drug substance. A bulk drug substance test volume of ~12 ml was selected, as this volume contains the equivalent amount of vector as 300 ml of crude harvest. Following US FDA guidance, 1 × 108 EOPC were also assayed. In order to test 12 ml of a single VP02 clinical bulk drug substance, each of three flasks of 1 × 107 293F-DCSIGN cells were treated with 4 ml of bulk drug substance (~1 × 1010 vector genomes, for a total of 3 × 1010 vector genomes assayed). In addition, two flasks of cells were treated with 1 × 1010 vector genomes + 1 × MID of MLV-4070A (inhibition control), another flask was left untreated (negative control), and three final flasks were treated with MLV-4070A alone, at one of two different input concentrations (positive controls) (Figure 7b). In the case of the EOPC assay, 1 × 107 EOPC were added to 1 × 107 293F-DCSIGN cells either alone (10 test flasks, for a total of 1 × 108 EOPC, as recommended by US FDA1) or with the MLV-4070A spike (two flasks, inhibition controls). Four control flasks each used 1 × 107 293F-DCSIGN cells mixed with 1 × 107 untransfected HEK293T cells: one was left untreated (negative control) and the other three were infected with MLV-4070A (positive controls) similar to the assay for bulk drug substance. For both the bulk drug substance and EOPC assays, F-PERT analysis was performed on cell culture supernatants harvested at passage 6 (Figure 7c and data not shown). The results demonstrated reliable amplification of the positive control in spiked controls and that all six large-scale production lots of VP02 and the corresponding EOPC had no detectable RCL.
Discussion
An RCL assay was developed to test clinical manufacturing lots of investigational product based on VP02, an E1001-enveloped lentiviral vector designed to target the DC-SIGN receptor on human DCs in vivo. The assay consists of an amplification phase in 293F-DCSIGN cells, followed by detection of RT activity in culture supernatant using the F-PERT assay. Replication-competent MLV-4070A virus is the assay positive control. This assay has been used to release six lots of VP02 for use in ongoing clinical trials in the field of cancer immunotherapy.
The choice of amplification cell line was a primary consideration in the design of this RCL assay. To date, RCL assays described for HIV-1-based vectors have typically utilized C8166-45 cells, which are highly permissive to HIV-1 infection.11,12,17 The C8166-45 cell line was derived by immortalization with human T-lymphotropic virus 1 (HTLV-1) and the presence of pre-existing retroviral sequences in this cell line poses potential drawbacks for its use in RCL testing. Reportedly, HTLV-1 Gag and Gag-Pol are not expressed in C8166-45 cells, because of a defect in Rex-1 (ref. 20). However, we typically observe higher levels of background RT-like activity from cell supernatants of uninfected C8166-45 cells in comparison with other cell types, which may represent the detection of extremely low-level (but nonzero) HTLV-1 RT activity. In addition, the presence of HTLV-1 provirus in the C8166-45 amplification cell line may complicate the investigation of any false-positive RCL test results. Despite these caveats, we evaluated a DC-SIGN expressing C8166-45 cell line (C8166-DCSIGN) in one of our five RCL assay design strategies (Table 1), given the widespread use of the parental cell line in current RCL testing methods for HIV-1-based lentiviral vectors.
The merit of each RCL assay design approach was assessed based on three evaluation criteria developed in collaboration with regulatory authorities (Figure 2b). Based on these evaluation criteria, the data presented in this report indicate that HEK293-derived cells are a more appropriate choice than C8166-derived cells for amplifying and detecting a putative RCL enveloped with E1001. The 293F-DCSIGN cells have some advantages over the traditional C8166-45 RCL assay cell line. For example, 293F-derived cells grow robustly in cell culture, in contrast to C8166-45 cells which have a slow doubling time and are highly sensitive to changes in cell density and culture medium pH—characteristics that can cause technical challenges in an assay wherein the cell culture phase is several weeks long. Moreover, a putative RCL derived from the HEK293T vector production system for VP02 would be highly likely to be HEK293 cell-tropic. HEK293 cells have previously been used to test for RCL in equine infectious anemia virus vector preparations enveloped with VSVG13, demonstrating the utility of this cell type in developing RCL assays for vectors with different backbones and envelopes.
