

Abstract
As an alternative to the transplantation of islets, a human liver cell line has been genetically engineered to reverse type 1 diabetes (TID). The initial liver cell line (Huh7ins) commenced secretion of insulin in response to a glucose concentration of 2.5 mmol/l. After transfection of the Huh7ins cells with human islet glucokinase, the resultant Melligen cells secreted insulin in response to glucose within the physiological range; commencing at 4.25 mmol/l. Melligen cells exhibited increased glucokinase enzymatic activity in response to physiological glucose concentrations, as compared with Huh7ins cells. When transplanted into diabetic immunoincompetent mice, Melligen cells restored normoglycemia. Quantitative real-time polymerase chain reaction (qRT-PCR) revealed that both cell lines expressed a range of β-cell transcription factors and pancreatic hormones. Exposure of Melligen and Huh7ins cells to proinflammatory cytokines (TNF-α, IL-1β, and IFN-γ) affected neither their viability nor their ability to secrete insulin to glucose. Gene expression (microarray and qRT-PCR) analyses indicated the survival of Melligen cells in the presence of known β-cell cytotoxins was associated with the expression of NF-κB and antiapoptotic genes (such as BIRC3). This study describes the successful generation of an artificial β-cell line, which, if encapsulated to avoid allograft rejection, may offer a clinically applicable cure for T1D.
Introduction
T1D is caused by the autoimmune destruction of insulin-producing pancreatic β-cells.1 Current treatment requires daily injections of insulin to control blood glucose levels or transplantation of insulin-secreting tissue. Since this latter strategy currently relies on a source of human tissue, it seems unlikely that there will ever be sufficient numbers of organs available to assist the number of insulin-dependent diabetics that require transplantation.2 Additionally, these patients would have to be under a chronic regimen of immunosuppressive drugs to prevent both rejection of the transplanted tissue and recurrent autoimmunity. The autoimmune destruction of islet cells could theoretically be overcome by genetically engineering an “artificial β-cell” (i.e., a nonislet cell) capable of synthesizing, storing, and secreting mature insulin in response to metabolic signals. As it is not a bona fide pancreatic β-cell, such a cell would not carry the complete suite of islet and β-cell antigens responsible for recurrent autoimmune reactions. Further, if artificial β-cells were engineered from the patient’s own cells, then the patient would be released from the need of daily insulin injections, and the debilitating complications of the disease, without the added complications that arise from lifelong immunosuppression.
Liver cells have been successfully used as the target cells for gene therapy in a number of successful studies,3–11 including the generation of artificial β-cells. Hepatocytes are known to play a crucial role in the intermediary metabolism, synthesis, and storage of proteins. Most importantly, liver cells express the high-capacity glucose transporter, GLUT 2 (ref. 12), and the glucose phosphorylation enzyme, glucokinase,13 which make up the key elements of the “glucose sensing system” that regulates insulin release from β-cells in response to changes in the external nutrient composition. These characteristics make hepatocytes attractive candidates for a gene therapeutic approach to curing T1D.
The ultimate goal for gene therapy to cure T1D will involve the direct delivery of the insulin gene, and other genes required for regulating the response to glucose, into a patient’s own cells (thereby circumventing issues of allograft rejection). The engineering of a β-cell from a non-β-cell precursor, such as hepatocytes, also has the advantage of putatively minimizing the magnitude of recurrent autoimmune responses, due to the absence of the entire suite of β-cell autoantigens expressed by pancreatic β-cells. However, currently, the most efficient vehicles available to deliver genes directly to the primary cells of an individual’s body are viral vectors, such as retroviral, lentiviral, or adenoviral.5–11 Presently, there are several hurdles surrounding the transfer of viral material with the available viral vector systems,14 such as logistics and safety concerns. This makes the development of an artificial β-cell line, which can control blood glucose levels and be delivered to the patient in an encapsulation system, perhaps a more attractive, and realistic, alternative than the direct delivery of genes to body tissues in vivo using viral vectors.
Well-designed encapsulation systems can protect the enclosed cells against exposure to the immune cells, notably cytotoxic T cells, which mediate allograft and autoimmune reactions. However, the encapsulated cells will still be exposed to immune mediators, such as proinflammatory cytokines, which played major roles in the initial autoimmune destruction of the β-cell population.15 For an artificial β-cell line to be a realistic cure for diabetes, it must fulfill two criteria: (i) the ability to secrete insulin in response to glucose in the physiological range, and (ii) the ability to retain viability in the presence of cytokines secreted during Thelper (Th)1/Th17 proinflammatory immune responses that precipitate autoimmune diabetes.16,17
The expression of GLUT2 and glucokinase (hexokinase IV), the predominant glucose transporter and glucose phosphorylating enzyme, respectively, is central to the ability of pancreatic β-cells, and insulinoma cell lines, such as INS-1 (ref. 18) and MIN6 (ref. 19), to respond to glucose in the physiological range. Other insulinoma cell lines, such as RIN 1046-38 cells,20 do not respond to glucose in the physiological range, and express only negligible/low levels of GLUT 2. Other studies have shown that the overexpression of GLUT 2 alone, or in combination with glucokinase, significantly improved the range of response to glucose of engineered insulin secreting cells.21,22 However, physiological glucose responsiveness cannot be achieved solely by the expression of the glucokinase and hexokinase enzymes. The ratio of glucokinase:hexokinase expression has been implicated in the maintenance of physiological glucose secretion.22,23 For example, an imbalance in the glucokinase:hexokinase ratio, in favor of hexokinase, enhances glycolytic flux at low glucose levels and consequently an inappropriate glucose-stimulated insulin secretion response results.22,23
Previously, our laboratory developed an artificial β-cell line, Huh7ins,4 which exhibited a number of desirable characteristics for potential use in the gene therapy of diabetes. Firstly, Huh7ins cells exhibited endogenous expression of the specific glucose transporter, GLUT 2, and the key glycolytic enzyme, glucokinase. Secondly, Huh7ins cells were able to synthesize, store and secrete insulin in response to glucose. Thirdly, the insulin produced by Huh7ins cells was stored in secretory granules that morphologically resembled those observed in pancreatic β-cells. Fourthly, Huh7ins cells expressed proinsulin convertase 1 and 2, which are the enzymes responsible for the conversion of proinsulin to insulin in pancreatic β-cells. Fifthly, Huh7ins cells responded to a glucose stimulus, via the activation of ATP-dependent K+ and calcium channels, akin to pancreatic β-cells.24 Finally, and perhaps most significantly, Huh7ins cells normalized blood glucose levels when transplanted into diabetic immune-incompetent (NOD/scid) mice, indicating that they retained the ability to secrete insulin in response to glucose following transplantation. However, after transplantation into diabetic recipients and subsequent reversion to normoglycemia, Huh7ins cells secreted insulin in response to subphysiological glucose concentrations (2.5 mmol/l), in contrast to pancreatic β-cells, which commenced insulin secretion at glucose concentrations of 4–5 mmol/l. Consequently, animals transplanted with Huh7ins cells developed chronic hypoglycemia. Therefore, before Huh7ins cells could be considered for potential clinical use, further molecular engineering strategies needed to be implemented to correct the range in which glucose stimulated insulin secretion.
The current study describes the re-engineering of Huh7ins cells to produce a novel cell line, termed Melligen, which stored insulin and secreted it in the physiological range and reversed diabetes in NOD/scid mice, without the induction of hypoglycemia. Melligen cells were also resistant to the toxic effects of proinflammatory cytokines that have been shown to induce the death of pancreatic β-cells and β-cell lines. The Melligen cells are the first truly glucose-responsive, nonpancreatic, insulin-secreting human cell line to be developed. As such Melligen cells warrant further investigation to assess their clinical potential to cure autoimmune diabetes.
Results
Gene transfer into Huh7ins cells
Huh7ins cells were stably transfected with the pIRESpuro3 vector carrying human glucokinase cDNA and 6 clones of Huh7ins cells expressing glucokinase were isolated and assayed for glucose-responsive insulin secretion. The transfectants remained stable for a period of greater than 6 months in continuous culture.
Insulin secretion and storage
In response to increasing concentrations of glucose (1–20 mmol/l), a dose–response curve for insulin secretion was generated for Huh7ins cell clones transfected with human islet glucokinase and compared with that for the parent Huh7ins cells (Figure 1a,b). It can be seen in Figure 1a that while glucose responsiveness commenced at 2.5 mmol/l in Huh7ins cells, it commenced in the physiological range (between 4–5 mmol/L) in the Huh7ins cells transfected with human islet glucokinase (Figure 1b). The actual amount of insulin secreted in response to glucose in the physiological range for clone 6 Huh7ins cells transfected with glucokinase, was also double that of the parent Huh7ins cells (also an advantageous development). Accordingly, clone 6 was used in all subsequent experiments, and the cells of this clone were designated Melligen cells. Glucose response assays performed using Huh7ins cells transfected with the empty vector revealed that their glucose responsiveness was not significantly different from the Huh7ins cells (results not shown). The exact concentration of glucose at which Melligen cells commenced glucose-responsive insulin secretion was determined to be 4.25 mmol/l (Figure 1c).
Figure 1.
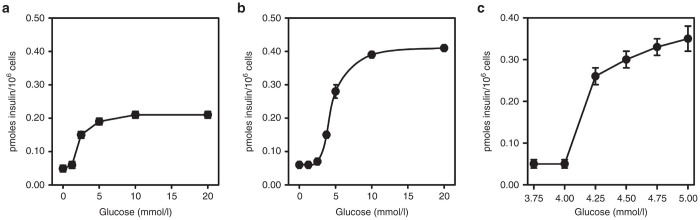
Insulin secretion from liver cell lines: Insulin secretion from (a) Huh7ins and (b) Melligen cells in response to increasing concentrations of glucose: 1.5–20 mmol/l. (c) Insulin secretion from Melligen cells in response to increasing concentrations of glucose: 3.75–5.0 mmol/l. Values are expressed as means ± SE (n = 6).
