

Abstract
Recombinant adeno-associated virus (rAAV) is currently the best vector for gene delivery into the skeletal muscle. However, the 5-kb packaging size of this virus is a major obstacle for large gene transfer. This past decade, many different strategies were developed to circumvent this issue (concatemerization-splicing, overlapping vectors, hybrid dual or fragmented AAV). Loss of function mutations in the DYSF gene whose coding sequence is 6.2kb lead to progressive muscular dystrophies (LGMD2B: OMIM_253601; MM: OMIM_254130; DMAT: OMIM_606768). In this study, we compared large gene transfer techniques to deliver the DYSF gene into the skeletal muscle. After rAAV8s intramuscular injection into dysferlin deficient mice, we showed that the overlap strategy is the most effective approach to reconstitute a full-length messenger. After systemic administration, the level of dysferlin obtained on different muscles corresponded to 0.5- to 2-fold compared to the normal level. We further demonstrated that the overlapping vector set was efficient to correct the histopathology, resistance to eccentric contractions and whole body force in the dysferlin deficient mice. Altogether, these data indicate that using overlapping vectors could be a promising approach for a potential clinical treatment of dysferlinopathies.
Introduction
Gene transfer is a promising way to treat recessive disorders. Adeno-associated virus (AAV) is an efficient and advantageous type of vector to deliver DNA into various tissues such as liver or skeletal muscle. AAV vectors are nonpathogenic, demonstrate very low levels of host genome integration, effectively transduce muscle fibers, and initiate long-term transgene expression. However, the packaging capacity of the AAV vector is limited to ~5 kb, preventing the encapsidation of large genes.1 To bypass this limitation, different strategies have emerged in the recent years for the in vivo expression of full-length large transgenes after rAAV delivery.
In the first described method called concatemerization-splicing,2–6 the reconstitution of full-length transgenes occurs following in vivo administration of two rAAV vectors. One vector is composed of a promoter, the 5’ portion of the transgene and a donor splicing signal sequence (5’ vector); and the second vector exhibits an acceptor splicing signal sequence, the remaining 3’ portions of the transgene, and a polyA sequence (3’ vector). After coinfection and conversion into a double strand, concatemerization occurs by intermolecular recombination mediated by inverted terminal repeat (ITR) sequences of the two rAAV genomes.7,8 Some of these events establish the desired transcriptional cassette such that the ITR concatemerization junction is removed by splicing, effectively generating a full-length messenger/protein from what was originally two distinctly transduced rAAV genomes. Another strategy based on overlap between two rAAV genomes carrying overlapping fragments has been proved efficient to reconstitute a split transgene by homologous recombination (HR).3,9 Interestingly, it has been shown that, HR was the underlying mechanism of obtaining a protein from the use of a rAAV cassette constituted by a full-length large cDNA (up to 8.9 kb) packaged into an AAV5 capsid.10–12 Indeed, in that case the capsid packages partial products from both ends of a full-length large cDNA and that the reconstitution happens by HR between overlapping regions of two vectors.13–15 As this strategy relies on the intracellular reassembly of genome fragments produced during the ineffective packaging of oversized AAV genomes, this vector context was termed “fragment AAV (fAAV)”.16 In addition to these methods, a combination of both concatemerization-splicing and HR referred as hybrid dual AAV vector system was also efficient in generating a large protein from a large cDNA.17,18 In that case, in addition to carry splicing sequences, the two AAVs include a homologous sequence.
Dysferlinopathies regroup different clinical presentations including limb girdle muscular dystrophy type 2B (LGMD2B), Miyoshi myopathy (MM), and distal anterior compartment myopathy (DMAT). They are caused by mutations in the DYSF gene, which encodes a 237 kDa tail-anchored membrane protein called Dysferlin. DYSF is composed of 55 exons spanning over 150-kb of genomic DNA and is expressed as a 6.2-kb cDNA predominantly present in skeletal muscle and in peripheral blood monocytes.19 Dysferlin belongs to the family of ferlins and is composed of seven C2 calcium-dependent phospholipid-binding domains, arrangement of specific FER and DYSF domains, and a transmembrane domain. A major role of dysferlin in muscle membrane repair was demonstrated.20 Besides this particular function, dysferlin was proposed to carry out additional roles associated with vesicle fusion events such as T-tubule integrity21 or trafficking of membrane-associated proteins.22
There is, to date, no effective therapy for dysferlinopathies. The application of gene therapy is complicated by the fact that the dysferlin cDNA exceeds the encapsidation capacity of the AAV vectors. Consequently, large-gene methods have started to be applied for these diseases. Indeed, two studies that used the concatemerization-splicing and “fragment” HR strategies demonstrated dysferlin full-length expression in vivo. The efficient expression of dysferlin was associated with restoration of membrane repair capacity of myofibers and restoration of physiologic function of muscles.12,23 Here, we present a comparative study to evaluate the relative efficiency of different large gene transfer methods using dysferlin as a model. In addition to the vectors for the concatemerization-splicing method previously generated and referred as Set 1,23 we constructed new two dual AAV vector sets (Sets 2 and 3) and a vector with the full-length cDNA to generate the “fragmented” packaged vector genomes (fAAV). To compare the efficiency of muscle transduction and level of expression of the full-length protein, we performed intramuscular (IM) injections of all the vectors into the Tibialis anterior (TA) muscle of dysferlin deficient mice (B6.A-Dysf prmd, also called Dysf prmd). From the comparison, the set of vectors driving the best expression of dysferlin was used for dose-effect and functional analyses after systemic administration.
Results
Packaging efficiency of the different strategies
To compare the different large gene transfer methods, we constructed several cassettes for full-length human dysferlin delivery using a single or a combination of two AAV vectors (Figure 1a). The first set of vectors (Set 1) was derived from a previously described pair of vectors, and generally has been termed concatemerization-dependent trans-splicing vectors.23 The 5’ cassette of this set is composed of a muscle-oriented promoter (C5-12), a chimeric intron β-globin, the first half of the dysferlin cDNA (from exon 1 to exon 28) and half the intron 28. The 3’ cassette carries the second half of intron 28 followed by the second half of the dysferlin cDNA. The second set of large gene delivery vectors (Set 2) was derived from the first set with the modification that the intron 28 was present in full in both cassettes. The consequence is that the two vectors overlap with 254 nt. The third vector combination (Set 3) is composed of overlapping vectors where the 5’ vector carries the dysferlin cDNA from exon 1 to the middle of exon 30 and the 3’ vector from the middle of exon 23 to exon 55, thereby exhibiting an overlap of 859 nt. Finally, a single vector with a full-length dysferlin cDNA cassette was constructed (fAAV vector). Important for our comparisons, all of these constructs carry the same promoter and regulatory sequences. In addition, the sequence immediately upstream of the dysferlin start codon was modified to correspond to a strong Kozak sequence. All cassettes were packaged into a rAAV2/8, an efficient serotype for skeletal muscle transduction and tittered by quantitative polymerase chain reaction (PCR) using conserved sequences within the AAV ITR.
Figure 1.

