

Abstract
Hypoxic non‐small cell lung cancer (NSCLC) is dependent on Notch‐1 signaling for survival. Targeting Notch‐1 by means of γ‐secretase inhibitors (GSI) proved effective in killing hypoxic NSCLC. Post‐mortem analysis of GSI‐treated, NSCLC‐burdened mice suggested enhanced phosphorylation of 4E‐BP1 at threonines 37/46 in hypoxic tumor tissues. In vitro dissection of this phenomenon revealed that Amyloid Precursor Protein (APP) inhibition was responsible for a non‐canonical 4E‐BP1 phosphorylation pattern rearrangement—a process, in part, mediated by APP regulation of the pseudophosphatase Styx. Upon APP depletion we observed modifications of eIF‐4F composition indicating increased recruitment of eIF‐4A to the mRNA cap. This phenomenon was supported by the observation that cells with depleted APP were partially resistant to silvestrol, an antibiotic that interferes with eIF‐4A assembly into eIF‐4F complexes. APP downregulation in dividing human cells increased the rate of global protein synthesis, both cap‐ and IRES‐dependent. Such an increase seemed independent of mTOR inhibition. After administration of Torin‐1, APP downregulation and Mechanistic Target of Rapamycin Complex 1 (mTORC‐1) inhibition affected 4E‐BP1 phosphorylation and global protein synthesis in opposite fashions. Additional investigations indicated that APP operates independently of mTORC‐1. Key phenomena described in this study were reversed by overexpression of the APP C‐terminal domain. The presented data suggest that APP may be a novel regulator of protein synthesis in dividing human cells, both cancerous and primary. Furthermore, APP appears to affect translation initiation using mechanisms seemingly dissimilar to mTORC‐1 regulation of cap‐dependent protein synthesis. J. Cell. Physiol. 230: 1064–1074, 2015. © 2014 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
Cell growth and proliferation are highly coordinated processes. A large amount of evidence supports a pivotal role for the phosphatidylinositol 3‐OH kinase (PI3K)/Akt/mTORC‐1 axis in the establishment of such a crosstalk. Studies showed that mutating several components of this signaling pathway influences both cell size and number, hence affecting organ size (Böhni et al., 1996; Leevers et al., 1996; Goberdhan et al., 1999). Likewise, inactivating mutations of genes leading to deregulated mTORC‐1 activity and failed metabolic checkpoints cause syndromes characterized by multiple, tumor‐like outgrowths in humans, such as Cowden syndrome (Liaw et al., 1997) and tuberous sclerosis (Brook‐Carter et al., 1994). Conversely, experimental knockout of a number of genes involved in PI3K activation and its downstream effectors causes organ hypoplasia and reduced body size in mice (Liu et al., 1993; Dummler et al., 2006). mTORC‐1 is a pivotal sensor of nutrient availability and stress conditions (Ellisen, 2005; Gwinn et al., 2008; Saqcena et al., 2013). In stress conditions the cell is generally under a dominant metabolic checkpoint that follows different mechanisms according to the severity and duration of such stimuli. Acute stress generally triggers phosphorylative inactivation of eIF‐2A and rapid translational reprogramming (Spriggs et al., 2010), while chronic stress usually leads to repression of mTORC‐1 activity through a number of mechanisms. The latter situation results in reduced 4E‐BP1 phosphorylation at a number of residues (T70, S65, T37/46) (Gingras et al., 2001). Hypophosphorylated 4E‐BPs sequester eIF‐4E in a conformation that prevents its association with eIF‐4G and ultimately the formation of a productive eIF‐4F assembly on the 5'‐end mRNA cap. In these conditions global protein synthesis is consequently suppressed. In conditions of severe hypoxia and nutrient deprivation (a typical situation in a hypoxic tumor microenvironment), inhibited mTORC‐1 activity is unable to prevent the formation of the autophagosome, a process mediated by hypophosphorylated ATG13 (Hosokawa et al., 2009).