The FDA places a great deal of importance on generating a positive control that is derived from the virus on which the vector system is based, where possible, and this posed a significant challenge to this development work. The most obvious assay positive control virus would consist of generating an HIV-1 provirus wherein gp160 is replaced with E1001; however, we felt that the potential (increased) skin-tropism of this virus posed an unacceptable risk to assay operators.15 Instead, we incorporated the E1001 envelope into replication-competent MLV, a retrovirus that is nonpathogenic to humans (MLV-E1001, Approach D). Although preliminary experiments demonstrated that MLV- and HIV-1-derived vector particles were equally capable of being pseudotyped with E1001 (provided in trans) in HEK293T cells (data not shown), replication of the chimeric MLV-E1001 virus failed following the first round of infection (Figure 6). These findings are consistent with the hypothesis that cis-acting sequences in the native MLV envelope glycoprotein, which modulate envelope protein synthesis and viral particle production, are not interchangeable between different members of the retrovirus family.21,22 The failure to generate a replication-competent MLV-E1001 virus by rational design further underlines the unlikelihood of spontaneous generation of an RCL within contemporary vector systems. It also indicates that generation/testing of a genotypic HIV-1-E1001 provirus would not necessarily have been successful, but had safety concerns been overridden.
Given the difficulties in engineering a positive control virus with an E1001 envelope, unmodified HIV-1 and MLV-4070A viruses were evaluated in HEK293-based cells. When 293F-DCSIGN cells were engineered to express CD4, the new cell line was permissive for HIV-1 entry, but viral amplification did not occur (Figure 4d). These results were somewhat surprising, given a previous report of HIV-1 replication in CD4-expressing HEK293 cells and may stem from differences in CD4 expression levels in the two cell lines examined.23,24 In contrast to the HIV-1 approach, no modifications were necessary to make 293F-DCSIGN cells compatible with MLV-4070A. This virus replicated robustly in the amplification cell line and was selected as the positive control for our RCL assay. We recognize that MLV-4070A does not directly model the likely genotype of a putative RCL generated by the VP02 vector system; however, in the context of an RCL assay, we believe that the selection of an appropriate amplification cell line is ultimately more important than the actual positive control virus used in the final assay.
A point of debate in the development of our RCL assay was whether or not to include the mannosidase-I inhibitor kifunensine in the final assay format. Kifunensine promotes high-mannose glycosylation of E1001 during VP02 manufacturing, which significantly enhances the binding of VP02 to the DC-SIGN receptor on target cells.3 When the RCL assay format was modeled in small scale, there was no net benefit of including kifunensine in the culture medium (see Supplementary Figure S2). It is likely that high-mannose glycosylation of DC-SIGN decreases affinity for E1001-enveloped vector in cells cultured continuously in the presence of kifunensine. We therefore considered the possibility of mixing in kifunensine-naive 293F-DCSIGN cells at each of the six cell passages during the RCL amplification phase. In principle, this would optimize the ability of a high-mannose, E1001-enveloped RCL (produced in the presence of kifunensine) to engage the receptor on an unmodified (kifunensine-naive) 293F-DCSIGN amplification cell. Although experiments with MLV-4070A demonstrated that efficient virus amplification can still be achieved (data not shown), adherence to such a procedure during a full-scale RCL assay was deemed impractical, given the resulting increase in culture handling time and potential risk of cross-contamination of flasks. Moreover, we reasoned that the final assay format must be able to detect E1001-enveloped RCLs that are capable of kifunensine-independent replication, given that this molecule is not present in patients. Considering this point in combination with the empirical data, we omitted kifunensine from the final RCL assay format.