The different approaches for adeno-associated virus (AAV)-mediated dysferlin gene expression. (a) Schematic representation of the four strategies designed for dysferlin expression. The dysferlin FL cDNA (human isoform 8) was split into two viral vectors to evaluate concatemerization-splicing (Set1), concatemerization-splicing plus overlapping vectors (Set 2) and overlapping (Set 3) approaches. Alternatively, a full-length dysferlin expression cassette was used to generate a fragmented rAAV preparation (fAAV vector). All cassettes were packaged into a rAAV2/8 vector using standard triple transfection protocol, following by CsCl Density gradient purification and Dot-Blot oriented selection of AAV-rich fractions. The size of each cassette from inverted terminal repeat (ITR) to ITR and the size of the homologous regions for sets 2 and 3 are mentioned. ITR are represented by large black arrows. Splicing sites (donor and acceptor) are indicated by grey dots. (b) Characterization of AAV vectors. The genomes were extracted from rAAV8 vector preparations (Set1, Set3, and fAAV batches) and analyzed by denaturing alkaline gel electrophoresis. Set 2 was omitted from the analysis since it is structurally related to Set 1. Upper panel: direct visualization of the gel. Lower panel: Southern blot hybridized with an ITR probe. (c) Alkaline Agarose Gel analysis of genomes isolated from fAAV Batch 1 and fAAV Batch 2 rAAV8 preparations. The fractions 2 to 7 from fAAV Batch 2 and pooled fractions of fAAV Batch 1 were analyzed by alkaline gel electrophoresis. The selected fractions corresponding to the largest genomes are indicated by the X symbol. (d) IM delivery of rAAV8 fAAV Batch 1 and fAAV Batch 2 was performed in the left TA muscle of 4-week-old Dysfprmd with an equal dose of each vector. Anti-dysferlin western blot was performed using Hamlet antibody (n = 4 for each batch). Actin was used for normalization.
To characterize the packaged genomes of these different sets, an alkaline gel electrophoresis in denaturing condition was performed on extracted genomes from the vector preparations (Figure 1b, upper panel). For all vectors included in the Sets 1 and 3, the major band was at the expected size, around 4.5 kb. The result was confirmed by Southern blot using an ITR probe (Figure 1b, lower panel). However, a different pattern was observed for packaged fAAV genomes which demonstrated a heterogeneous ladder of DNA species from 1 to 4.5 kb in size (Figure 1b). In that case, truncated genome fragments are packaged, with a preference for smaller species, when the genome exceeds the AAV packaging capacity. Following this observation, a new batch of fAAV vector was prepared. In contrast to the first one which contained all the fractions collected during the second round of CsCl density gradients during purification, this second batch contained only the most dense fractions of the CsC1 gradient that are enriched in the largest packaged genomes (Figure 1c). Next, fAAV Batch 1 and Batch 2 were injected into the TA muscle of Dysfprmd male mice (n = 4) at an equivalent dose (9e8 viral genome (vg) per muscle) to compare their relative efficiency. The expression of dysferlin was evaluated 1 month later at protein levels (Figure 1d). Not surprisingly, it was observed that the enrichment of fAAV Batch 2 with the largest packaged genomes allows a more efficient expression of dysferlin (Figure 1d).
Comparison of dysferlin expression of different therapeutic strategies in vivo after IM injection
To compare the transduction efficiency of the different approaches, IM injection of rAAV2/8 preparations of set of vectors were performed in TA muscles of 1-month-old Dysfprmd male mice (n = 4) at the dose of 1.3e10 vg per muscle. It should be noted that this model has a moderate myopathology with the first pathological signs being visible at 2 months of age. Therefore, at the time of injection, the Dysfprmd TA muscles show no signs of pathology. The expression of dysferlin was compared 1 month later by qRT-PCR and Western blot analysis. For quantification of the concatamerization-splicing event for Sets 1 and 2, the qRT-PCR analysis was performed with a pair of primers (F.exon28, R.exon29) coupled with a specific probe (P.Exon28-29) designed to overlap the Exon 28–29 junction. For Set 3 and fAAV, due to the large homologous regions shared between cassettes, it was not possible to design a qRT-PCR amplicon that can be specific of reconstituted dysferlin. The results showed that Set 1 led to a similar level of mRNA compared to the human control, while an approximate 50% reduction was observed using the Set 2 vectors (Figure 2a).
Figure 2.

Comparison of transgene expression after intramuscular injection. The left TA muscle of 1-month-old Dysfprmd mice was injected with 1.3e10 vg of each Set of vectors (ratio 1:1). The right TA muscle was injected with PBS as a control (n = 3–4/group). (a) Dysferlin mRNA level was measured by quantitative real-time polymerase chain reaction (qRT-PCR) using a specific probe and primer set specific of concatemerization-splicing and intronic HR vectors with primers flanking exon28-29 junction and junction overlapping probe. Dysferlin mRNA levels were normalized by acidic ribophosphoprotein (Rplp0) mRNA level. Data are significantly different compared to untreated deficient mice. Data are given as mean ± SEM; ***P < 0.001. (b) Western blot analyses were performed with N-terminal (Romeo) and C-terminal (Hamlet) domain specific antibodies directed against dysferlin and normalized by an anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody. (c) Immunohistofluorescence for detection of dysferlin using Hamlet antibody. The difference in proportion of dysferlin between the sarcolemma and the internal membrane compartment after transfer is something that is always observed whatever the strategy. The reason underlying this particularity is still unclear. Scale bar = 200 µm. (d) Colocalization staining was performed using Romeo antibody in red (upper panel) and RyR antibody in green (middle panel). Merge images showing colocalization of dysferlin and RyR are presented in the lower panel. Scale bar = 30 µm. Inset: control with secondary antibody alone in WT.
Western blots performed using N-terminal (Romeo) or C-terminal (Hamlet) dysferlin antibodies revealed the presence of a band at the expected size (237 kDa) in all injected muscles that was absent in phosphate-buffered saline (PBS) control-treated mice (Figure 2b). The western blot showed substantial differences in the expression level from each vector Set. Sets 1 and 2 were somewhat similar considering the variability of western blot quantification with both antibodies, yielding the lowest levels of Dysferlin (Figure 2 and Table 1). In contrast, both Set 3 and fAAV vectors mediated enhanced Dysferlin transduction with Set 3 demonstrating an approximate 4–10-fold increase compared to Batch 2 fAAV (Figure 2; Table 1).
Table 1. Quantification of dysferlin western-blot performed with Hamlet and Romeo antibodies and expressed in percentage compared to WT.
| WT | NI | Set1 | Set2 | Set3 | fAAV | |
|---|---|---|---|---|---|---|
| N-ter Romeo Ab | 100% | 5% | 27% | 40% | 664% | 60% |
| SEM | ± 0 | ± 1.3% | ± 10.3 | ± 8.4 | ± 228% | ± 27.5% |
| C-ter Hamlet Ab | 100% | 1.34% | 28.74% | 17.76% | 378.83% | 41.67% |
| SEM | ± 0 | ± 0.7% | ± 11.3% | ± 3.1% | ± 128% | ± 23.8% |
The blots were performed in duplicate with n = 4.
SEM, standard error of the mean.
Immunohistofluorescence was performed on muscle sections for each of the above conditions using the Hamlet dysferlin antibody (Figure 2c). A dysferlin staining that correlated with the WB quantification can be observed. The lowest expression levels was obtained for Sets 1 and 2, while Set 3 showed a high proportion of positive fibers and a high level of expressed protein in transduced fibers. A colabelling of dysferlin and RyR, a protein of the sarcoplasmic reticulum in close proximity with T-tubules, was also performed, showing no difference between the conditions (Figure 2d).