The storage of insulin in Huh7ins cells and Melligen cells was measured and no differences in insulin storage were observed: 8.5 ± 0.7 pmoles insulin/106 cells (n = 3) for Huh7ins cells transfected with the empty vector and 8.7 ± 0.9 pmoles insulin/106 cells (n = 3) for Melligen cells.
Reversal of diabetes in NOD/scid mice
Transplanted Melligen cells coalesced into a mass at the site of grafting and grafts were visible under the skin 1–2 weeks after the cells were transplanted. All 16 diabetic mice that were transplanted with either Melligen (n = 8) or Huh7ins (n = 8) cells reverted to normoglycemia within 19 days post-transplantation. Blood glucose levels of animals transplanted with Melligen cells remained normoglycemic (5.2–5.7 mmol/l) until removal of the graft at day 27 post-transplantation. By comparison, diabetic animals that were transplanted with Huh7ins cells became hypoglycemic from day 21 (3.1 ± 0.4 mmol/l on day 19 decreasing to 2.6 ± 0.7 mmol/l on day 27) (Figure 2a). Removal of the Huh7ins or Melligen grafts at day 27 resulted in an immediate increase in the blood glucose levels to hyperglycemic values (Figure 2a). These results indicated that reversal of hyperglcemia was not attributable to regeneration of pancreatic β-cells.
Figure 2.
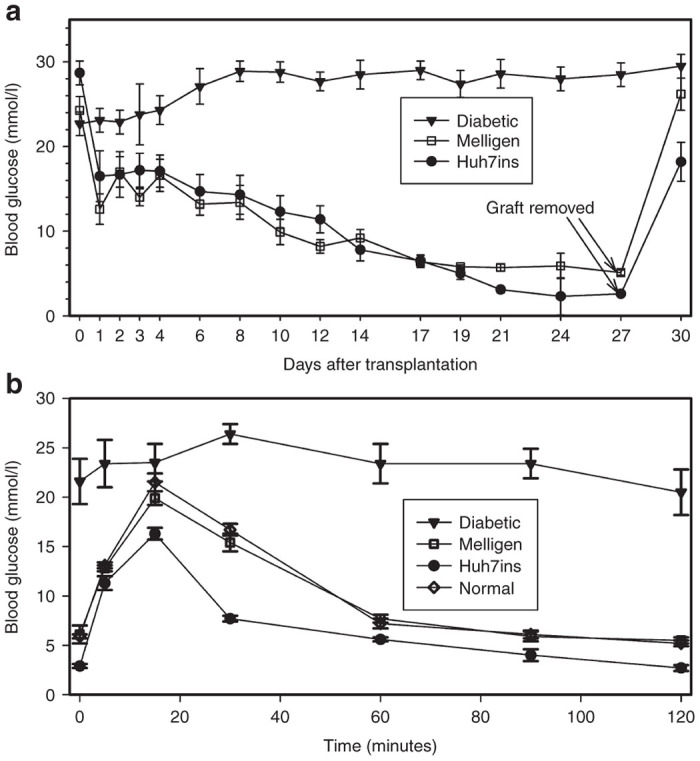
Reversal of diabetes in NOD/scid mice following transplantation of Huh7ins and Melligen cells: (a) Blood glucose levels of NOD/scid mice transplanted with Huh7ins and Melligen cells and diabetic controls. Grafts of Huh7ins and Melligen cells were removed at 27 days. (b) Intraperitoneal glucose tolerance test in mice transplanted with Huh7ins and Melligen cells, normal and diabetic controls. Values are expressed as means ± SE (n = 8).
The fasting blood glucose levels of mice transplanted with Huh7ins cells were significantly lower than untreated normal controls (Figure 2b). During the intraperitoneal glucose tolerance tests, the glucose dose–response curve for mice transplanted with Huh7ins cells peaked at significantly lower blood glucose levels (16.3 ± 0.6 mmol/l, P < 0.01) when compared to normal untreated animals (21.5 ± 0.9 mmol/l), and by 120 minutes blood glucose levels had returned to hypoglycemic levels (2.6 ± 0.3 mmol/l). Importantly, there was no significant difference between the intraperitoneal glucose tolerance test results for animals transplanted with Melligen cells and the untreated normal controls (Figure 2b). A scatter plot of 18 hours fasting blood glucose values for the animals in Figure 2b (Supplementary Figure S1) shows little variation in the groups of transplanted animals.
After grafts of Melligen and Huh7ins cells were excised, it was determined if alternative cells were producing insulin, or other pancreatic hormones. Figure 3a shows extensive staining for insulin (75.3 ± 6.5% of total graft cells) and a small amount of staining for glucagon (1.1 ± 0.1% of total graft cells) and somatostatin (0.1 ± 0.02% of total graft cells) in the Melligen cell graft. Less than 1% of cells showed expression of all three hormones. The remainder of the tissue that did not stain for either hormone, was likely connective tissue. By comparison, insulin was present in the Huh7ins cell graft (70 ± 5.35% of total graft cells), but no staining for somatostatin and glucagon was seen in grafts removed from animals transplanted with Huh7ins cells (results not shown). Not surprisingly, extensive staining of insulin, glucagon, and somatostatin was seen in the pancreas from normal control animals (Figure 3b). As expected, insulin staining was minimal in the pancreas of diabetic animals that were engrafted with Melligen cells (Figure 3c) or in untreated diabetic animals. Electron microscopy revealed the presence of cytoplasmic granules (270–330 nm in diameter) in Melligen cell grafts (Figure 3d), which were of a similar size and appearance to those in pancreatic β-cells.25 Immunoelectron microscopy of insulin-producing cells in the grafts indicated that insulin was stored in the granules (Figure 3e).
Figure 3.
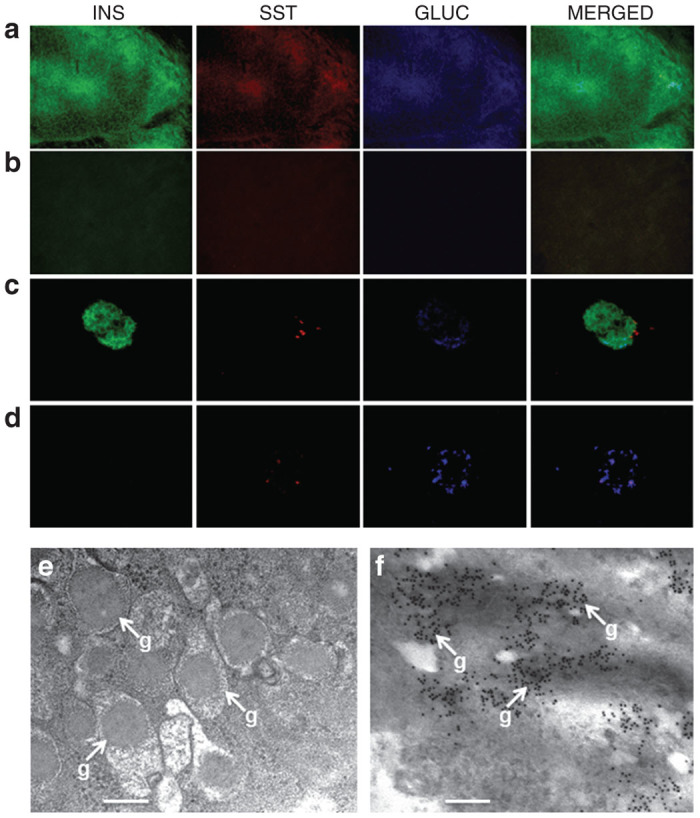
Expression of pancreatic hormones following reversal of diabetes in NOD/scid mice following transplantation of Melligen cells. (a–c) Photomicrographs of triple anti-insulin (INS), anti-somatostatin (SST), and anti-glucagon (GLUC) staining of (a) Melligen graft, (b) normal mouse pancreas, and (c) diabetic mouse pancreas; original magnification, 400×. (d) Transmission electron micrograph showing secretory vesicles with dense granules (g) (bar = 250 nm) in Melligen grafts, 30 days after transplant. (e) Immunoelectron micrograph showing localization of insulin in Melligen grafts, 30 days after transplant (bar = 300 nm).
Glucokinase and hexokinase enzyme activities
To examine the ratio of glucokinase: hexokinase expression in Melligen cells, glucokinase protein and enzymatic activity in Huh7ins and Melligen cells were measured. Qualitative western analysis showed the presence of human glucokinase in Melligen, Huh7ins, and MIN6 cells, but there was an upregulation in expression levels of glucokinase protein in the Melligen cells (Figure 4). The qualitative changes in protein expression, as determined from western analyses, may be correlated with estimates of glucokinase enzymatic activity determined in extracts from the cell lines. Huh7, Huh7ins, and Huh7ins-empty vector cells contained 16 ± 1.2, 24.2 ± 2.3, and 25.4 ± 2.6 U/g protein of glucose phosphorylating activity, respectively when assayed at 20 mmol/l glucose (Figure 5a) in the absence of glucose-6-phosphate. This activity was reduced to 3.0 ± 0.5, 4.8 ± 0.4, and 4.7 ± 0.5 U/g protein, respectively, when the assay was conducted in the presence of 10 mmol/l glucose-6-phosphate (Figure 5b). This finding was consistent with the premise that most (~80%) of the enzymatic activity was contributed by low Km glucose-6-phosphate sensitive hexokinases in the Huh7 cell lines. By comparison, Melligen cells had a significantly higher (P < 0.01) level of glucose-phosphorylating capacity, as compared to the other cell lines, of 34.6 ± 3.4 U/g protein when measured in the absence of glucose-6-phosphate, and this correlated with the increased protein concentration seen in the western analysis. More importantly, in the presence of glucose-6-phosphate, Melligen cells exhibited a threefold enhancement in activity: 20.6 ± 2.7 U/g protein, over the Huh7ins cells (4.8 ± 0.1 U/g protein). Therefore, the Melligen cells had hexokinase activity that represented 42% of the total glucose phosphorylating capacity, as compared to 80% in the other cells, and, therefore, had significantly more of the activity contributed by glucose-6-phosphate-insensitive glucokinases.