A suitable RCL assay readout method must be equally sensitive in detecting the assay positive control virus and an RCL. To date, RCL assays for HIV-1-based vectors have employed either p24 ELISA or quantitative PCR (qPCR) as the endpoint detection method.11,12,17 p24 ELISA does not recognize the MLV capsid protein and therefore was not compatible with detecting our assay positive control. The established psi–gag qPCR method for RCL detection was also incompatible with our assay format because sequences in the VP02 packaging construct have been codon-optimized to substantially reduce psi–gag recombination.17,25 Therefore it is likely that any RCL, should it arise, would occur via alternate, unpredictable recombination events for which it would be difficult to design qPCR primers. Furthermore, these and other RCL sequences that could be targeted for qPCR amplification (e.g., E1001) are not shared with our assay control virus (MLV-4070A), making qPCR an inappropriate stand-alone assay readout method. In contrast, any viable retrovirus (RCL or positive control virus) will always contain active RT enzyme, making the F-PERT assay the best available detection method for our RCL assay. However, no method is fool-proof. Cellular DNA polymerases and endogenous RTs present in conditioned cell culture medium can contribute to high background signal in the F-PERT assay.13,14,26,27 Therefore, it is prudent to have a strategy in place for follow-up testing in the event that a vector test article yields a positive assay result.
Other examples of RCL assays in the literature have demonstrated vector entry into assay cells and robust replication of the positive control virus.11–13,17,28 The study described here has added an additional criterion in the development of an RCL assay: the ability of the assay cells to release functional vector particles that model the putative RCL. This criterion is the best approximation for determining whether a vector system-derived RCL would amplify in a particular assay cell line and was of particular importance to regulatory authorities. Notably, C8166-based cells (Approach A) failed to meet this criterion, a result that was unexpected. Prior RCL assay development using this cell line has assumed that it is capable of assembling infectious HIV-1-based particles with non-native (non-gp120) envelope glycoproteins. However, our data suggests that this may not be universally true. In conclusion, this work identifies important questions to consider in developing next-generation RCL assays, or adapting a pre-existing RCL assay for use with new vector pseudotypes.
Materials and Methods
Cell lines
The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: C8166-45 (Cat#404) from Dr. Robert Gallo.29 HEK293T cells were obtained from ATCC (Manassas, VA). The DC-SIGN sequence expressed in C8166-DCSIGN, 293F-DCSIGN, and 293F-DCSIGN-CD4 cells is codon-optimized. FreeStyle 293-F (referred to herein as 293F) cells were obtained from Life Technologies (Grand Island, NY). All stable cell lines were subjected to limiting dilution cloning using a selectable marker; where applicable, clones were screened by surface staining for DC-SIGN, CXCR4, or CD4 (all antibodies from eBiosciences, San Diego, CA). The 293F-based cell lines were grown in Dulbecco’s modified Eagle medium (high glucose) supplemented with 10% fetal bovine serum, 2mM l-glutamine, and 1% nonessential amino acids. C8166-based cells were grown in CD Hybridoma Medium (Life Technologies) supplemented with 10% fetal bovine serum and 8mM l-glutamine.
Viruses
The infectious strain of HIV-1 used in this study was based on NL4-3. HIVPC: Constructed entirely from synthetic DNA fragments, this proviral DNA encodes knock-out mutations within vif, vpr, vpu, and nef (pMK4-3ΔA4). The vif ORF was modified as in what follows: T-to-C mutations at positions 5042, 5063, and 5086 were generated to disrupt potential translation initiation codons; in addition, codons at positions 5125–5127 and 5431–5433 were replaced with premature stop codons (ATG>TAA and CCT>TAA, respectively) (all positions are relative to the NL4-3 R-to-R sequence). Within the vpr ORF, the initiator methionine was replaced with CCC at positions 5559–5561 and the codon at positions 5733–5735 was replaced with a TAA stop codon. The vpu ORF was modified to replace the initiator methionine at positions 6061–6063 with CCC and the codon at positions 6178–6180 was replaced with a TAA stop codon. The nef ORF was modified as follows: the initiator methionine at positions 8787–8789 was replaced with CCC, the ATG sequence at positions 8844–8846 was replaced with a TAA stop codon to stop potential downstream translation initiation, and the GGC sequence at positions 9141–9143 was replaced with a TAA stop codon. HIVPC-ID: An integrase-defective version of HIVPC was generated by the sequential insertion into pMK4-3ΔA4 of fragments synthesized to mutate Integrase (Asp64Val, corresponding to the codon at positions 4419–4421; cloned into the AgeI/SalI sites [2.3kb]) and delete the 3′-PPT (cloned into the BlpI/NcoI sites [1.7kb]). HIVΔEnv(GFP): The GFP ORF was cloned into pMK4-3ΔA4 using the BlpI/XhoI sites in the nef ORF, followed by deletion of sequence between NdeI and NheI sites in the env ORF.