Systemic injection of the HR vectors
Given the efficient performance of Set 3 vectors, as well as the additional complications moving fAAV into the clinic, Set 3 vectors were used to reconstitute dysferlin after a systemic injection in the tail vein of 1-month-old Dysfprmd mice (n = 3) at the dose of 9e13 vg/kg.
One month after injection, a collection of muscles (Tibialis anterior (TA), Psoas (Pso), Triceps brachii (Tri), Deltoid (Del), Gastrocnemius (Ga), Gluteus (Glu), Biceps brachii (Bi), Quadriceps femoris (Qua)), the heart, and the liver were sampled for analysis. RNA extracted from treated mice was converted to cDNA and the region including the vector overlap was sequenced. The sequenced region was 100% identical to the dysferlin transcript encoded by the transgene cassettes, confirming that the dysferlin expression originated from the vectors (data not shown). We observed efficient full-length dysferlin expression in all treated muscles compared to C57BL/6 mice (Figure 3a). Dysfprmd mice treated with Set 3 vectors demonstrated positive dysferlin expression by immunostaining using a monoclonal antibody against dysferlin compared to noninjected mice, with some fibers presenting stronger expression of dysferlin than other. In wild-type mice, dysferlin was mainly localized at the sarcolemma and, at a reduced level, in the cytoplasm associated to T-tubules.24 In treated deficient mice, labelling of the newly synthesized dysferlin was strong both at the sarcolemma and in the cytosol (Figure 3b).
Figure 3.

Systemic administration of HR-based vectors transduces effectively the muscles of dysferlin deficient mice and improves the phenotypes of the muscles. Set3 rAAV8 (9e13 vg/kg) was delivered in the tail vein of 1-month-old Dysfprmd mice (n = 3/group). (a) Detection of transgenic dysferlin (237 kDa) by Hamlet western blot in all sampled muscles. Bi, Biceps brachii; Del, Deltoid; Ga, Gastrocnemius; Glu, Gluteus; Pso, Psoas; TA, Tibialis anterior; Tri, Triceps brachii; Qua, Quadriceps femoris. (b) Immunostaining performed with a dysferlin C-terminal (Hamlet) domain specific antibody in TA muscle compared to WT muscle. No signal was detected in phosphate-buffered saline–injected deficient mice or with secondary antibody control. Scale bar = 40 µm. (c) Representative area of hematoxylin-phloxine-saffron counterstained muscle sections of Glu, Pso, and TA muscles from WT, untreated, and treated deficient mice. Scale bar = 50 µm. (d) Centralized nuclear fibers (CNF) index. Values are expressed in CNF per mm2 of muscle section. Data are significantly different compared to untreated deficient mice. Data are given as mean ± SEM; n = 3 for each group; **P < 0.01; *P < 0.05.
The histological state of treated mice was evaluated in hematoxylin-phloxine-saffron–stained sections (Figure 3c). The expression of dysferlin was associated with a decrease of centralized nuclear fibers in all collected muscles compared to the noninjected deficient mice (Figure 3d). Importantly, even though a significant level of full-length dysferlin protein was expressed with this high dose (9e13 vg/kg), a full phenotypic restoration of dysferlin deficient mice was not reached after one month of expression since some level of centronucleation remained (Figure 3c,d). In addition, expression of dysferlin was checked in other organs. WB analyses showed a consequent level of expressed protein into the heart and a weak expression in liver tissue (Figure 4a). No cardiac or hepatic histopathological abnormalities were observed in treated mice (Figure 4b) and no increased activity level of seric enzymes (alanine (ALAT) and aspartate (ASAT) aminotransferase) was observed between untreated and treated deficient mice (Figure 4c).
Figure 4.
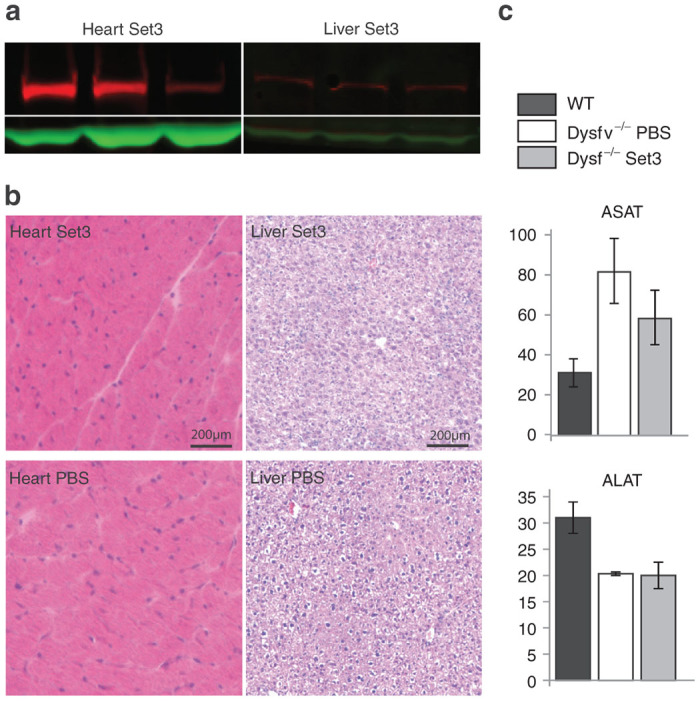
Ectopic transgene expression and assessment of enzymatic activity in serum. (a) Western blot of heart and liver tissues sampled from mice after Set3 systemic injection. Detection was performed with Hamlet antibody and GAPDH was used as a loading control. Three samples are shown. (b) Histology of heart and liver of injected mice (upper panels) compared to phosphate-buffered saline–injected control (lower panels). No abnormality was noticed in any of the samples. Scale bar = 200 µm. (c) Quantification of enzymes, ASAT and ALAT. Units are expressed in U/l. No significant difference was observed between treated and untreated mice.
A dose of 4.5e13 vg/kg of the Set 3 allowed long-term functional rescue
Because of the absence of a full histological recovery upon following vector mediated dysferlin restoration/overabundance (Figure 3), we reasoned that there was the possibility that the quantity of dysferlin might be too high since it was previously shown that overexpression of dysferlin was associated with toxicity.25 Therefore, we performed experiments with different amounts of vectors to determine the required minimal dose of dysferlin to improve the pathology using the Set 3 vectors. Three doses, corresponding to half (4.5e13 vg/kg), a third (3e13 vg/kg), or a quarter (2.3e13 vg/kg) of the initial dose (9e13 vg/kg), were tested by systemic administration in the tail vein in 1-month-old Dysfprmd mice (n = 4/group) and were analyzed 1 month or 1 year postinjection.