In several solid tumors, including NSCLC, hypoxic tissues require Notch signaling for survival or for hypoxia‐induced proliferation (Chen et al., 2007; Eliasz et al., 2010; Xing et al., 2011; Zou et al., 2013). In a previous study we found that Notch inhibition through administration of γ‐secretase inhibitors (GSI) can target hypoxic NSCLC specifically, thus reducing its volume and related markers in an orthotopic NSCLC model (Liang et al., 2012). The γ‐secretase complex cleaves a number of different proteins in their transmembrane domain (Hemming et al., 2008). There is no stringent sequence or site specificity for cleavage, and the intracellular domain that results from γ‐secretase cleavage seems predominantly determined by the stability of the cleavage product according to the N‐end rule (Bachmair et al., 1986). Among the most studied γ‐secretase substrates is the Amyloid Precursor Protein, or APP. APP is an extremely pleiotropic, single pass transmembrane protein involved in numerous cellular functions, none of which are definitively considered to be APP's main role (Müller and Zheng, 2012). Its overall structure and cleavage pattern (ADAM10 and γ‐secretase sequential cleavage) are reminiscent of Notch receptors, although the APP intracellular domain (AICD) is much smaller compared to Notch proteins (∼6kDa). Currently, it is unclear whether APP (which is ubiquitously expressed as multiple splice variants, all of which undergo extensive post‐translational modification) is a receptor or a ligand. It has two homologues, APLP‐1 and APLP‐2. APP has been linked to intra/extracullular matrix interactions, axonal functions, iron transport, and cytoskeleton communications. Some soluble, extracellular portions (APPsα) seem to have growth factor‐like properties, while AICD seems to affect transcription (alongside cofactor Fe65) after its recruitment to membrane‐bound APP. Cleavage of APP generates an AICD/Fe65 complex that translocates to the nucleus, where it affects transcription alongside factor Tip60 (Cao and Südhof, 2004). However, it is important to underscore that the mechanisms through which AICD can regulate transcription are still opaque (Beckett et al., 2012).
A few studies have implicated APP in tumorigenesis. More specifically, APP seems to promote cell growth through activation of extracellular signal‐regulated protein kinase (ERK), an event also observed in neuronal cells (Nizzari et al., 2007). Additionally, APP, or its fragments, appears to interact with Akt signaling (Lee et al., 2009).
This study tests the hypothesis that γ‐secretase inhibition could re‐induce mTORC‐1 activity in a quiescent hypoxic tumor environment. In vitro studies disproved this hypothesis and unexpectedly evidenced a role for APP in regulating protein synthesis independently of mTORC‐1.
Materials and Methods
Unless otherwise specified, all studies are shown 48 h after experimental manipulation. Lists of siRNAs, antibodies and oligonucleotides used in this study are reported in Supplementary Material Table S1, Table S2, and Table S3, respectively.
Rationale of APP silencing
In the experiments shown here we used three different siRNAs against APP: one from Santa Cruz Biotechnologies; Santa Cruz, CA (# sc‐29677), and two from Qiagen (Valencia, CA), referred to as siRNA9 (# SI02780281) and siRNA10 (# SI02780288). Both the Santa Cruz Biotechnology and the Qiagen siRNA10 were effective in downregulating APP, and primary experiments were performed using sc‐29677 or SI02780288 indifferently. Since APP has ten putative splice variants, siRNAs potentially targeting different splice variants to different extents show occasional lack of reproducibility. The Qiagen siRNA10 targets the common 3'‐end common to all splice variants and equally downregulates all of them. siRNA9 was not effective in sufficiently reducing either the APP mRNA or protein expression levels. Some experiments performed here were confirmed using shRNAs to APP cloned in pLKO.1 (#TRCN0000011043 and #TRCN0000006707, Sigma‐Aldrich, St. Louis, MO). We provide a detailed representative downregulation experiment for APP in Figure 2B and C. We routinely confirmed APP downregulation in all experiments. Blots are omitted from figures to avoid unnecessary redundancy.
Figure 2.

APP depletion causes 4E‐BP1 phosphorylation pattern rearrangements. A: Immunoblot of the specified proteins and phosphoproteins in total cell lysates obtained from the indicated cell lines after transfection with either a control siRNA or control plasmid (c), and cells transfected with siRNA to APP (siAPP) or with a plasmid encoding AICD (AICD). Artificial APP downregulation in NSCLC cells using siRNA: B: Q‐PCR. Columns represent the average of four independent experiments (one in each cell line A549, H1299, H1437, and H1650); bars represent S.D. The mRNA abundance for cells transfected with control siRNA (c) was arbitrarily set to 1 at 48 h after transfection. C: Representative immunoblot at the specified time‐points after siRNA transfection (cell line H1299). Virtually identical results were obtained in all cells tested. For the results shown here we used siRNA to APP 10 (Qiagen). The band visible in siAPP lanes is either non‐specific or, less probably, the highly APP homolog APLP‐2. That band was not affected by GSI treatment, and its intensity or presence was not consistently reproducible in our immunoblots. For these reasons we tend to consider it a non‐specific band. D: Immunoblot of the specified proteins in total cell lysates obtained from cell line H1299 after exposure to the indicated concentrations of GSI. DMSO‐treated cells were transfected with either control (c) or siRNA to APP. E: Immunoblot of the specified proteins and phosphoproteins in total cell lysates obtained from cell line H1299 48 h after transfection with either a control siRNA, or with a siRNA to APP. Similar results were obtained in multiple experiments and in cell line A549.