A molecular clone of MLV-4070A (pAMS) was obtained from ATCC (catalog no. 45167). MLV-E1001: A fragment of pAMS encoding the 4070A ORF was replaced with a 4.6-kb synthetic fragment encoding the 3′-end of MLV-4070A pol and the E1001 ORF. This fragment encoded an A-to-G mutation at position 5777 (nts from R region of MLV-4070A) to disrupt the 4070A env initiator methionine, while maintaining the correct Integrase amino acid sequence in the overlapping pol ORF.
Viral stocks were produced by transient transfection of HEK293T cells with proviral plasmids. Typically, cells were seeded into tissue culture vessels at 6.1 × 104 cell/0.175 ml complete media per cm2 at 24 hours prior to transfection. For transfection, ~10 ng total plasmid DNA was mixed with 0.044 μl Lipofectamine 2000CD in a total volume of 1.2 μl Opti-Mem (Life Technologies) per cm2 of tissue culture vessel.
Vectors
The VP02 vector genome was modified to encode GFP downstream of the ubiquitin promoter; all other VP02 vector components are as described earlier.3 GFP-expressing VP02 reporter vector was produced by transient transfection of HEK293T cells with the five vector component plasmids, using Lipofectamine 2000 (Life Technologies) and total plasmid DNA at a ratio of 2:1 (μl Lipofectamine:μg DNA). Alternatively, as noted in the figure legend, cells were transfected using calcium phosphate. Where indicated, kifunensine (Glycosyn, Gracefield, New Zealand) was added to the culture media at 5 hours post-transfection to a final concentration of 1 μg/ml. VP02 vector was harvested at 2 or 3 days post-transfection and clarified using a 0.45-μm filter (Figure 3b,c).
To study release of transduction-competent vector particles in Figure 3b,c, the E1001 ORF was cloned into an LNCX-based MLV vector containing the CMV promoter (MLV-CMV-E1001).30 VSVG-pseudotyped MLV-CMV-E1001 vector was generated by cotransfection of HEK293T cells with plasmids encoding vector genome, VSVG (phGK),31 and MLV gag/pol (pHIT60).32 VSVG-pseudotyped HIVΔEnv(GFP) (Figures 3b,c and 5b) was generated by cotransfection of vector genome and VSVG plasmids. At 18 hours post-transfection, sodium butyrate was added to a final concentration of 10 mM for 5 hours, followed by media replacement. VSVG-pseudotyped vector was harvested at 2 days post-transfection and passed through a 0.22–0.45 μm filter.
F-PERT assay
A modified F-PERT assay to that previously reported was used.26,33
Analysis of positive control virus replication fitness
The F-PERT assay was used to analyze the replication fitness of each positive control virus in the cell line of interest. The indicated target cell type was seeded in multiwell dishes. Input virus was normalized by F-PERT assay, with titers reported in relation to a virus standard of known infectious units (IU). At 5 hours postinfection, input inoculum was replaced with fresh medium alone or containing kifunensine at 1 μg/ml, where noted. Thereafter, cells were passaged every ~3 days at a split ratio of 1 : 6. F-PERT analysis was performed on samples of culture supernatant fluid taken at each passage. In Figure 5b, a modified protocol was followed. Cells were inoculated with 10-fold serial dilutions of HIVΔEnv(GFP) positive control vector pseudotyped with VSVG envelope. At 18 hours post-transduction, input vector was replaced with fresh medium containing kifunensine at 1 μg/ml, where noted. Every 2–3 days thereafter, supernatant was transferred without filtration onto freshly seeded cells in a 48-well plate and a sample was saved for F-PERT analysis. Media replacement was performed as before, 18 hours following each supernatant transfer.