Blood and the most affected muscles in Dysf prmd (TA, Pso, and Glu) were sampled for dysferlin analysis 1 month after the Set 3 AAV vector administration. We detected the expected 237 kDa band of the dysferlin protein in all injected groups by WB analysis (Figure 5a). A quantification using vinculin as a loading control was performed in Glu, Pso, and TA muscles samples (Figure 5b). The efficiency of expression of dysferlin presented variations according to the muscles. The 4.5e13 vg/kg dose led to a level of dysferlin in TA corresponding to the endogenous level of dysferlin in a wild-type (WT) muscle. For the same dose, compared to the WT condition, the quantity of expressed dysferlin was decreased twofold in the Pso muscle and approximately twofold elevated in the Glu muscle (Figure 5b). The dysferlin expression corresponding to two lowest injected doses (3e13 or 2.3e13 vg/kg) was lower as expected. On the muscle sections, the level of transduction is related to the quantity of injected vectors (Figure 5c, upper panel). Inflammatory infiltrates was checked by immunohistofluorescence using CD8 and CD11b antibodies. No positive CD8 cell was observed in any conditions (data not shown). CD11b labeling showed clusters of positive cells in noninjected deficient mice whereas in treated mice, only few remaining cells were detected in all treated samples, suggesting a decrease of inflammation after rAAV delivery (Figure 5c, lower panel).
Figure 5.

Determination of dose effect after systemic administration of HR-based vectors. Three doses (4.5e13, 3e13, and 2.3e13 vg/kg) corresponding to a half, a third, or a quarter of the maximum dose of Set3 vectors were injected in the tail vein in 1-month-old Dysfprmd mice (n = 4/group). Endpoint analysis occurred at 28 days post-rAAV delivery. (a) Western blot of TA muscles comparing the relative quantity of dysferlin in treated muscles. Dysferlin quantification was normalized by vinculin. (b) Western blot quantification of dysferlin, normalized by vinculin in analyzed muscles. Values are given in arbitrary unit (A.U.). Data are significantly different compared to WT mice. Data are given as mean ± SEM; n = 6 for Glu and Pso in WT and n = 4 for the others; ***P < 0.001. (c) Immunofluorescence analysis of muscles injected with the three doses. Upper panel: DAPI counterstained anti-dysferlin (Romeo) staining performed on muscle sections of untreated and treated muscles of dysferlin deficient mice. Lower panel: 4’,6-diamidino-2-phenylindole (DAPI) counterstained anti-CD-11b staining to detect inflammatory response. White arrows indicate positive cells/cluster of cells. Scale bar = 50 µm. (d) Centralized nuclear fibers (CNF) index of Glu, Pso, and TA treated muscles (number of centronucleated fibers per mm2). Values are expressed in CNF per mm2 of muscle section. (e) Level of creatine kinase in serum of treated and untreated deficient mice compared to normal mice. Units are expressed in U/l. (f) Quantification of Evans Blue-positive fibers per mm2 on muscle sections of treated and untreated deficient mice compared to normal mice, 3 days after LSI assay. Data are significantly different compared to untreated deficient mice. Data are given as mean ± SEM; n = 4 for each group; ***P < 0.001; **P < 0.01; *P < 0.05.
At the histological level, the presence of dysferlin was associated with a decrease of centralized nuclear fibers in TA and Glu muscles, but not in the Pso muscle (Figure 5d). This could be explained by the fact that the expression of dysferlin in the Pso muscle of treated mice did not reach physiological levels. After treatment with Set 3 vectors (Figure 1), the creatine kinase (CK) level was measured in serum. Regardless of the administered dose, the level of CK was lower than that determined in the nontreated mice, with a level very close to the WT condition for the highest injected dose (4.5e13 vg/kg dose; Figure 5e).
To evaluate the functional rescue of muscles after rAAV dysferlin treatment using Set 3 vectors, a functional eccentric assay, named large-strain injury (LSI), was performed for the three doses.26,27 This assay was designed to evaluate the ability of the TA muscle to recover from eccentric exercise and intrinsically, the sarcolemma ability to respond to this exercise. The LSI assay was performed in vivo in the TA muscle of Set 3 treated mice. The membrane damage was induced by 15 repetitive eccentric contractions and muscles were collected 3 days later. Evans Blue dye (EBD) was injected the day before muscle harvest to stain the sensitive-to-damage fibers. As we previously reported, the dysferlin-deficient muscles showed a very significant increase in EBD positive fibers compared to WT mice.28 In all muscles treated with Set 3 vectors, the abundance of EBD positive fibers was significantly decreased by at least fivefold (Figure 5f).
The long-term outcome was analyzed in mice injected with a quarter or a half of the maximal dose after 1 year. The dysferlin protein level obtained with the half dose is close to the WT one whereas it is lower and almost absent in some mice injected with a quarter of the maximum dose (Figure 6a,b). An increase of the muscle weight was observed with both doses (Figure 6c). At the histological level, the positive effect of the injection can be clearly observed with a nice correlation with the level of dysferlin obtained after injection (Figure 6d). The 1/2 dose histology is close to the WT condition while, with the ¼ dose, the mice with less dysferlin present the worst histology. Finally, assessment of the global force of the mice was performed using the escape test (Figure 6e). The data showed a very significant restoration of the global force for the highest dose (0.141 N/g) versus WT (0.149 N/g) and compared to Dysfprmd (0.108 N/g). These values indicated that the mice injected with ½ dose reached 95% of the WT force and therefore correspond to an improvement of 81% compared to the Dysfprmd mice (72% of the WT value).
Figure 6.

Long-term improvement of dysferlin-deficient mice with HR-based vectors. Half or the quarter of the maximum doses of Set3 vectors were injected in the tail vein of 1-month-old Dysfprmd mice (n = 4/group) to evaluate the long-term phenotype improvement. Endpoint analysis occurred at 1-year post-rAAV delivery. Data are given as mean ± SEM; n = 4; *P < 0.05; **P < 0.01; ***P < 0.001. (a) Western blot of Qua muscles performed with Hamlet antibody and comparing the relative quantity of dysferlin in treated muscles. Dysferlin quantification was normalized by α-actin. (b) Quantification of the western blot, showing a level with the ½ dose reaching nearly the WT level. Values are given in AU. (c) Weight of the Qua muscles showing increases with both doses that are significant compared to the phosphate-buffered saline (PBS)-injected Dysfprmd mice. (d) Representative area of hematoxylin-phloxine-saffron–stained muscle sections of Qua muscles from treated deficient mice compared to WT- and PBS-injected deficient mice. The order from left to right for the two doses is the same than in the western blot in (a), showing that the less improved muscles correspond to the mice with less dysferlin (i.e., the two middle muscles of the ¼ dose). Scale bar = 100 µm. (d) Assessment of the whole body force 1 year after injection, showing that mice injected with ½ dose reached 95% of the WT force (0.141 versus 0.149 N/g for the WT and compared to 0.108 N/g for the PBS-injected deficient mice). n = 4.
All together, these data show that, the highest administered dose of Set 3 vectors (4.5e13 vg/kg) allowed the restoration of physiological levels of dysferlin in treated mice, leading to an improvement of the dystrophic phenotype including restoration of the whole body force nearly to the WT level.