Figure 4.
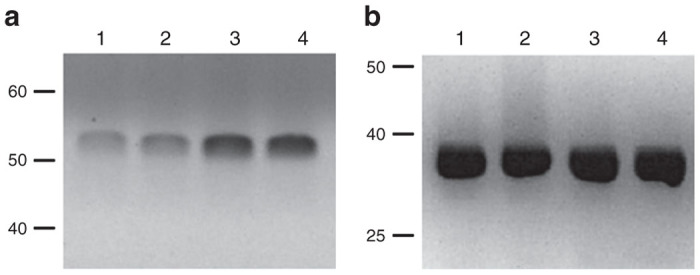
Protein expression of glucokinase in liver cell lines. Western blot analysis for (a) human glucokinase in Huh7ins cells (lane 1), Huh7 ins cells with pIRlSpuro3 vector only (lane 2), Melligen cells (lane 3), and human islets (lane 4) and (b) GADPH. Human glucokinase generated a band at 52 kDa and GADPH a band of 36 kDa. GADPH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 5.
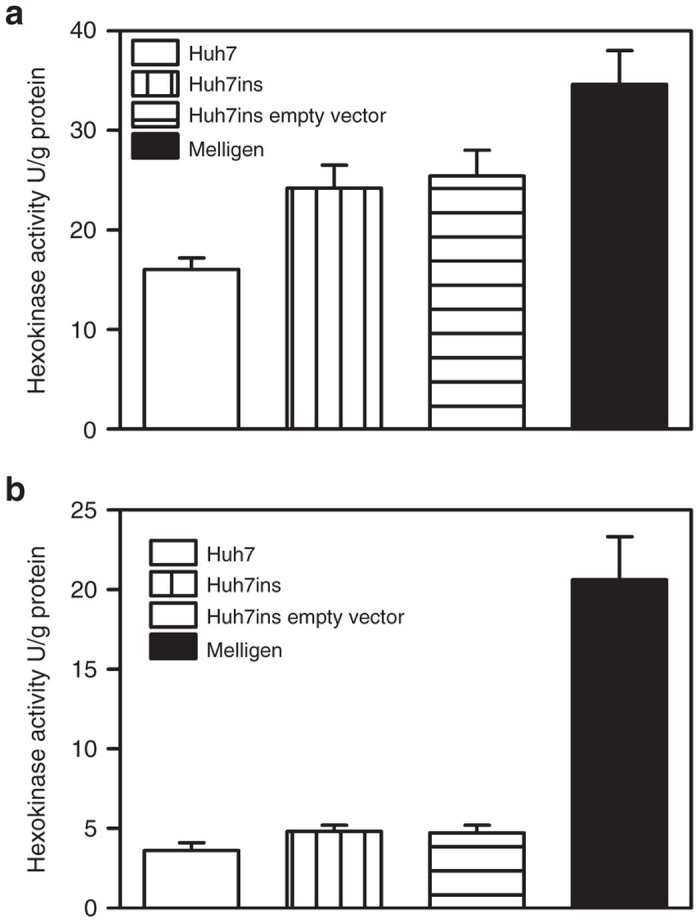
Enzyme activity of glucokinase in liver cell lines. Glucokinase activity of Huh7, Huh7ins, Huh7ins (with pIRlSpuro3 vector only), and Melligen cells in the presence of (a) 20 mmol/l glucose and (b) 20 mmol/l glucose plus 10 mmol/l glucose-6-phosphate. Mean ± SEM, n = 6.
Resistance of Huh7ins and Melligen cells to cytokines
Due to the important role of proinflammatory cytokines in the destruction of pancreatic β-cells and transplanted islets, cell viability was assessed to establish the cytotoxic effect of various cytokines on Huh7ins, Melligen cells, and the parent cell line, Huh7. After establishing the optimized cytokine concentrations that reduced the viability of MIN6 β-cells, these experimental parameters were applied to the three liver cell lines.
Significant differences were seen in the susceptibility of MIN6 cells and the human liver cell lines to cytokine-induced toxicity from day 3 (P < 0.05). By day 3, cytokine-treated MIN6 cells had viabilities of 72 ± 5%, while untreated MIN6 cells grew at an exponential rate (100 ± 1%) over the 10 days of the experiment. Treatment of MIN6 cells with the triple cytokine culture medium (TCCM) for 10 days caused a significant further decrease in cell viability to 15 ± 4.5% when compared to untreated cells 100 ± 1% (P < 0.05). When exposed to the same cytokine combination for 10 days, Huh7 cells showed no significant decrease in cell viability, as compared to untreated cells. Similarly, in experiments using Huh7ins and Melligen cells, there was no significant decrease in cell viability after exposure to the triple cytokines over 10 days (results not shown).
Insulin secretion and storage
Cytokine-treated MIN6 cells secreted significantly less insulin (7 ± 3 pmol/well) than untreated MIN6 cells (39 ± 2 pmol/well) from day 1 of the incubation (P < 0.05) and throughout the entire period studied. By day 10, cytokine-treated MIN6 cells had secreted (13.6 ± 2.7 pmol/well); by comparison, untreated cells had secreted 53.6 ± 8.0 pmol/well. In contrast, Huh7ins and Melligen cells coincubated with cytokines secreted amounts of insulin that were not significantly different to their untreated controls. For example, Melligen cells treated with TCCM secreted 20.8 ± 0.6 pmol/well at day 10, as compared with 21.9 ± 1.0 pmol/well secreted by untreated cells.
As the cells proliferated, insulin storage steadily increased in untreated MIN6 cells over the 10 days of the experiment. After exposure of MIN6 cells to the cytokine cocktail, insulin content was significantly diminished after day 1, as compared to the control MIN6 cells (P < 0.05). The difference between the amounts of stored insulin in the treated and untreated MIN6 cells continued to be significant throughout the remaining days of the experiment with untreated and treated cells reaching 81.9 ± 0.5 pmol/106 cells and 21.2 ± 1.7 pmol/106 cells by day 10, respectively. In contrast, Huh7ins and Melligen cells treated with the cytokine combination did not show a significant difference in insulin storage, as compared to the untreated Huh7ins and Melligen cells. Untreated Melligen cells, for example, stored 8.0 ± 0.2 pmol/106 cells, as compared with 8.9 ± 0.3 pmol/104 cells for TCCM-treated cells at day 10.
After 10 days of cytokine treatment, MIN6 cells showed a significant decrease in insulin response to a glucose stimulus when compared to the control MIN6 cells (P < 0.05). Insulin secretion of untreated MIN6 cells in response to 20 mmol/l glucose increased more than fivefold over 1 hour when compared to basal levels (4 ± 2 pmol/well/hours) (P < 0.05) (Figure 6a). Treated MIN6 cells did not release significantly higher levels of insulin to the glucose stimulus above basal levels (P > 0.05) (Figure 6a). This difference may be attributable to reduced viability after 10 days of coincubation with cytokines, because control cells continued in the log growth phase, as indicated by the trypan blue exclusion assays (results not shown).
Figure 6.

The effect of cytokine treatment on glucose responsiveness in MIN6, Huh7ins, and Melligen cells. Glucose responsiveness was determined using 20 mmol/l glucose stimulus with and without cytokine treatment for (a) MIN6 cells, (b) Huh7ins cells, and (c) Melligen cells after 10 days incubation with triple cytokine culture medium (TCCM). The 10-day cytokine treatment did not affect (d) Huh7ins and Melligen cell responsiveness to glucose in the millimolar range with and without the TCCM treatment. Results are expressed as mean ± SE, n = 3 independent experiments.
The effect of the proinflammatory cytokines treatment on glucose-responsiveness was also determined for the Huh7ins and Melligen cells. Untreated Huh7ins and Melligen cells showed a five- and sevenfold increase in insulin secretion respectively, when stimulated with 20mmol/l glucose, with return to basal levels of insulin secretion upon removal of the glucose stimulus (Figure 6b,c). Huh7ins and Melligen cells incubated with cytokines for 10 days showed the same increase in insulin secretion upon the 20 mmol/l glucose stimulus as the untreated cells with a return to basal levels of secretion within 1 hour after stimulation (Figure 6b,c). There was no significant difference in the amount of insulin secreted by the TCCM treated Huh7ins and Melligen cells during the glucose stimulus (P > 0.05). This data indicated that Huh7ins and Melligen cells retained their ability to respond to a glucose stimulus during and after 10 days of cytokine treatment. Basal insulin secretion samples collected during the course of the experiment also showed no significant increase in amounts of insulin secreted compared to controls (P > 0.05, results not shown).
To determine if TCCM treatment affected the glucose response curve between the critical levels of 0–4.5 mmol/l glucose, Huh7ins and Melligen cells were cultured for 10 days with and without cytokines. At day 10, the cells were stimulated with increasing concentrations of glucose in basal medium. Huh7ins cells commenced secretion of increased amounts of insulin to glucose at the subfasting level of 2.5 mmol/l glucose, while Melligen cells secreted increased amounts of insulin in response to 4.5 mmol/l glucose (Figure 6d). In both cell lines, there was no significant difference observed between cytokine-treated and untreated cells at any glucose concentration (P > 0.05).
Nitric oxide production
The modified Griess reaction was used to determine nitric oxide levels, and hence iNOS activity. The iNOS enzymatic activity was estimated by measurements of nitrite accumulation in tissue culture medium, during a 48h exposure to TCCM. MIN6 cells treated with TCCM showed significantly (P < 0.05) increased nitrite production at both the 24 (10.3 ± 0.5 µmol/l nitrite) and 48 hours (17.9 ± 3.4 µmol/l nitrite) time points compared to untreated cells with 8.6 ± 0.1 µmol/l nitrite at 24 hours and 9.7 ± 0.2 µmol/l nitrite at 48 hours (n = 5). By comparison, the human liver cell lines showed no significant difference (P > 0.05) in levels of nitrite between treated and untreated cells at any time point. For example, the Melligen cells produced 9.0 ± 0.2 µmol/l nitrite product following 48 hours treatment with TCCM, compared with 8.6 ± 0.1 µmol/l nitrite (n = 5) in the untreated Melligen cells over the same time period.