Quantification of transduction by GFP fluorescence
Following transduction by GFP reporter-encoding vectors, 1 × 104 cells per sample were analyzed by flow cytometry using either a FACSVerse or FACSCalibur (BD Biosciences, India). Cells were stained with TO-PRO-3 (Life Technologies); dead cells and auto-fluorescent events were out-gated using FSC/SSC profiling and comparison with appropriate cell controls. GFP events were gated as subpopulations of the resulting events and presented as percentage of live cells. To titer GFP vector stocks, this percentage was related back to the number of transduced target cells, to calculated GFP-transducing units/ml (GFP-TU/ml). VSVG- and E1001-enveloped GFP vectors were titrated on 293F-DCSIGN or C8166-DCSIGN cells, where appropriate.
Clinical lot RCL assay
To initiate the assay, 225 cm2 tissue culture flasks were each seeded with 1 × 107 viable 293F-DCSIGN cells. The following day, three test article flasks were each inoculated with 4 ml of VP02 bulk drug substance (~1 × 1010 vector genomes) and supplemented with culture medium to a final volume of 23 ml each. Following US FDA recommendation,1 a total of 12 ml bulk drug substance was tested for each manufacturing lot, equivalent to the number of vector particles contained within 300 ml crude harvest material from a manufacturing subrun (this is the theoretical minimum test volume that allows 95% detection confidence, assuming an RCL concentration of 0.01 IU/ml). Control flasks were as follows: (i) negative control comprised 1 × 107 293F-DCSIGN cells treated with polybrene alone (8 μg/ml) (Sigma-Aldrich, St. Louis, MO); (ii) two positive controls with 1 × 107 293F-DCSIGN cells and 3.9 IU of MLV-4070A positive control virus (1 × MID [98% infection rate]) and polybrene; (iii) one positive control with 1 × 107 293F-DCSIGN cells and 39 IU of MLV-4070A positive control virus (10 × MID) and polybrene; (iv) two spiked test article flasks were identical to the test article flasks, except that 3.9 IU of MLV-4070A positive control virus was also included. On the day following inoculation, all flasks were topped up with culture medium to 46 ml total medium per flask; all subsequent passages used this same medium volume. At 3 days postinoculation, ~1/4 of the cells in each flask were passaged into fresh flasks of equivalent size (P1). Passages were continued in a similar fashion until a total of six passages were complete. After the final passage, cell supernatant was filtered (0.22 μm) and analyzed by F-PERT assay.
Cocultivation RCL assay
For the EOPC test material, the producer cells from all the culture vessels used for vector production were harvested 1 day following the vector harvest and cryopreserved. To initiate a cocultivation assay, vials of EOPC were thawed at 37ºC and cell viability was determined. Each of 10 test article flasks was inoculated with 1 × 107 viable EOPC mixed with 1 × 107 293F-DCSIGN assay target cells in a 225 cm2 flask in the presence of 8 μg/ml polybrene. Control flasks were as follows: (i) negative control with 107 293F-DCSIGN assay cells mixed with 107 HEK293T cells in a flask treated with polybrene alone; (ii) two positive controls with 1 × 107 293F-DCSIGN cells mixed with 1 × 107 HEK293T cells in a flask treated with 3.9 IU of MLV-4070A positive control virus (1 × MID [98% infection rate]) and polybrene; (iii) one positive control with 1 × 107 293F-DCSIGN cells mixed with 1 × 107 HEK293T cells in a flask treated with 39 IU of MLV-4070A positive control virus and polybrene; (iv) spiked test article was identical to the test article flasks, except that 3.9 IU of MLV-4070A positive control virus was also included. Two passages were continued as for the clinical lot RCL assay, at which point supernatant was harvested from the P2 cocultured cells and inoculated onto fresh 225 cm2 flasks seeded 1 day prior with 107 293F-DCSIGN cells. Following this second inoculation, four additional passages were continued as before for a total of six passages. After the final passage, cell supernatant was filtered (0.22 μm) and analyzed by F-PERT assay.