Discussion
Several studies reported the development of strategies to overcome the packaging limitation of wild type AAV vectors (5-kb) for the transfer of large transgenes.2,3,6,29–32 Despite an early suggestion that some serotypes may be tolerant for encapsidation of larger genomes,10,11 it appears that all these strategies rely on intermolecular recombination, exploiting the fact that the AAV genomes are recognized as targets for host DNA repair pathways.1,16 Depending on the case, this event could happen between ITR sequences during concatemerization,2,3,5,6,23,29–37 between overlapping sequences of two distinct cassettes,3,15,36,38–41 or between common regions of fragments of the same initial cassette for fragmented/oversized.10–16,36 The recombination could, in addition, be coupled with a splicing process as in hybrid vectors.17,18,36,39
In the literature, these vector strategies for large gene transfer were evaluated in different contexts, with different genes of varying sizes, and in different cell types and/or target tissues, including skeletal muscle3,16,18,39 or in the retina.15,16,36 In this study, we undertook a comparison in skeletal muscle using the 6.2-kb long dysferlin cDNA as a model and the AAV8 capsid. This study built on our previous results demonstrating reconstitution of dysferlin expression by concatemerization-splicing vectors (Set 1),23 albeit with a modest efficiency following systemic administration. In this comparative study, the overlapping AAV large gene transduction strategy (Set 3) was identified as the most potent approach for dysferlin restoration.
The overlapping strategy has been previously used in skeletal muscle to transfer either β-galactosidase by IM injection or minidystrophin following both IM3,18,38,39 and intravenous injection (IV) administration.40,41 Early experiments3,38 were performed using AAV2 which explained the low efficiency of transduction, but using the alternative serotypes such as AAV6 or AAV9 the efficiency was relatively high. In particular, 80% of fibers expressed the LacZ transgene after IM transfer of an overlapping vector set using an 872 bp alkaline phosphatase coding sequence (AP) overlapping sequence38 and 90% positive fibers were observed after IM transfer of a vector set with a 400 bp overlap of dystrophin sequence.39 The same vector set led to 35–40% positive fibers following IV administration40 and demonstrated therapeutic efficiency.41 In our study, we demonstrated that full-length dysferlin protein was restored and over-expressed (0.5- to 2-fold) compared to the WT level, depending on the muscle following systemic delivery of our overlapping vector set (Set 3, Figures 3, 5 and 6). Furthermore, restoration of histological deficiencies and whole-body force were observed to reach nearly the WT condition with a dose 4.5e13 vg/kg (Figure 6). Altogether, these results reinforce that the AAV overlapping vector large gene delivery strategy is of great interest to target the skeletal muscle for the treatment of muscular dystrophies.
In previous works, the overlapping vector strategy was used to transfer eye-disease–related genes, MYO7A or ABCA4, to the retina following subretinal injection.15,36 Of note, these vectors efficiently transduced retinal pigment epithelial cells but not neuronal photoreceptors (PR), suggesting that the nature of the cells can impact the efficiency of large transgene reconstruction.36 It was proposed that the lack of overlapping vector transduction efficiency in the PR is a consequence of the low levels of HR in postmitotic neurons.42 Downregulation of the HR factors is one hallmark associated with the exit from the cell cycle and terminal differentiation in neurons42 and muscle fibers,43 a fact that may underlay the permissivity of these tissues to AAV large gene transduction. However, the absence of canonical HR in nondividing cells seemingly contradicts the decent efficiency of the different AAV vector genome reconstruction approaches observed in skeletal muscle and, therefore, suggests that the necessary repair factors, and or mechanism, remain uncharacterized. In fact, whereas AAV transduction seems to rely on the inhibition of the MRN (Mre11, Rad50, Nbs1) complex,43–45 it has been shown that reconstitution of full-length transcript from fAAV genomes was dependent on the DNA strand-annealing protein Rad51C,16 a protein of the HR pathway that was shown to be less abundant in brain tissue than in skeletal muscle.46 Therefore, the differences of how the cellular machinery process AAV vectors in muscle and neurons, despite both being postmitotic cells, might depend on these DNA repair factors or others that remained to be determined. Beside this point, several studies pinpoint the importance of the nature of the overlapping sequence for the reconstitution efficiency in overlapping or hybrid strategies. For example, a recent study performed in the skeletal muscle observed a significant difference in efficiency of four different set of dual AAV vectors that have identical expression cassette except for the region of overlap.47
Interestingly, adding an overlapping region of 254 bp to the splicing process in the Set 2 did not led to increase efficiency (Figure 2a, compares Set 1 and Set 2). The overlapping sequence present in Set 2 has not been evaluated per se for its recombinogenic properties but it was shown in previous studies that combining splicing and HR is not always beneficial. Addition of two different overlapping sequences, a ~300 bp “highly recombinogenic” AP fragment or a 77 bp fragment of AK (derived from the F1 phage genome in a study performed with two different transgenes (MYO7A and ABCA4) let to different outcomes36). In vitro and in vivo, the introduction of the AK sequence had a small positive effect, while introduction of the AP sequence introduction was very deleterious, although it was only evaluated in vitro.36 Therefore, combining an AAV trans-splicing strategy with a homologous fragment can impair or enhance the large gene transduction efficiency in a manner that requires further characterization.18,36 It remains possible that particular sequence combinations may influence the ITR intermolecular recombination event, the HR of the overlapping sequences, and/or alter the splicing process, resulting in decreased efficiency.
Consistent with earlier reports, our data further confirmed that the “fragmented” full-length large gene transduction strategy relies on partial genome encapsidation and HR of the transduced fragments.10–14,16,36 Importantly, this notion is further supported by the demonstration that the transduction efficiency was directly related to the quality and size of the encapsidated vector genome fragments and not on the overall production titer. Therefore, careful attention must be exercised when using fAAV preparations to exclude the nontransducing species, which is dependent up on the transgene size, to reduce the possibility of tittering genome fragments incompetent for transduction (Figure 1). In fact, differences in fAAV vector tittering may account for the performance variations when comparing the efficiencies of large gene transduction strategies.16,48 Although the transduction efficiency of fAAV-dysferlin was approaching that of the Set 3 vectors (Figure 2c; Table 1), we chose not to pursue this strategy for the systemic studies due to clinical safety concerns as the fAAV genomes cannot be clearly defined and are difficult to titer accurately due to contamination by nontransducing species.16 Regarding these packaged species, a ladder pattern was observed indicative of fragmented genomes of various sizes (Figure 1b). Additionally, it has been shown that the termination of packaging during AAV genome encapsidation is a relaxed process and thus, likely generates heterogeneous 5’ ends, perhaps in a sequence specific manner, a phenomenon that requires further characterization prior to consideration of fAAV as a therapeutic.49
The restoration of dysferlin levels, the significant improvement of dysferlin deficient muscle histology, the phenotypic recovery after an eccentric contraction protocol and restoration of the whole body force present one year after injection are very promising data for the use of AAV-mediated transfer of dysferlin cDNA as a therapeutic strategy for dysferlinopathy. This point is of increasing interest as there is evidence that it might be beneficial to transfer the full-length dysferlin, rather than a truncated dysferlin variant termed minidysferlin.28,50 First, dysferlin is composed of many unique domains that can support precise functions as suggested by their various and specific protein partners. This point suggests dysferlin may not be as amenable to domain deletions as has been noted for dystrophin, for which strategies relying on mini- or micro-dystrophin gene therapy were shown to be functional even in large animal models.51,52 Second, even though a naturally occurring minidysferlin has been identified in a patient presenting a mild dystrophic phenotype50; the data in animal models suggest that not all of the functions are recovered after gene transfer of this particular form.28 In particular, even if the membrane repair function was restored after rAAV transduction, the gene transfer was not associated with correction of the dysferlin deficient muscle histology or with the recovery of resistance to eccentric contractions.50 This suggests that some functions supported by the missing dysferlin domains are required to fully restore the phenotype of dysferlin deficiency.