RT-PCR and qRT-PCR analysis
Pancreatic hormones and β-cell transcription factors.
As expected, analysis of Melligen cells by RT-PCR showed the presence of human islet glucokinase, which was not detected in the parent cell lines (Figure 7a). By comparison, expression levels of other transcription factors and hormones that were examined, with the exceptions of glucokinase and the pancreatic hormone somatostatin (which was only detected in Melligen cells), did not change. Interestingly, glucagon was detected in all liver cell lines and pancreatic polypeptide was only detected in human islet tissue. Examination of several key factors by qRT-PCR indicated that there was a 2.9 ± 0.9-fold (n = 8) increase in Pdx-1 expression, a 2.4 ± 0.4-fold (n = 8) increase in GLUT 2 expression, a 10.4 ± 1.7-fold (n = 8) increase in islet glucokinase expression and a 7.8 ± 0.8-fold (n = 8) increase in somatostatin expression in Melligen cells, as compared to Huh7ins. In contrast, there were no differences in expression levels of insulin, glucagon, Neurod 1 or Neurogenin 3 between Huh7ins and Melligen cells. Expression of islet glucokinase and insulin in the Melligen cells was maintained following transplantation (Figure 7b).
Figure 7.
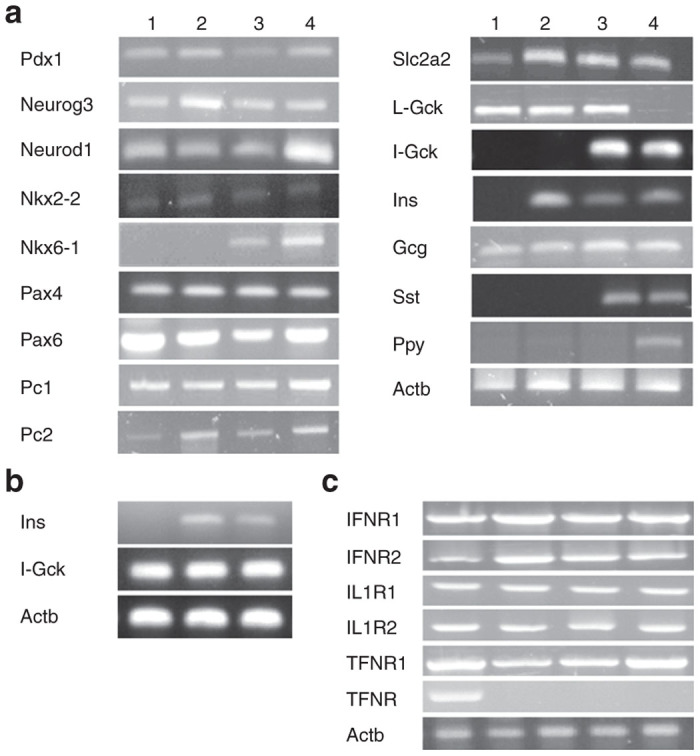
β-cell transcription factors and pancreatic hormones expressed in liver cell lines. (a) RT-PCR detection of mouse β-cell transcription factors (Pdx1, Neurod1, Neurog3, Nkx2-2, Nkx6-1, Pax 4, Pax 6, MAFA, MAFB), the pancreatic endocrine hormones: insulin (Ins), somatostatin (Sst), glucagon (Gcg) and pancreatic polypeptide (Ppy); GLUT 2 (Slc2a2) and liver glucokinase (L-Gck); islet glucokinase (I-Gck), insulin proconvertases 1 and 2 (Pc1 and Pc2); exocrine marker p48 and Actb in Huh7 cells (lane 1), Huh7ins cells (lane 2), Melligen cells (lane 3) and human islets (lane 4). (b) RT-PCR analysis of human insulin and human islet glucokinase in transplanted Huh7ins cells (lane 1), transplanted Melligen cells (lane 2) and human islets (lane 3); (c) RT-PCR analysis of cytokine receptor expression, IFNR1, IFNR2, IL1R1, IL1R2, TNFR1 and TNFR2, in human islets (lane 1), Huh7 cells (lane 2), Huh7ins cells (lane 3), and Melligen cells (lane 4). RT-PCR, real-time polymerase chain reaction.
Cytokine receptor expression.
To establish if the observed resistance of the Melligen cells to the proinflammatory cytokine cocktail was due to the absence of cytokine receptors, RT-PCR was performed using cDNA generated from RNA isolated from human islets (positive control), Huh7, Huh7ins, and Melligen cells. Expression of all the tested cytokine receptors was detected, with the exception of TNFR2 (which was not expressed in the liver cell lines), and confirmed in all cells (Figure 7c).
Genes associated with the NF-ĸB pathway.
NF-ĸB exerts different effects on cell survival and death depending upon the phenotype of the cell. For example, in β-cells, increased expression levels of NF-ĸB promote decreased viability while in hepatocytes NF-ĸB acts as a survival factor. After treating the cells for 4 hours with TCCM, RT-PCR was performed, using human islet cells as a positive control. Expression levels of the inhibitors of NF-ĸB, IκBα, IκBβ, and IκBϵ were detected in all cell lines and islets. qRT-PCR analyses indicated that all inhibitors were downregulated in Melligen cells: IκBϵ was downregulated by 0.55 ± 0.01-fold (P < 0.05) and IκBα and IκBβ were also significantly downregulated by 0.57 ± 0.01-fold and 0.66 ± 0.10-fold, respectively (P < 0.05) (n = 3).
Transcriptomics
Exposure of Melligen cells to proinflammatory cytokines modulated gene expression levels at 1 and 24 hours after exposure. Using the Partek Genomic Suite, PCA mapping of the Gene Array data showed that the greater differences in gene expression levels occurred 24 hours after exposure to cytotoxic cytokines.
After 1 hour exposure of Melligen cells to the cytokines, 17 of the 18 genes found to be differentially expressed, were significantly upregulated by twofold or more. The upregulated genes included the chemokine (C-X-C motif) ligand family, CXCL1, CXCL2, CXCL3, and CXCL10 (Supplementary Table S2). The expression of CXCL10 was found to be upregulated by the greatest degree with a 19-fold upregulation. Interferon regulatory factor 1 (IRF1), induced by IFN-γ, was also significantly upregulated. Genes typically induced by IL-1β included TIFA and PTX3. Putative NF-κB target genes upregulated in the Melligen cells at 1 hour cytokine treatment included baculoviral inhibitor of apoptosis protein repeat-containing 3 (BIRC3) and TNFα-induced protein 3 (TNFAIP3) with the highest significance level. The only gene significantly (P < 0.05) downregulated at 1 hour cytokine treatment, compared to 1 hour untreated cells, was NFKBIA (also known as IκBα). While these candidates are of putative biological significance, given their influence on cell viability, their potential role in Melligen cell survival is yet to be elucidated.
RNA transcript levels of different genes were also determined after 24 hours of cytokine treatment of the Melligen cells, with untreated cells used as a control. Exposure of the Melligen cells to the same combination of cytokines for 24 hours significantly modified the expression of 148 genes. The 148 genes identified are presented in Supplementary Table S3. The large majority of the modified genes (140 of 148) were upregulated (P < 0.05) upon cytokine treatment and mainly represented genes known to be inducible by IFN-γ, TNF-α, and IL-1β.
To further validate the results of the microarray analysis, five genes were selected for validation of the observed gene expression changes by qRT-PCR. These were genes not previously found to be differentially expressed in the cytokine-treated parental cell line Huh7, as determined in the literature. These genes were also involved in the highest scoring networks established by ingenuity pathway analysis (results not included).
qRT-PCR analysis was performed on five of the genes detected by the gene array to be upregulated by more than twofold and with a significance level of P < 0.05. The five genes, BIRC3, CASP1, IRF1, SOD2, and STAT1, were selected from the cascades identified above and on the basis of their potential involvement in the resistance of these cells to cytokine toxicity. The genes selected were also genes that had been shown to play pivotal roles in the effects of, and signaling pathways regulated by, IFN-γ, TNF-α, and IL-1β singly and in combination in the β-cell death occurring in T1D (Program, IPA). IRF1 and BIRC3 were also upregulated at both 1 and 24 hours.
The results obtained by qRT-PCR confirmed the upregulation of BIRC3, CASP1, IRF1, SOD2, and STAT1 by the 24 hours cytokine treatment of Melligen cells. Following normalization with β-actin mRNA levels, there was a significant (P < 0.05) upregulation of BIRC3 by 11.5 ± 2.4-fold (n = 8) and this was the highest recorded fold change in gene expression of the five genes selected. It was found that CASP1, IRF1, SOD2, and STAT1 were upregulated by 2.0 ± 0.4, 5.4 ± 1.3, 4.5 ± 0.4, and 2.4 ± 0.3-fold, respectively. To determine if the absence of the insulin and glucokinase gene expression in the liver cell line regulated the five selected genes to the same level, Huh7 cells were treated with the TCCM for 24 hours. mRNA was extracted and used to quantitate expression of the genes of interest by qRT-PCR. The results showed that BIRC3, CASP1, IRF1, and SOD2 were significantly upregulated (P < 0.05) in the Melligen cells, as compared to the Huh7 cells, but no significant difference was found for STAT1 gene expression between the cell lines.
Discussion
If artificial β-cells are to be a viable alternative for pancreatic β-cell replacement in T1D, they must be endowed with the physiological characteristics of pancreatic β-cells; namely biologically active insulin production, insulin storage and glucose-dependent insulin secretion, and resistance to the cytotoxic effects of proinflammatory cytokines. The Huh7ins cell line possessed some of these characteristics, but they were responsive to low physiological glucose concentrations (2.5 mmol/l). In order to produce a more clinically viable cell, we investigated the effect of stable expression of the human islet glucokinase transgene, which resulted in the production of the Melligen cells.