Acknowledgments
The authors thank Kenneth Cornetta (Indiana University School of Medicine, IN, United States) for insightful scientific discussions. B.K.-C., B.A.T., S.U.T., C.J.N., D.J.C., L.Z., B.J.M., T.K., S.H.R., and W.R.G. are at least one of the following: a current employee of Immune Design (IMDZ) and a holder of stock and/or options in IMDZ. D.C.F., L.M., H.J.S., L.J.E.P., K.A.M., P.A.R., and J.E.M. are at least one of the following: a current employee of Oxford BioMedica (OXB), a holder of stock, and/or options in OXB.
References
- FDA (2010) Briefing Document—Testing for replication competent retrovirus (RCR)/lentivirus (RCL) in retroviral and lentiviral vector based gene therapy products—Revisiting current FDA recommendations. U.S. Food and Drug Administration; pp 1–18. [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen SU, Kelley-Clarke B, Nicolai CJ, Cassiano LA, Nelson LT, Slough MM. Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol Ther. 2014;22:575–587. doi: 10.1038/mt.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (2006) Guidance for Industry—Supplemental guidance on testing for replication competent retrovirus in retroviral vector based gene therapy products and during follow-up of patients in clinical trials using retroviral vectors. U.S. Food and Drug Administration; pp 1–13. [DOI] [PubMed] [Google Scholar]
- Odegard JM, Kelley-Clarke B, Tareen SU, Campbell DJ, Flynn PA, Nicolai CJ. Virological and preclinical characterization of a dendritic cell targeting, integration-deficient lentiviral vector for cancer immunotherapy. J Immunother. 2015;38:41–53. doi: 10.1097/CJI.0000000000000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein AD, Tropea JE, Mitchell M, Kaushal GP. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem. 1990;265:15599–15605. [PubMed] [Google Scholar]
- Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol. 2008;26:326. doi: 10.1038/nbt1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, Bayer M, Ma H, Samulski J, Li C, McCown T. Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Mol Ther. 2011;19:547–556. doi: 10.1038/mt.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornetta K, Yao J, Jasti A, Koop S, Douglas M, Hsu D. Replication-competent lentivirus analysis of clinical grade vector products. Mol Ther. 2011;19:557–566. doi: 10.1038/mt.2010.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escarpe P, Zayek N, Chin P, Borellini F, Zufferey R, Veres G. Development of a sensitive assay for detection of replication-competent recombinant lentivirus in large-scale HIV-based vector preparations. Mol Ther. 2003;8:332–341. doi: 10.1016/s1525-0016(03)00167-9. [DOI] [PubMed] [Google Scholar]
- Miskin J, Chipchase D, Rohll J, Beard G, Wardell T, Angell D. A replication competent lentivirus (RCL) assay for equine infectious anaemia virus (EIAV)-based lentiviral vectors. Gene Ther. 2006;13:196–205. doi: 10.1038/sj.gt.3302666. [DOI] [PubMed] [Google Scholar]
- Sastry L, Xu Y, Duffy L, Koop S, Jasti A, Roehl H. Product-enhanced reverse transcriptase assay for replication-competent retrovirus and lentivirus detection. Hum Gene Ther. 2005;16:1227–1236. doi: 10.1089/hum.2005.16.1227. [DOI] [PubMed] [Google Scholar]
- EMEA 2005. Guideline on Development and Manufacture of Lentiviral Vectors. European Medicines Agency, Evaluation of Medicines for Human Use, Committee for medicinal products for human use (CHMP), pp 1–8.