In summary, our results demonstrate that overlapping AAV vector transduction is a leading approach to safely restore full-length dysferlin into the deficient skeletal muscle resulting in correction of the dystrophic phenotype.
Materials and Methods
Plasmid constructs
The full-length human dysferlin isoform 8 (NM_003494.3) cDNA was used in all cassettes of this study. The two cassettes for the concatemerization-splicing strategy (Set 1), previously described in ref. 23, were composed as follows: the Set1 5’cassette includes a synthetic muscle-oriented promoter C5.12, a chimeric intron (β-globin), a consensus Kozak sequence (GCCACC), the exons 1 to 28 of dysferlin and the half intron 28 of dysferlin and the Set1 3’ cassette includes the second half of intron 28, the exons 29 to 55 and a SV40 polyadenylation signal sequence. The cassettes Set2 were derived from Set1 5’ and 3’ cassettes with the modification that the full length intron 28 of dysferlin was added in the place of the half intron 28 in both plasmids. The two cassettes for the HR strategy, named Set3, were composed as follows: Set3_5’ includes the C5.12 promoter, a chimeric intron (β-globin), a consensus Kozak sequence (GCCACC) and the exons 1 to 30 and Set3_3’ includes the exons 24 to 55 and a SV40 polyadenylation signal sequence. The region of homology between the two Set3 cassettes is of 859pb. A plasmid containing the C5.12 promoter, a chimeric intron (β-globin), a consensus Kozak sequence, the full-length dysferlin cDNA and a SV40 polyadenylation signal sequence was used for fAAV construct. All cassettes were pAAV2 and verified by sequencing.
Production of recombinant AAV
All recombinant AAV2/8 used in this study were produced using a triple transfection protocol and purified by two sequential cesium chloride (CsCl) density gradients. At the end of second centrifugation, 11 fractions of 500 µl were recovered from the CsCl Density Gradient tube and purified through dialysis in 1× PBS solution (ThermoScientific, Illkirch, France). For the Fragment batch 2 preparation, only fractions 2 and 3 of the second gradient were pooled for the dialysis. Therefore, rAAVs used for systemic administration were enriched in large fragments. The fractions were analyzed by dot blot to determine those containing rAAV genomes. The viral genome number (vg) of each preparation was determined by quantitative real-time PCR-based titration method using primers and probe corresponding to the ITR region of the AAV vector genome.53
Alkaline agarose gel electrophoresis
An aliquot of each rAAV sample containing 6e10 vg was treated with DNase (Millipore, Molsheim, France) to remove any unprotected DNA. The capsids were degraded during 10 minutes at 72 °C in the presence of proteinase K (Macherey-Nagel, Hoerdt, France). DNA was extracted with High Pure Viral Nucleic Acid kit and eluted in 50 µl of elution buffer (Roche, Boulogne-Billancourt, France). All samples were denatured for 5 minutes at 95 °C and then immediately kept on ice for 5 minutes. To 15 µl of each sample, 3.1 µl of ethylenediaminetetraacetic acid (Roche) of 100 mmol/l were added; 3 µl of loading buffer 6× composed of 300 mmol/l NaOH, 6 mmol/l ethylenediaminetetraacetic acid, 18% ficoll, 0.5% bromocresol green and 0.25% xylene cyanol; and 1 µl of GelRed 20X (Biotium, Hayward, CA). The size-separation of samples was performed on an alkaline agarose gel (0.8% of agarose, 50 mmol/l NaOH and 1 mmol/l ethylenediaminetetraacetic acid) at 4 °C overnight at 18V. The GeneRuler DNA Ladder Mix (ThermoScientific) was used to determine the approximate molecular weights (MW) in kb of DNA in each of the bands. After 30 minuteswash in TAE (Tris base, Acetic acid, EDTA (ethylenediaminetetraacetic acid)) ×, the gel was stained for 5H using GelRed diluted 1/10,000 in TAE 1×. A photograph of gel was performed for direct visualization of bands. The gel was transferred using the iBlot Dry Blotting System (ThermoScientific) onto a nylon membrane which was hybridized with a specific ITR probe labeled with the kit AlkPhos direct (Amersham Biosciences, GE Healthcare, Velizy-Villacoublay, France). Detection was performed on a fluorescence Storm scanner (100 µm, 650 V).
Animal models, vector administration, and functional evaluation
All procedures on animals were performed in accordance with the directive of 24 November 1986 (86/609/EEC) of the Council of the European Communities and were approved by Genethon’s ethics committee under the numbers CE11-013 and A-CE11-013. C57BL/6 mice were purchased from Charles River Laboratories (Les Oncins, France). B6.A-Dysfprmd (Dysfprmd) mice were obtained and bred at Généthon’s animal facility.
All experimental protocols were performed on 4-week-old male mice. IM injections were performed into the left TA muscle with 30 µl (1.3e10 vg per TA) of the rAAV viral preparation (alone or in equal proportion of each vector according to the vector set) or PBS (ThermoScientific). Systemic administrations were performed by tail vein injections of 500 µl with different quantities of viral genome (9e13, 7.25e13, 4.5e13, 3e13, or 2.3e13 vg/kg) of a mix of the Set3rAAV viral preparations or PBS. The endpoint was 28 days or 1 year after injection.
The repetitive large-strain injury (LSI test) was performed in vivo on TA muscle.26,27 All mice were received an intraperitoneal injection with EBD (1 mg/g of body weight), 2 days after the LSI test. The mice were sacrificed 24 hours after EBD injection and the TA muscles were removed and quickly frozen in liquid nitrogen-cooled isopentane.
Assessment of the whole body force by escape test
Global activity of mice was evaluated by escape test. Mice were placed on a platform facing the entrance of a tube that was 30 cm long. A cuff was wrapped around the tail and connected to a fixed force transducer. In response to a gentle pinching of the tail, mice tried to escape within the tube. A short peak of force was recorded by the force transducer and the test was performed five times. Maximal peak and the average of the five peaks normalized to animal body weight are reported.
RNA extraction and quantitative RT-PCR analysis
Total RNA was extracted from muscles lysates by the Trizol method (Life Technologies). The Free DNA kit (ThermoScientific) was used to remove residual DNA from the samples. One microgram of total RNA was reverse-transcribed using random hexamers and the Verso cDNA kit (ThermoScientific). Quantitative RT-PCR was performed with following primers and TaqMan probe: Exon28F: CTCAACCGGGCTGTCGAT, Exon29R: GTCGGTGTGTGTAGTACATCTTCTCA and Exons28/29 Probe: CAAGGCTGGGAG. The RH approaches (Set 3 and fAAV) do not allow designing specific probe due to the large homologous region shared between 5’ and 3’ parts. To validate the recombination, a RT-PCR was performed from RNA extracted from muscle of mice injected with both vectors using primers flanking the region of homology and the product was sequenced. Total RNA from an adult skeletal muscle served as quantification control (Stratagene, Agilent Biotechnologies, Les Ulis, France). The ubiquitous acidic ribosomal phosphoprotein (Rplp0) was used to normalize the data across samples.