In pancreatic β-cells the low expression of hexokinase results in low glycolytic flux and low ATP/ADP levels at glucose levels <2.5 mmol/l,26,27 which maintains a low basal rate of insulin secretion and prevents hypoglycemia. At glucose levels >2.5 mmol/l, β-cells phosphorylate glucose via the high-affinity glucokinase, which correlates with the increase in the ATP/ADP ratio of 5–10 mmol/l,27 and allows the β-cell to maintain blood glucose levels as they are very sensitive to blood glucose levels near the normal fasting glucose range. Figure 1 clearly shows that the expression of human islet glucokinase in Huh7ins cells (Melligen cells) resulted in a clear shift in the glucose dose–response curve to the right. The Melligen cells commenced secretion of insulin to glucose at 4.25 mmol/l, rather than 2.5 mmol/l for the Huh7ins cells. This shift in the glucose dose–response curve is undoubtedly related to the noticeable shift in the glucokinase to hexokinase ratio clearly seen in the glucose phosphorylation studies. In the presence of glucose-6-phosphate, Melligen cells exhibited a threefold enhancement in activity (Figure 5b), over the activity in Huh7ins cells and other liver cell lines tested. In Melligen cells, the greater proportion of the total glucose phosphorylating capacity (58%) was provided by glucose-6-phosphate insensitive glucokinases, rather than hexokinase, as seen in the Huh7 and Huh7ins cells. Studies have shown that high glucose concentrations in isolated islets cause increased glucokinase activity and parallel increases in the capacity of glucose-stimulated insulin release.28 While insulin storage in the Melligen cells was not altered, they secreted twice as much insulin to glucose over the course of the dose–response curve.
Animals transplanted with Melligen cells maintained normoglycemia from day 19 onwards and exhibited a normal glucose response curve, which was not significantly different to that observed for nondiabetic animals. By comparison, animals transplanted with Huh7ins cells became hypoglycemic from day 21 onwards and the glucose tolerance curve peaked at significantly lower levels when compared to animals transplanted with Melligen cells or untreated, nondiabetic controls, returning to hypoglycemic levels by 120 minutes. The development of hypoglycemia and abnormal glucose tolerance seen in this study after diabetic animals were engrafted with Huh7ins cells, and in our previous study,4 is most likely attributed to their secretion of insulin in response to glucose at subnormal fasting levels (2.5 mmol/l). However, it is likely that if the Melligen cells were maintained for an extensive period in vivo, they would continue to proliferate and eventually render the animals susceptible to hypoglycemia due to the constitutive insulin secretion from the mass of cells. Any potential clinical application of Melligen cells to cure T1D would require microencapsulation technology to ensure that the cells are contained at the site of administration, due to their tumorigenic nature causing continual cell proliferation, and to protect them from interaction with the cellular arm of the immune system. Given that Melligen cells have been shown to be resistant to the cytotoxic effects of the cytokines responsible for mediating β-cell death in T1D, it suggests that protecting them from cellular contact with autoimmune mediators may be sufficient to ensure their continued viability post-transplant, however further studies are required to assess this. Of critical importance, it was noted that the human insulin and human islet glucokinase transgenes, expressed under the control of the cytomegalovirus promoter in both constructs, were stably expressed for the duration of the in vivo experiment using the NOD/scid mouse model. Thus, Melligen cells were able to retain their ability to function as artificial β-cells akin to pancreatic β-cells largely via the continued expression of glucokinase.
Melligen cells maintained a β-cell-like phenotype, as is seen in the Huh7ins cells,4,29 by developing secretory granules of similar appearance and size to those of pancreatic β-cells together with a regulated insulin secretory mechanism. This has undoubtedly arisen from the expression of insulin against a background of endogenous expression of pancreatic cell transcription factors and β-cell proteins that are present in the dedifferentiated parental Huh7 cell line. Numerous studies have shown pancreatic transdifferentiation of liver cells upon expression of β-cell transcription factors and we have shown spontaneous expression of β-cell transcription factors, pancreatic transdifferentiation, and reversal of diabetes in several animal models following the lentiviral delivery of furin cleavable insulin.9,11,30 We have previously described the presence of GLUT2, liver glucokinase, PC1 and PC2, and the pancreatic β-cell transcription factors, Pdx1, NeuroD1, Neurogenin 3, Pax4, Pax6, Nkx6.1, in Huh7ins cells.4,29 As expected, human islet glucokinase was expressed in the Melligen cells, at both the RNA and protein levels, but was not expressed by the parental cell lines. There was little difference in the pattern of expression of β-cell transcription factors by Melligen cells and Huh7ins cells, except for significant upregulation of expression levels of Pdx1 and GLUT2 and the expression of somatostatin and Pax6 in Melligen cells. The increased expression of GLUT 2 is likely related to the overexpression of the islet glucokinase and the shift in the balance of the glucokinase to hexokinase ratio in the Melligen cell line. Triple immunofluorescent staining also showed the presence of the three pancreatic hormones, insulin, glucagon, and somatostatin, in Melligen cells, while only insulin protein was detected in Huh7ins cells after transplantation. These data suggest that Melligen cells possess a more pancreatic-prone phenotype, as compared to Huh7ins cells. The reason for this observation is unknown, possibly the cells have undergone further transdifferentiation during the transfection process and we are currently investigating possibilities in our laboratory. Extensive pancreatic transdifferentiation has been reported on lentiviral transduction of liver cells9,11,30 and when liver cells are exposed to toxins31 and high levels of glucose.32 By comparison to human insulin (75% of the cells in the graft), the levels of human glucagon and somatostatin were extremely low (1 and 0.1% respectively) and it is doubtful that they had any significant biological effect by comparison to the normal production of these hormones from the mouse pancreas.
The proinflammatory cytokines used in this study have established roles in the induction of β-cell death and metabolic dysfunction in primary human islet cells, both in vivo and in vitro.33,34 The results from the MTT viability assay and the insulin storage and secretion data over an extended time period indicated that the cocktail of proinflammatory cytokines exerted no significant effect on the viability, insulin secretion and storage, and the glucose-responsive insulin secretion of the Huh7ins and Melligen cell lines. By comparison, the control pancreatic cell line, MIN6, was significantly affected, and following 10 days exposure was no longer capable of secreting insulin in response to glucose. These results corroborate an earlier study of the effect of cytokines on another insulin-secreting cell line, HEPG2ins/g.3 The resistance of the Huh7ins and Melligen cells to the detrimental effects of proinflammatory cytokines was not likely attributable to decreased expression of their cognate receptors as the cytokine receptors were shown to be expressed, at least at the RNA level.
Cytokines, such as IFN-γ, TNF-α, and IL-1β, stimulate iNOS expression and nitric oxide production leading to β-cell damage and death. In contrast to MIN6 cells, Huh7, Huh7ins, and Melligen cells did not produce increased levels of NO at either 24 or 48 hours. Similarly, Chang et al.35 identified downregulated iNOS gene expression in cytokine-treated rat hepatocytes. Therefore, the time-points for microarray analysis in this study were chosen from the previous studies,35–37 which indicated that early and relevant changes in the gene expression profile of insulin-secreting cells were particularly evident at both 1 and 24 hours after cytokine exposure.
Cytokine treatment for 6 and 24 hours, as shown by Cardozo et al.38, confirmed the up- and downregulation of several functional genes in primary rat pancreatic β-cells, including those involved in metabolic function, such as GLUT2 and glucokinase. Similarly, the microarray study by Sarkar et al.36, performed on human pancreatic islets, showed metabolic perturbations after treating the islets with IFN-γ, TNF-α, and IL-1β. The changes induced by the cytokines include changes in expression of genes involved in glucose transport and responsiveness, GLUT2, and glucokinase. However, in addition to the upregulation of proapoptotic genes, as seen in a study by Cardozo et al.39, there have also been reports of an upregulation of antiapoptotic genes, including BIRC3 and TNFAIP3, in other studies.36,37 In the present study, metabolic mechanisms involving GLUT2 and glucokinase molecular expression were not altered in Melligen cells at 1 or 24 hours, suggesting that these cells may be protected against the defects in glucose metabolism that cytokines otherwise cause in primary rat and human β-cells at these specific time points. Overall, the microarray analyses revealed that 148 genes were differentially expressed and were putatively regulated by the cytokine cocktail of IFN-γ, TNF-α, and IL-1β after 24 hours of treatment with only 18 genes differentially expressed after 1 hour treatment. Some of these genes are of immunological significance, and, accordingly, their upregulation may have contributed to the ability of Melligen cells to resist the cytotoxic effects of proinflammatory cytokines; however, this will require further investigation.
To further elucidate the mechanism underlying the resistance of the Melligen cells to the cytokine treatment in the current study, the effect of cytokine attack in liver cells both in vivo and in vitro at a gene level was determined from the literature. In acute liver failure or acute viral hepatitis, hepatocytes are exposed to elevated levels of several proinflammatory cytokines. These cytokines, notably TNF-α, can activate both cell survival and apoptotic pathways in primary rat hepatocytes.39 In primary rat hepatocytes treated with IFN-γ, TNF-α and IL-1β, it has been demonstrated that the expression of members of the IAP family are regulated by NF-κB. This family includes BIRC3, cIAP1, X chromosome-linked IAP (XIAP), Survivin11, and Livin12 (ref. 40). As determined by a microarray on HepG2 cells, with TNF-α treatment for 6 hours, BIRC3 was induced by the cytokine associated with NF-κB and the overexpression of BIRC3 was shown to inhibit apoptosis in this liver cell line.41 Together, these studies identify pathways, at a gene level, that have been shown to protect liver cells against cytokine induced cell death. Supporting the prevalent hepatocyte phenotype maintained by the Melligen cells, the results from this study showed an increased expression of BIRC3 after both 1 and 24 hours cytokine treatment, which may have contributed toward protection against cytokine-induced cell death.