- Ebina H, Kanemura Y, Suzuki Y, Urata K, Misawa N, Koyanagi Y. Integrase-independent HIV-1 infection is augmented under conditions of DNA damage and produces a viral reservoir. Virology. 2012;427:44–50. doi: 10.1016/j.virol.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Sastry L, Xu Y, Johnson T, Desai K, Rissing D, Marsh J. Certification assays for HIV-1-based vectors: frequent passage of gag sequences without evidence of replication-competent viruses. Mol Ther. 2003;8:830–839. doi: 10.1016/j.ymthe.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Arnolds KL, Lares AP, Spencer JV. The US27 gene product of human cytomegalovirus enhances signaling of host chemokine receptor CXCR4. Virology. 2013;439:122–131. doi: 10.1016/j.virol.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque K, Finzi A, Binette J, Cohen EA. Role of CD4 receptor down-regulation during HIV-1 infection. Curr HIV Res. 2004;2:51–59. doi: 10.2174/1570162043485086. [DOI] [PubMed] [Google Scholar]
- Bhat NK, Adachi Y, Samuel KP, Derse D. HTLV-1 gene expression by defective proviruses in an infected T-cell line. Virology. 1993;196:15–24. doi: 10.1006/viro.1993.1450. [DOI] [PubMed] [Google Scholar]
- Logg CR, Baranick BT, Lemp NA, Kasahara N. Adaptive evolution of a tagged chimeric gammaretrovirus: identification of novel cis-acting elements that modulate splicing. J Mol Biol. 2007;369:1214–1229. doi: 10.1016/j.jmb.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nack U, Schnierle BS. Replacement of the murine leukemia virus (MLV) envelope gene with a truncated HIV envelope gene in MLV generates a virus with impaired replication capacity. Virology. 2003;315:209–216. doi: 10.1016/s0042-6822(03)00519-1. [DOI] [PubMed] [Google Scholar]
- Lama J, Mangasarian A, Trono D. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr Biol. 1999;9:622–631. doi: 10.1016/s0960-9822(99)80284-x. [DOI] [PubMed] [Google Scholar]
- Reil H, Höxter M, Moosmayer D, Pauli G, Hauser H. CD4 expressing human 293 cells as a tool for studies in HIV-1 replication: the efficiency of translational frameshifting is not altered by HIV-1 infection. Virology. 1994;205:371–375. doi: 10.1006/viro.1994.1655. [DOI] [PubMed] [Google Scholar]
- Tareen SU, Nicolai CJ, Campbell DJ, Flynn PA, Slough MM, Vin CD. A rev-independent gag/pol eliminates detectable psi-gag recombination in lentiviral vectors. Biores Open Access. 2013;2:421–430. doi: 10.1089/biores.2013.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold BA, Hepler RW, Keller PM. One-step fluorescent probe product-enhanced reverse transcriptase assay. Biotechniques. 1998;25:98–106. doi: 10.2144/98251st06. [DOI] [PubMed] [Google Scholar]
- Pyra H, Böni J, Schüpbach J. Ultrasensitive retrovirus detection by a reverse transcriptase assay based on product enhancement. Proc Natl Acad Sci USA. 1994;91:1544–1548. doi: 10.1073/pnas.91.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley DC, Bannister R, Leroux-Carlucci MA, Evans NE, Miskin JE, Mitrophanous KA. Development of an equine-tropic replication-competent lentivirus assay for equine infectious anemia virus-based lentiviral vectors. Hum Gene Ther Methods. 2012;23:309. doi: 10.1089/hgtb.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahuddin SZ, Markham PD, Wong-Staal F, Franchini G, Kalyanaraman VS, Gallo RC. Restricted expression of human T-cell leukemia-lymphoma virus (HTLV) in transformed human umbilical cord blood lymphocytes. Virology. 1983;129:51–64. doi: 10.1016/0042-6822(83)90395-1. [DOI] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–982, 984. [PMC free article] [PubMed] [Google Scholar]
- Farley DC, Iqball S, Smith JC, Miskin JE, Kingsman SM, Mitrophanous KA. Factors that influence VSV-G pseudotyping and transduction efficiency of lentiviral vectors-in vitro and in vivo implications. J Gene Med. 2007;9:345. doi: 10.1002/jgm.1022. [DOI] [PubMed] [Google Scholar]
- Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995;23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohll JB, Mitrophanous KA, Martin-Rendon E, Ellard FM, Radcliffe PA, Mazarakis ND. Design, production, safety, evaluation, and clinical applications of nonprimate lentiviral vectors. Methods Enzymol. 2002;346:466–500. doi: 10.1016/s0076-6879(02)46072-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.