Western blot analysis and enzyme-linked immunosorbent assay
Proteins were extracted from muscles, heart, or liver by cell lysis buffer (Pierce RIPA buffer, Thermo Scientific) and proteases inhibitors (Complete PIC, Roche) after homogenization with a FastPrep-24 (MP Biomedicals, Illkirch, France) in the Lysing Matrix A tubes (MP Biomedicals). Protein concentrations were determined by the BCA Protein Assay kit (Pierce, Thermo Scientific). The samples were prepared following the NuPAGE Tris-Acetate Gel protocol (Life Technologies). The proteins (100–120 µg) were separated using a NuPAGE precast 3–8% Tris-Acetate gel (ThermoScientific) and then transferred to nitrocellulose membrane (Life Technologies) with iBlot Dry Blotting system (Life Technologies) using the P3 program for 8.5 minutes. Detection of dysferlin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), or vinculin were performed using standard Odyssey protocol with primary specific monoclonal anti-dysferlin antibody: Mouse NCL-Hamlet (Novocastra, Leica Biosystems, Nanterre, France) diluted 1/200, Rabbit Romeo (Epitomics, Burlingame, CA) diluted 1:200, Rabbit anti–GAPDH (FL-335) antibody (Santa Cruz Biotechnology, Heidelberg, Germany) diluted 1:500, Mouse anti-Vinculin (Sigma) diluted 1: 500 and anti-actin; Rabbit (H-300; SantaCruz) diluted 1:500 or Mouse (ab11003; Abcam, Paris, France) diluted 1:500. Blot was revealed using the Odyssey secondary antibody goat anti mouse (GAM) 680, goat anti rabbit (GAR 680 and GAR 800 diluted 1:5,000, scanned. Band density was quantified using the Image Studio Lite 4.0 software (LI-COR Biosciences, Lincoln, NE). The integrated density of dysferlin was normalized by the vinculin’s, GAPDH’s or actin’s one.
Histology and immunohistochemistry
Cryosections (8–10-mm thickness) were prepared from frozen skeletal and cardiac muscles and formalin-fixed paraffin-embedded liver. Histological staining was performed with hematoxylin-phloxine-saffron.
Immunodetection was performed with the monoclonal antibody dysferlin-Hamlet (NCL-Hamlet, diluted 1:50) as described previously28 or with the monoclonal dysferlin-Romeo (ab124684; Abcam) diluted 1:50. Mouse anti-CD11b (550282; BD Biosciences; Le Pont de Claix, France); diluted 1:40 was used to detect inflammatory cells and RyR (ab2868; abcam) diluted 1:200 was used to perform colocalization of dysferlin in T-tubules. Sections were mounted with DAPI-Fluoromount-G (Southern Biotech, Birmingham, AL) and examined under a confocal microscope Leica TCS SP2.
Evans Blue dye positive fibers were revealed by fluorescence excitation at 633 nm on a Leica confocal fluorescent microscope on cryosections treated 10 minutes in frozen acetone. Sections were mounted with Fluoromount-G (Southern Biotech) before the observation. The cartography of the whole section was performed using a motorized stage at an original 40x magnification and using the software Cartograph (Microvision, Evry, France). The ratio of the area corresponding to the Evans Blue positive cells versus the whole area of the section was measured using Image J in the red channel.
Serum enzyme measurement
CK, ALAT, and ASAT activities in serum were measured with VITROS Slides (Ortho-Clinical Diagnostics, Rochester, NY).
Statistical analysis
Data for each group are represented as the means plus standard error of the mean. Differences between groups, compared one versus one, were determined using the Student’s t-test. Statistical significance was defined as P < 0.001 (***), P < 0.01 (**), and P < 0.05 (*).
Acknowledgments
This work was funded by the Association Française contre les Myopathies. Marina Pryadkina was a recipient of a MRT PhD grant. We would like to thank Marc Bartoli and Marie Chapoton (Univ. Méditerranée, Marseille) for helpful discussion and Denis Furling and Arnaud Klein (Institute of Myology, Paris) for experimental help. In addition, support was also provided in part through an Unrestricted Grant from Research to Prevent Blindness to the UNC Department of Ophthalmology.
The authors declare no conflict of interest.
References
- Hirsch ML, Agbandje-McKenna M, Samulski RJ. Little vector, big gene transduction: fragmented genome reassembly of adeno-associated virus. Mol Ther. 2010;18:6–8. doi: 10.1038/mt.2009.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H, Storm TA, Kay MA. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver in vivo. J Virol. 2000;74:9451–9463. doi: 10.1128/jvi.74.20.9451-9463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Yue Y, Engelhardt JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol Ther. 2001;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- Nakai H, Fuess S, Storm TA, Meuse LA, Kay MA. Free DNA ends are essential for concatemerization of synthetic double-stranded adeno-associated virus vector genomes transfected into mouse hepatocytes in vivo. Mol Ther. 2003;7:112–121. doi: 10.1016/s1525-0016(02)00034-5. [DOI] [PubMed] [Google Scholar]
- Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, Chamberlain JS. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol. 2005;23:1435–1439. doi: 10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Long C, Bostick B, Duan D. Efficient whole-body transduction with trans-splicing adeno-associated viral vectors. Mol Ther. 2007;15:750–755. doi: 10.1038/sj.mt.6300081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Yan Z, Yue Y, Engelhardt JF. Structural analysis of adeno-associated virus transduction circular intermediates. Virology. 1999;261:8–14. doi: 10.1006/viro.1999.9821. [DOI] [PubMed] [Google Scholar]
- Halbert CL, Allen JM, Miller AD. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat Biotechnol. 2002;20:697–701. doi: 10.1038/nbt0702-697. [DOI] [PubMed] [Google Scholar]
- Allocca M, Doria M, Petrillo M, Colella P, Garcia-Hoyos M, Gibbs D. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118:1955–1964. doi: 10.1172/JCI34316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Yue Y, Duan D. Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome > or = 8.2 kb. Mol Ther. 2010;18:75–79. doi: 10.1038/mt.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose WE, Clark KR, Griffin D, Malik V, Shontz KM, Montgomery CL. Homologous recombination mediates functional recovery of dysferlin deficiency following AAV5 gene transfer. PLoS One. 2012;7:e39233. doi: 10.1371/journal.pone.0039233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Nakai H, Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther. 2010;18:87–92. doi: 10.1038/mt.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes VS, Boye SE, Louie CM, Boye S, Dyka F, Chiodo V. Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther. 2013;20:824–833. doi: 10.1038/gt.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch ML, Li C, Bellon I, Yin C, Chavala S, Pryadkina M. Oversized AAV transductifon is mediated via a DNA-PKcs-independent, Rad51C-dependent repair pathway. Mol Ther. 2013;21:2205–2216. doi: 10.1038/mt.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Duan D. Efficient transgene reconstitution with hybrid dual AAV vectors carrying the minimized bridging sequences. Hum Gene Ther. 2011;22:77–83. doi: 10.1089/hum.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Lai Y, Duan D. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol Ther. 2008;16:124–130. doi: 10.1038/sj.mt.6300322. [DOI] [PubMed] [Google Scholar]