Under proinflammatory conditions, the insulin-secreting Melligen cells function efficiently and resist cytokine-induced cell death. Depending on cell type, the effects of NF-κB upregulation have been shown to be both proapoptotic or antiapoptotic. The dichotomy of NF-κB may be the mechanism by which the Melligen cells are protected compared to the β-cells that are otherwise susceptible to the proinflammatory cytokines. The microarray data showed that indeed there are genes differentially expressed after cytokine treatment and that some of these genes are also differentially expressed in the Melligen predecessor, Huh7. Furthermore, the gene array established that glucose metabolism in the Melligen cells was not affected by the cytokine treatment at either 1 or 24 hours treatment indicating desirable cell function notwithstanding the presence of an autoimmune attack. Although the Melligen cells have become more β-cell like, they respond to cytokine treatment more as liver cells and not as β-cells since they have the ability to inhibit apoptotic function based on their gene expression profile. Comparing the five candidate genes by real-time qRT-PCR, results revealed that the presence of the β-cell autoantigen (insulin) and the presence of glucokinase do affect the Melligen cells at a molecular level in response to proinflammatory cytokines.
The ability of the Melligen cells to maintain viability in a clinical environment will need to be confirmed by further in vivo studies, utilizing an appropriate encapsulation system that will protect the cells from destruction by the immune system and protect the recipient from the tumorigenic nature of the cell. Strategies such as the future engineering of a suicide gene, which would, on activation, destroy transplanted cells in a clinical setting, are also a consideration. The utilization of Melligen cells or other nonpancreatic artificial β-cells as a cure for T1D will have an advantage over current insulin therapy regimes, in that the new technology will avoid the autoimmune process and mimic the natural physiological secretion of insulin from the pancreas, delivering the insulin first to the liver and then to the peripheral tissue in response to a glucose load, whereas the insulin delivered by injections or pumps reaches the peripheral tissue first and not the portal circulation. This should both reduce complications by reducing glucose excursions and narrowing the range of glucose concentrations tissues such as the eyes and kidneys are exposed to. This technology would also have the advantage over studies that deliver β-cell transcription factors to the liver, avoiding the need for the use of viral vectors and issues such as exocrine differentiation of liver tissue resulting in hepatitis-like syndromes7 and abnormal glucose tolerance seen in a number of studies.42 Thus, Melligen cells represent a truly regulated insulin-secreting liver cell line that responds to glucose in the physiological range and provides important information regarding the minimal molecular machinery required to generate an artificial β-cell.
Materials and Methods
Plasmid construction and transfection
Human islet glucokinase cDNA contained in the pBluescript commercial vector was a gift from Dr M. Alan Permutt, the University of Washington (St Louis). The human islet glucokinase cDNA was isolated from pBluescript SK+ by digestion with EcoR1. The 2733 bp fragment containing the human islet glucokinase cDNA was inserted into the pIRESpuro3 expression vector (Clontech, Clayton, Australia) at the EcoR1 site (971–972 bp).
Huh7ins cells were transfected with the pIRESpuro3-glucokinase construct, or the pIRESpuro3 vector alone (Huh7ins-empty vector), using Effectene transfection reagent, following the manufacturer’s instructions (Qiagen, Doncaster, Australia). The eukaryocidal antibiotic puromycin (1.1 μg/ml) was added to the cultures 24 hours after transfection. Culture medium plus drugs were changed every 2–3 days and after 14 days of selection, colonies were isolated and expanded. The cells containing the pIRESpuro3-glucokinase construct will hereafter be referred to as Melligen cells.
Cell culture
Huh7ins cells were cultured as monolayers in Dulbecco’s modified Eagle’s medium (Life Technologies, Mulgrave, Australia) supplemented with 10% (v/v) fetal calf serum (Life Technologies), and 0.55 mg/ml G418 (Life Technologies), in 5% CO2, as previously described.4 Additionally, 1.1 μg/ml puromycin (Life Technologies) was added to the Melligen cell culture medium. MIN6 cells19 were cultured as monolayers in Dulbecco’s modified Eagle’s medium containing 20% (v/v) fetal calf serum in 5% CO2 in air. Donor pancreas tissue was collected, with permission (Human Ethics Committee, Westmead Hospital, Sydney), and human islets were extracted from the whole donor pancreas by a modified Edmonton method, using an aphaeresis unit and were separated on density gradients.43 Islets were frozen in liquid nitrogen until use.
Acute stimulation of insulin secretion
Before stimulation, 5 × 106 cells (Huh7, Huh7ins, Huh7ins-empty vector, and Melligen cells) were plated into six-well plates and grown overnight. Tissue culture plates were thoroughly washed with basal medium (phosphate-buffered saline (PBS) containing 1 mmol/l CaCl2 supplemented with 20 mmol/l 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 mg/ml bovine serum albumin, and 1.25 mmol/l glucose) to remove all traces of culture medium and fetal calf serum. To stabilize the basal secretion of insulin, monolayers were incubated in the basal medium (pH 7.4) for two consecutive periods of 1 hour. Dose–response curves were constructed after monolayers were exposed to increasing glucose concentrations (1.25–20 mmol/l) for 1 hour. Glucose was dissolved in basal medium, and basal medium alone was used as a control.
Insulin secretion and storage
Huh7ins-empty vector cells and Melligen cells were washed with PBS, and harvested by trypsinization. Insulin was extracted overnight in acid/ethanol, as previously described.4 Levels of human insulin were measured using an Eliza kit (EZHI-14K, Merck Millipore, Bayswater, Australia) which has no cross-reactivity with human proinsulin and mouse insulin secreted by MIN6 cells was measured with the EZRMI-12K kit (Merck Millipore).
Treatment of cells with cytokines
The recombinant human (h) cytokines, TNFα, IL-1β, and hIFNγ, and murine (m) IFNγ (as IFNγ is species-specific) were purchased from Biosource Int. (Camarillo, CA) TNFα and IL-1β were dissolved in PBS containing 0.1% (w/v) bovine serum albumin to obtain stock solutions of 1.1 × 106 and 5 × 105 U/ml respectively. IFNγ was dissolved in 10 mmol/l acetic acid containing 0.1% bovine serum albumin to obtain a stock solution of 1 × 105 U/ml. The murine glucose-responsive MIN6 cell line19 was used as the control pancreatic β-cell line to confirm the efficacy of the cytokines. In order to establish the optimal cytokine concentration, initial experiments used serial dilutions of single cytokine concentrations of mIFN-γ, TNF-α, and IL-1β on MIN6 cells. The effective doses required to destroy half the MIN6 cell population (ED50) indicated on the respective manufacturer’s data sheet for each cytokine constituted the most dilute concentration used in generating each cell death curve. After establishing the concentrations to be used, the appropriate TCCM was made fresh by diluting the stock solutions of TNF-α, IL1β, and IFNγ with culture medium to obtain final concentrations of 1,000 U/ml, 374 U/ml, and 1,000 U/ml, respectively.
Due to the different proliferation rates of the MIN6 cells and the liver cell lines, the seeding density for the various cell lines was adjusted accordingly to ensure exponential growth of the cells during the timeframe of the experiment. In order to examine cell viability, the MTT assay44 was used in a 96-well format. Briefly, Huh7, Huh7ins, and Melligen cells were plated at 1 × 103 cells per well (as they proliferated at the same rate, Supplementary Figure S2) and MIN6 cells were plated at 2 × 103 cells per well as MIN6 cells proliferated at a slower rate than the liver cell lines. The cells were allowed to adhere to the bottom of the wells overnight (37 °C/5% CO2). TCCM was added daily and cells were assayed for viability on days 1, 2, 3, 6, 8, and 10. To examine insulin secretion and storage in these cell lines, the cells were plated in 12 well plates at a seeding density of 5×103 cells per well for Huh7ins and Melligen cells and 2×104 cells per well for MIN6 cells. To examine glucose responsiveness Huh7ins and Melligen cells were plated in 6 well plates at 1×104 per well and MIN6 cells in 12 well plates at 2×104 cells per well. The TCCM was added thereafter daily to the treated wells (in triplicate) and fresh media added to the control wells (also in triplicate). For each cell line, insulin secretion was measured on days 1, 2, 3, 5, 8, and 10. Chronic insulin secretion was determined by collecting media daily from each plate and pooling with media samples collected on preceding days. The volume of media removed from each well daily was recorded and the ELIZA was used to measure chronic insulin secretion. Cells were harvested to examine insulin storage at day 10. Plates of cells treated identically with TCCM were used to determine glucose responsiveness at day 10.
Nitric oxide levels in MIN6 cells and liver cell lines treated with TCCM for 24 and 48 hours were examined by the modified Griess reaction, as previously described.45
RT-PCR analyses
For RT-PCR analyses, Huh7, Huh7ins, Huh7ins-empty vector, and Melligen cells were snap-frozen in liquid nitrogen. Human islets were used as the positive control. Total RNA was extracted using Trizol (Invitrogen, Mulgrave, Australia) and samples were treated with DNase I (Invitrogen). Expression levels of insulin, other pancreatic hormones (glucagon, somatostatin, pancreatic polypeptide), islet and liver glucokinase, and several β-cell transcription factors (Pdx-1, Neurod1, Neurog3, Pax4, Pax6, Nkx2.2, Nkx6.1) were determined. The expression levels of insulin and islet glucokinase were also examined in the transplanted cells. Expression levels of cytokine receptors (IFNR1, IFNR2, IL1R1, IL1R2, TNFR1 TNFR2) and those genes associated with the NF-ĸβ (NF-ĸB inhibitors: IκBα, IκBβ, and IκBϵ), Fas and iNOS signaling pathways were examined in the cell samples ± TCCM treatment for changes in gene expression. Human islets were used throughout as the positive control. Sequences of the primers used are shown in Supplementary Table S1.
Aliquots of cDNA were used as the template for real-time qRT-PCR using the SYBR GreenER qPCR Supermix (Invitrogen) and a real-time detection system (Eppendorf Realplex2, Eppendorf, Hamburg, Germany). Real-time qRT-PCR reactions contained either primers specific to the gene of interest or the reference gene, β2-microglobulin (Supplementary Table S1). Amplification efficiencies for each primer set were determined to be similar. Data were analyzed using the 2−∆∆t method where ∆∆CT = (Ct, Target − Ct, Reference)control tissue − (Ct, Target − Ct, Reference)sample tissue. The fold change in the target gene expression, normalized to β-actin and relative to the expression in the control tissue, was calculated for each sample. The mean and standard error of fold changes in expression for each gene were then determined.
Transcriptomics analysis
Melligen cells were cultured in media alone or treated with TCCM for either 1 or 24 hours and RNA was isolated using the RNeasy Kit (Qiagen, Doncaster, Australia), according to the manufacturer’s instructions. Preparation and labeling of cDNA, hybridization, and scanning of the Human Genome ST 1.0 arrays were performed according to the manufacturer’s protocol (Affymetrix, Mulgrave, Australia). The microarray procedure (labeling reactions, hybridization procedures, reading the signals, and normalization) was performed at the Clive and Vera Ramaciotti Centre for Gene Function Analysis (University of New South Wales, Australia).
Data obtained from microarray analyses were imported into GeneSpring 6.1 software (GeneSpring 6.1, Silicon Genetics, Redwood City, CA). The fold changes were filtered in the dataset using P value < 0.01 and a signal-to-noise ratio >2 in preparation for use in ANOVA statistical analysis. Data analysis was performed using Partek Genomics Suite 6.2 software (Partek, St. Louis, MO).
Additional filtering (≥ 2-fold change) was applied to identify genes of interest, which were further analyzed using Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, CA). Those genes with known gene symbols and their corresponding expression values were uploaded into the software. Each gene symbol was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. Networks of these genes were algorithmically generated based on their connectivity and assigned a score. The score takes into account the number of focus genes in the network and the size of the network to approximate how relevant this network is to the original list of focus genes.
Canonical pathways analysis identified the pathways, from the IPA library of pathways, which were most significant in the input data set. The significance of the association between the data set and the canonical pathway was determined based on two parameters: (i) a ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway, and (ii) a P value calculated using Fisher’s exact test determining the probability that the association between the genes in the data set and the canonical pathway was due to chance alone. The expression of selected genes (BIRC3, CASP1, IRF1, SOD2, and STAT1) were further validated by qRT-PCR. Sequences are listed in Supplementary Table S1.
Western blot analysis
Melligen, Huh7ins, Huh7ins-empty vector cells, and human islets were centrifuged (200 g for 5 minutes). The cell pellets were suspended in buffer I (Tris 10 mmol/l, 20 mmol/l NaH2PO4, 1.0 mmol/l ethylenediaminetetraacetic acid, 0.1 mmol/l phenylmethanesulfonyl fluoride, 10 µg/ml pepstatin, 10 µl/ml leupeptin, pH 7.8), after which the freeze (−70 °C, 10 minutes)—thaw (37 °C, 10 minutes) cycle was applied three times. The cells were then incubated for 20 minutes at 4 °C. Supernatants were collected and protein concentrations were determined using the Micro BCA protein Assay Reagent Kit Life Technologies.
Protein samples (15 μg/30 µl) were electrophoresed on 10% polyacrylamide gels for western blot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as a loading control (Life Technologies). Separated proteins were transferred to a nitrocellulose membrane (Merck Millipore, MA), which was blocked in PBS containing with 5%w/v skim milk powder (overnight at 4 °C). After washing (three times, 10 minutes) with PBS containing 0.05%v/v Tween 20, nitrocellulose membranes were incubated with primary rabbit anti-human glucokinase antibody (1/1,000 dilution) (Santa Cruz Biotechnology, Dallas, TX) for 2 hours at room temperature (RT), then washed again three times with PBS (0.05% Tween 20). The nitrocellulose membrane was then incubated with a secondary polyclonal (donkey) anti-rabbit horseradish peroxidase IgG conjugated antibody (1/800 dilution) (Sigma-Aldrich, Sydney, Australia) for 1 hour at RT. After washing three times with PBS (0.05% Tween20), glucokinase protein expression (52 kDa) was visualized using Super Signal Chemiluminescence Substrate (Quantum Scientific, Milton, Australia). The membrane was then stripped with 10% sodium dodecyl sulfate, 0.5 mol/l Tris HCl (pH 6.8) and β-mercaptoethanol and the incubation and detection process was repeated with the loading control: anti-GADPH (36kDa) (Merck/Millipore, MA; 1/2,000).
Glucokinase and hexokinase enzyme activity
Glucose phosphorylation was measured in cell homogenates by following the conversion of [U-14C] glucose to [U-14C] glucose-6-phosphate as previously described.46 The individual activities of glucokinase and hexokinase were determined by performing duplicate assays, either in the presence or absence of 10 mmol/l glucose-6-phosphate, which inhibited low Km hexokinase activity.47
Transplantation of cells
Male NOD/scid mice were obtained from the Australian Research Council (ARC) Facility (Perth, Western Australia). Maintenance and experimental manipulations were performed in accordance with the National Health & Medical Research Council of Australia principles of laboratory care and regulations of the Australian Research Council were approved by the University of Technology Sydney. Diabetes was induced by multiple injections of streptozotocin (STZ; 75 mg/kg body weight/day for 5 consecutive days). Animals were considered diabetic if blood glucose levels were >20 mmol/l on three separate occasions. After treatment, animals were monitored for body weight and blood glucose levels.
Animals were in groups of eight for each treatment. The Huh7ins and Melligen cells (1 × 107) were initially aspirated into polyethylene tubing (internal diameter 0.48 mm; external diameter 0.96 mm) attached to a 23-gauge needle placed on a Hamilton syringe. The end of the tubing was then inserted into the subcutaneous tissues above the right scapula to allow injection of the cells into the mice. Blood glucose levels and weight of the mice were monitored every 1–3 days, in the late morning. Insulin was not administered to the mice at any time. Grafts were removed from the mice one week after the blood glucose levels were normalized (4–5 mmol/l). Blood glucose levels of the mice were monitored thereafter with an increase expected if the graft had been responsible for reversal of the diabetes. Intraperitoneal glucose tolerance tests (2 g glucose/kg after an 18-hour fast) were carried out on the transplanted animals at 26 days together with diabetic and normal controls.
Microscopic analysis
For immunofluorescence microscopy, frozen sections (6–8 μm) of transplanted cells and pancreas were prepared. For triple staining of insulin, glucagon, and somatostatin, the primary antibodies were mouse anti-human insulin (1:100; BioGenex, Fremont, CA), goat anti-glucagon and goat anti-somatostatin (1:200; Santa Cruz, Dallas, TX). The secondary antibodies were anti-mouse IgG, and anti-goat IgG (1:200; Vector, Burlingame, CA). The fluoresceins were fluorescein avidin D, AMCA avidin D and rhodamine avidin D (1:200; Vector). The staining procedure followed the fluorescein M.O.M kit (FMK–220, Vector). Between application of the primary antibodies against insulin and glucagon and glucagon and somatostatin, the avidin/biotin blocking kit (sp-200; Vector) was used, following the manufacturer’s instructions. All cells in 10 random fields were scored. Data were expressed as the number of positive cells per mm2 of tissue.
For electron microscopy, tissue was fixed and processed as previously described.9 For insulin immunoelectron microscopy, a postembedding immunogold procedure was used. Tissues and cells were embedded in LR White (ProSciTech, Thurigawa, Australia) and labeling procedures were performed as previously described.9
Statistical analysis
Data are represented as mean values ± standard error. Comparisons between two groups were analyzed using Student’s t-test. Comparisons between more than two groups were analyzed using general linear model analysis of variance (SPSS 17.0.0, SPSS, 2008, Armonk, NY). Repeated-measures analyses were used where appropriate. P values less than 0.05 were considered to be statistically significant.
Acknowledgments
We would like to thank A/Prof Wayne Hawthorne, Westmead Millennium Institute, Westmead Hospital Sydney for providing human islets and Richard Limburg for IT support. Financial support: Diabetes Australia Research Trust, Rebecca L. Cooper Medical Research Foundation and the University of Technology Sydney.
A.M.S. and C.T. are inventors on patent “Cells genetically modified to comprise pancreatic islet glucokinase and uses thereof WO 2009021276 A1, Ann M. Simpson and Chang Tao, European patent: EP20080782908, Australian patent: AU 2008/001160, United States of America patent: US12/672,832. The University of Technology Sydney has licensed the above patent family to PharmaCyte Biotech, Inc.
The first two authors contributed equally to this work.