- Anderson LV, Davison K, Moss JA, Young C, Cullen MJ, Walsh J. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum Mol Genet. 1999;8:855–861. doi: 10.1093/hmg/8.5.855. [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Klinge L, Harris J, Sewry C, Charlton R, Anderson L, Laval S. Dysferlin associates with the developing T-tubule system in rodent and human skeletal muscle. Muscle Nerve. 2010;41:166–173. doi: 10.1002/mus.21166. [DOI] [PubMed] [Google Scholar]
- Lek A, Evesson FJ, Sutton RB, North KN, Cooper ST. Ferlins: regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair. Traffic. 2012;13:185–194. doi: 10.1111/j.1600-0854.2011.01267.x. [DOI] [PubMed] [Google Scholar]
- Lostal W, Bartoli M, Bourg N, Roudaut C, Bentaïb A, Miyake K. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum Mol Genet. 2010;19:1897–1907. doi: 10.1093/hmg/ddq065. [DOI] [PubMed] [Google Scholar]
- Ampong BN, Imamura M, Matsumiya T, Yoshida M, Takeda S. Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. 2005;24:134–144. [PubMed] [Google Scholar]
- Glover LE, Newton K, Krishnan G, Bronson R, Boyle A, Krivickas LS. Dysferlin overexpression in skeletal muscle produces a progressive myopathy. Ann Neurol. 2010;67:384–393. doi: 10.1002/ana.21926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JA, Lovering RM, Roche R, Ru LW, Reed PW, Bloch RJ. Extensive mononuclear infiltration and myogenesis characterize recovery of dysferlin-null skeletal muscle from contraction-induced injuries. Am J Physiol Cell Physiol. 2010;298:C298–C312. doi: 10.1152/ajpcell.00122.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JA, Lovering RM, Bloch RJ. Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport. 2008;19:1579–1584. doi: 10.1097/WNR.0b013e328311ca35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lostal W, Bartoli M, Roudaut C, Bourg N, Krahn M, Pryadkina M. Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy. PLoS One. 2012;7:e38036. doi: 10.1371/journal.pone.0038036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zhang Y, Duan D, Engelhardt JF. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc Natl Acad Sci USA. 2000;97:6716–6721. doi: 10.1073/pnas.97.12.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich SJ, Auricchio A, Hildinger M, Glover E, Maguire AM, Wilson JM. Efficient trans-splicing in the retina expands the utility of adeno-associated virus as a vector for gene therapy. Hum Gene Ther. 2003;14:37–44. doi: 10.1089/10430340360464697. [DOI] [PubMed] [Google Scholar]
- Duan D, Yue Y, Yan Z, Engelhardt JF. A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat Med. 2000;6:595–598. doi: 10.1038/75080. [DOI] [PubMed] [Google Scholar]
- Sun L, Li J, Xiao X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat Med. 2000;6:599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- Chao H, Sun L, Bruce A, Xiao X, Walsh CE. Expression of human factor VIII by splicing between dimerized AAV vectors. Mol Ther. 2002;5:716–722. doi: 10.1006/mthe.2002.0607. [DOI] [PubMed] [Google Scholar]
- Xu Z, Yue Y, Lai Y, Ye C, Qiu J, Pintel DJ. Trans-splicing adeno-associated viral vector-mediated gene therapy is limited by the accumulation of spliced mRNA but not by dual vector coinfection efficiency. Hum Gene Ther. 2004;15:896–905. doi: 10.1089/hum.2004.15.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Yue Y, Liu M, Duan D. Synthetic intron improves transduction efficiency of trans-splicing adeno-associated viral vectors. Hum Gene Ther. 2006;17:1036–1042. doi: 10.1089/hum.2006.17.ft-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani I, Colella P, Sommella A, Iodice C, Cesi G, de Simone S. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. 2014;6:194–211. doi: 10.1002/emmm.201302948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo T, Popplewell L, Athanasopoulos T, Dickson G. Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum Gene Ther. 2014;25:98–108. doi: 10.1089/hum.2013.164. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Duan D. Viral serotype and the transgene sequence influence overlapping adeno-associated viral (AAV) vector-mediated gene transfer in skeletal muscle. J Gene Med. 2006;8:298–305. doi: 10.1002/jgm.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Duan D. Novel mini-dystrophin gene dual adeno-associated virus vectors restore neuronal nitric oxide synthase expression at the sarcolemma. Hum Gene Ther. 2012;23:98–103. doi: 10.1089/hum.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yue Y, Li L, Hakim CH, Zhang K, Thomas GD. Dual AAV therapy ameliorates exercise-induced muscle injury and functional ischemia in murine models of Duchenne muscular dystrophy. Hum Mol Genet. 2013;22:3720–3729. doi: 10.1093/hmg/ddt224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom GL, Gregorevic P, Allen JM, Chamberlain JS. Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol Ther. 2011;19:36–45. doi: 10.1038/mt.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res. 2007;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Lovric J, Mano M, Zentilin L, Eulalio A, Zacchigna S, Giacca M. Terminal differentiation of cardiac and skeletal myocytes induces permissivity to AAV transduction by relieving inhibition imposed by DNA damage response proteins. Mol Ther. 2012;20:2087–2097. doi: 10.1038/mt.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi VW, McCarty DM, Samulski RJ. Host cell DNA repair pathways in adeno-associated viral genome processing. J Virol. 2006;80:10346–10356. doi: 10.1128/JVI.00841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RA, Palacios JA, Cassell GD, Adam S, Giacca M, Weitzman MD. The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J Virol. 2007;81:12936–12945. doi: 10.1128/JVI.01523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosanjh MK, Collins DW, Fan W, Lennon GG, Albala JS, Shen Z. Isolation and characterization of RAD51C, a new human member of the RAD51 family of related genes. Nucleic Acids Res. 1998;26:1179–1184. doi: 10.1093/nar/26.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lostal W, Kodippili K, Yue Y, Duan D. Full-length dystrophin reconstitution with adeno-associated viral vectors. Hum Gene Ther. 2014;25:552–562. doi: 10.1089/hum.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyka FM, Boye SL, Chiodo VA, Hauswirth WW, Boye SE. Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum Gene Ther Methods. 2014;25:166–177. doi: 10.1089/hgtb.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Chen L, Dederich D, Dong B, He J, Steinmann KE. Native molecular state of adeno-associated viral vectors revealed by single-molecule sequencing. Hum Gene Ther. 2012;23:46–55. doi: 10.1089/hum.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn M, Wein N, Bartoli M, Lostal W, Courrier S, Bourg-Alibert N. A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathy. Sci Transl Med. 2010;2:50ra69. doi: 10.1126/scitranslmed.3000951. [DOI] [PubMed] [Google Scholar]
- Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S, Dickson G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med. 2011;13:497–506. doi: 10.1002/jgm.1602. [DOI] [PubMed] [Google Scholar]
- Shin JH, Pan X, Hakim CH, Yang HT, Yue Y, Zhang K. Microdystrophin ameliorates muscular dystrophy in the canine model of duchenne muscular dystrophy. Mol Ther. 2013;21:750–757. doi: 10.1038/mt.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoli M, Poupiot J, Goyenvalle A, Perez N, Garcia L, Danos O. Noninvasive monitoring of therapeutic gene transfer in animal models of muscular dystrophies. Gene Ther. 2006;13:20–28. doi: 10.1038/sj.gt.3302594. [DOI] [PubMed] [Google Scholar]