References
- Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986;314:1360–1368. doi: 10.1056/NEJM198605223142106. [DOI] [PubMed] [Google Scholar]
- Paty BW, Ryan EA, Shapiro AM, Lakey JR, Robertson RP. Intrahepatic islet transplantation in type 1 diabetic patients does not restore hypoglycemic hormonal counterregulation or symptom recognition after insulin independence. Diabetes. 2002;51:3428–3434. doi: 10.2337/diabetes.51.12.3428. [DOI] [PubMed] [Google Scholar]
- Simpson AM, Marshall GM, Tuch BE, Maxwell L, Szymanska B, Tu J. Gene therapy of diabetes: glucose-stimulated insulin secretion in a human hepatoma cell line (HEP G2ins/g) Gene Ther. 1997;4:1202–1215. doi: 10.1038/sj.gt.3300527. [DOI] [PubMed] [Google Scholar]
- Tuch BE, Szymanska B, Yao M, Tabiin MT, Gross DJ, Holman S. Function of a genetically modified human liver cell line that stores, processes and secretes insulin. Gene Ther. 2003;10:490–503. doi: 10.1038/sj.gt.3301911. [DOI] [PubMed] [Google Scholar]
- Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–572. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- Ber I, Shternhall K, Perl S, Ohanuna Z, Goldberg I, Barshack I. Functional, persistent, and extended liver to pancreas transdifferentiation. J Biol Chem. 2003;278:31950–31957. doi: 10.1074/jbc.M303127200. [DOI] [PubMed] [Google Scholar]
- Kojima H, Fujimiya M, Matsumura K, Younan P, Imaeda H, Maeda M. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- Sapir T, Shternhall K, Meivar-Levy I, Blumenfeld T, Cohen H, Skutelsky E. Cell-replacement therapy for diabetes: Generating functional insulin-producing tissue from adult human liver cells. Proc Natl Acad Sci USA. 2005;102:7964–7969. doi: 10.1073/pnas.0405277102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, O’Brien BA, Swan MA, Koina ME, Nassif N, Wei MQ. Long-term correction of diabetes in rats after lentiviral hepatic insulin gene therapy. Diabetologia. 2007;50:1910–1920. doi: 10.1007/s00125-007-0722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsner M, Terbish T, Jörns A, Naujok O, Wedekind D, Hedrich HJ. Reversal of diabetes through gene therapy of diabetic rats by hepatic insulin expression via lentiviral transduction. Mol Ther. 2012;20:918–926. doi: 10.1038/mt.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, O’Brien BA, Byrne MR, Ch’ng E, Gatt PN, Swan MA. Long-term reversal of diabetes in non-obese diabetic mice by liver-directed gene therapy. J Gene Med. 2013;15:28–41. doi: 10.1002/jgm.2692. [DOI] [PubMed] [Google Scholar]
- Permutt MA, Koranyi L, Keller K, Lacy PE, Scharp DW, Mueckler M. Cloning and functional expression of a human pancreatic islet glucose-transporter cDNA. Proc Natl Acad Sci USA. 1989;86:8688–8692. doi: 10.1073/pnas.86.22.8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhouse S., Horecker BL, Stadtman ER.Current Topics in Cellular Regulation Academic Press; New York, San Francisco, London, 19761–50. [Google Scholar]
- Bouard D, Alazard-Dany D, Cosset FL. Viral vectors: from virology to transgene expression. Br J Pharmacol. 2009;157:153–165. doi: 10.1038/bjp.2008.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos P, Spasojevic M, Faas MM. Treatment of diabetes with encapsulated islets. Adv Exp Med Biol. 2010;670:38–53. doi: 10.1007/978-1-4419-5786-3_5. [DOI] [PubMed] [Google Scholar]
- Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov. 2014;13:278–289. doi: 10.1038/nrd4231. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Mandrup-Poulsen T. A choice of death–the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. 2001;44:2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Clark SA, Quaade C, Constandy H, Hansen P, Halban P, Ferber S. Novel insulinoma cell lines produced by iterative engineering of GLUT2, glucokinase, and human insulin expression. Diabetes. 1997;46:958–967. doi: 10.2337/diab.46.6.958. [DOI] [PubMed] [Google Scholar]
- Moore HP, Walker MD, Lee F, Kelly RB. Expressing a human proinsulin cDNA in a mouse ACTH-secreting cell. Intracellular storage, proteolytic processing, and secretion on stimulation. Cell. 1983;35 2 Pt 1:531–538. doi: 10.1016/0092-8674(83)90187-3. [DOI] [PubMed] [Google Scholar]
- Hohmeier HE, BeltrandelRio H, Clark SA, Henkel-Rieger R, Normington K, Newgard CB. Regulation of insulin secretion from novel engineered insulinoma cell lines. Diabetes. 1997;46:968–977. doi: 10.2337/diab.46.6.968. [DOI] [PubMed] [Google Scholar]
- Newgard CB. Cellular engineering and gene therapy strategies for insulin replacement in diabetes. Diabetes. 1994;43:341–350. doi: 10.2337/diab.43.3.341. [DOI] [PubMed] [Google Scholar]
- Simpson AM, Swan MA, Lui GJ, Tao C, O’Brien BA, Ch’ng E.Insulin trafficking in a glucose responsive engineered human liver cell line is regulated by the interaction of ATP-sensitive potassium channels and voltage-gated calcium channels. In: Molina FM (Ed) Gene Therapy: Tools and Potential Applications ISBN 980-953-307-690-9 2013703–726.
- Orci L. Macro- and micro-domains in the endocrine pancreas. Diabetes. 1982;31 6 Pt 1:538–565. doi: 10.2337/diab.31.6.538. [DOI] [PubMed] [Google Scholar]
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem. 1997;272:18572–18579. doi: 10.1074/jbc.272.30.18572. [DOI] [PubMed] [Google Scholar]
- Detimary P, Dejonghe S, Ling Z, Pipeleers D, Schuit F, Henquin JC. The changes in adenine nucleotides measured in glucose-stimulated rodent islets occur in beta cells but not in alpha cells and are also observed in human islets. J Biol Chem. 1998;273:33905–33908. doi: 10.1074/jbc.273.51.33905. [DOI] [PubMed] [Google Scholar]
- Liang Y, Najafi H, Smith RM, Zimmerman EC, Magnuson MA, Tal M. Concordant glucose induction of glucokinase, glucose usage, and glucose-stimulated insulin release in pancreatic islets maintained in organ culture. Diabetes. 1992;41:792–806. doi: 10.2337/diab.41.7.792. [DOI] [PubMed] [Google Scholar]
- Lutherborrow MA, Appavoo M, Simpson AM, Tuch BE. Gene expression profiling of HUH7-ins: lack of a granulogenic function for chromogranin A. Islets. 2009;1:62–74. doi: 10.4161/isl.1.1.8994. [DOI] [PubMed] [Google Scholar]
- Gerace D, Ren B, Hawthorne WJ, Byrne MR, Phillips PM, O’Brien BA. Pancreatic transdifferentiation in porcine liver following lentiviral delivery of human furin-cleavable insulin. Transplant Proc. 2013;45:1869–1874. doi: 10.1016/j.transproceed.2013.01.051. [DOI] [PubMed] [Google Scholar]
- Shanmukhappa K, Mourya R, Sabla GE, Degen JL, Bezerra JA. Hepatic to pancreatic switch defines a role for hemostatic factors in cellular plasticity in mice. Proc Natl Acad Sci USA. 2005;102:10182–10187. doi: 10.1073/pnas.0501691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li S, Hatch H, Ahrens K, Cornelius JG, Petersen BE. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci USA. 2002;99:8078–8083. doi: 10.1073/pnas.122210699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey ST, Arvelo MB, Hasenkamp W, Bach FH, Ferran C. A20 inhibits cytokine-induced apoptosis and nuclear factor kappaB-dependent gene activation in islets. J Exp Med. 1999;190:1135–1146. doi: 10.1084/jem.190.8.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes. 2004;53:113–121. doi: 10.2337/diabetes.53.1.113. [DOI] [PubMed] [Google Scholar]
- Chang K, Lee SJ, Cheong I, Billiar TR, Chung HT, Han JA. Nitric oxide suppresses inducible nitric oxide synthase expression by inhibiting post-translational modification of IkappaB. Exp Mol Med. 2004;36:311–324. doi: 10.1038/emm.2004.42. [DOI] [PubMed] [Google Scholar]
- Sarkar SA, Kutlu B, Velmurugan K, Kizaka-Kondoh S, Lee CE, Wong R. Cytokine-mediated induction of anti-apoptotic genes that are linked to nuclear factor kappa-B (NF-kappaB) signalling in human islets and in a mouse beta cell line. Diabetologia. 2009;52:1092–1101. doi: 10.1007/s00125-009-1331-x. [DOI] [PubMed] [Google Scholar]
- Liuwantara D, Elliot M, Smith MW, Yam AO, Walters SN, Marino E. Nuclear factor-kappaB regulates beta-cell death: a critical role for A20 in beta-cell protection. Diabetes. 2006;55:2491–2501. doi: 10.2337/db06-0142. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Heimberg H, Heremans Y, Leeman R, Kutlu B, Kruhøffer M. A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J Biol Chem. 2001;276:48879–48886. doi: 10.1074/jbc.M108658200. [DOI] [PubMed] [Google Scholar]
- Cardozo AK, Berthou L, Kruhøffer M, Orntoft T, Nicolls MR, Eizirik DL. Gene microarray study corroborates proteomic findings in rodent islet cells. J Proteome Res. 2003;2:553–555. doi: 10.1021/pr034029o. [DOI] [PubMed] [Google Scholar]
- Lopes M, Kutlu B, Miani M, Bang-Berthelsen CH, Størling J, Pociot F. Temporal profiling of cytokine-induced genes in pancreatic β-cells by meta-analysis and network inference. Genomics. 2014;103:264–275. doi: 10.1016/j.ygeno.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Schoemaker MH, Ros JE, Homan M, Trautwein C, Liston P, Poelstra K. Cytokine regulation of pro- and anti-apoptotic genes in rat hepatocytes: NF-kappaB-regulated inhibitor of apoptosis protein 2 (cIAP2) prevents apoptosis. J Hepatol. 2002;36:742–750. doi: 10.1016/s0168-8278(02)00063-6. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–238. doi: 10.1056/NEJM200007273430401. [DOI] [PubMed] [Google Scholar]
- van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–245. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- Tabiin MT, Tuch BE, Bai L, Han X, Simpson AM. Resistance of insulin-secreting hepatocytes to the toxicity of human autoimmune cytokines. Autoimmunity. 2001;17:229–242. doi: 10.1006/jaut.2001.0539. [DOI] [PubMed] [Google Scholar]
- Kuwajima M, Newgard CB, Foster DW, McGarry JD. The glucose-phosphorylating capacity of liver as measured by three independent assays. Implications for the mechanism of hepatic glycogen synthesis. J Biol Chem. 1986;261:8849–8853. [PubMed] [Google Scholar]
- Ferber S, BeltrandelRio H, Johnson JH, Noel RJ, Cassidy LE, Clark S. GLUT-2 gene transfer into insulinoma cells confers both low and high affinity glucose-stimulated insulin release. Relationship to glucokinase activity. J Biol Chem. 1994;269:11523–11529. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.