Chemicals and plasmids
γ‐secretase inhibitor MRK‐003 was a gift of Merck & Co. (Whitehouse Station, NJ). 5 μM MRK‐003 was used in most experiments; exceptions are reported in figures and/or legends. Torin 1 (1‐[4‐[4‐(1‐Oxopropyl)‐1‐piperazinyl]‐3‐(trifluoromethyl) phenyl]‐9‐(3‐quinolinyl)‐benzo[h]‐1,6‐naphthyridin‐2(1H)‐one) (Tocris Bioscience, Bristol, UK) was dissolved in DMSO and used at a final concentration of 0.250 μM. Roscovitine (Cell Signaling) was dissolved in DMSO and used at a final concentration of 20 μM. UO126 (Selleck Chemicals, Huston, TX) was dissolved in DMSO, and used at a final concentration of 10 μM. Silvestrol (Medchemexpress LLC, Princeton, NJ) was dissolved in DMSO and used at a final concentration of 0.04 μM and 0.1 μM. Homoharringtonine (Tocris) was dissolved in DMSO and used at a final concentration of 0.1 μM. Insulin in solution (Sigma–Aldrich) was diluted in sterile PBS and used at the specified concentrations. pcDNA3‐RLUC‐POLIRES‐FLUC was a gift from Nahum Sonenberg (McGill University, Montreal); pCDF1‐MCS1‐EF1‐cop GFP expressing the APP C‐terminal 59 aa was a gift from Dr. Xiao Z.C. (Institute of Molecular and Cell Biology, Singapore); full length APP 695 in pCAX vector was from Addgene (Cambridge, MA). We used the empty plasmids as transfection controls.
In vivo studies
The Loyola Institutional Animal Care and User Committee approved all studies. NOD.CB17‐Prkdcscid/J mice (Jackson Laboratories, USA) were injected via the lateral tail vein with NSCLC cell lines. Details of mouse models and treatments are reported elsewhere (Eliasz et al., 2010).
Cell culture and transfection
A549, H1299, H1650, H1437, and WI38 were from ATCC (Manassas, VA). Me16 were a gift from Dr. Harvey I. Pass (New York University). HaCaT cells were provided by Dr. Mitchel Denning (Loyola University Chicago). Cells were cultured in RPMI‐1640 supplemented with 10% fetal bovine serum (FBS) under 0.5% O2, 5.0% CO2, 94.5% N2 in a Coy CleanSpot glove‐box incubator (Coy Laboratory Products, Grass Lake, MI). Transfection of siRNA was performed using electroporation as previously described (Chen et al., 2007). Transfection of DNA was performed primarily using electroporation, or Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer protocols. Cell lines and cultures were fingerprinted using the GenePrint fluorescent STR system (Promega, Madison, WI). Absence of mycoplasma contamination was monitored using the MycoSensor QPCR assay kit (Stratagene, La Jolla, CA).
Antibodies and protein analysis
Western blot analysis was performed as previously described (Chen et al., 2007). The amount of total cell lysate loaded was experimentally determined to establish a linear range for the assay for each protein assayed (from 5 to 50 μg total cell lysate). Whenever possible, transfer nitrocellulose membranes were sliced according to suitable molecular weight ranges and simultaneously hybridized with different antibodies to reduce artifacts deriving from multiple stripping of primary antibodies procedures. Immunoprecipitations were performed according to manufacturer protocols. Lysates were incubated with specific primary antibodies at the concentrations recommended by manufacturers overnight at 4°C, followed by 1 h incubation with Protein A/G Magnetic Beads (Thermo Fisher Scientific Inc., Waltman, MA). After elution, proteins were resolved by SDS‐PAGE, and transferred to Hybond‐C membranes (Amersham, Cleveland, OH).
Immunofluorescence was performed as previously described (Eliasz et al., 2010) on either 8 μm‐thick, flash frozen tissue slides or on cells seeded in glass immunochemistry slides. Primary antibody concentrations were optimized in pilot experiments. Secondary antibodies were conjugated with either Alexa Fluor 488 or Alexa Fluor 568. For tissue slides, autofluorescence was quenched using the sodium borohydride protocol (http://www.uab.edu/highresolutionimaging/autofluorescence). Nuclei were visualized using DAPI staining. The Golgi apparatus was visualized using rhodamine labeled wheat germ agglutinin (Vector Laboratories, Burlingame, CA). Slides were mounted using fluorescence mounting medium (Dako, Troy, MI). Images were acquired at room temperature using EVOS FL Cell Imaging System (Life Technologies Corp., Carlsbad, CA).
Cap binding assay and rate of global protein synthesis
Cap binding assay was performed 48 and 72 h after transfection with control siRNA or siRNA against APP as previously described (Villalonga et al., 2009). Cells were lysed and incubated with either m7‐GTP‐Sepharose 4B beads (GE Healthcare, Cleveland, OH) or with unmodified Sepharose 4B beads (negative control).
The rate of global protein synthesis was measured using the Click‐iT AHA Protein Synthesis kit (Invitrogen) according to the manufacturer's instruction. The incorporation of AHA was analyzed using a BD FACS Canto II instrument (Becton Dickinson, Franklin lakes, NJ). Flow cytometry data was analyzed by FlowJo software.
For AHA incorporation in the presence or absence of silvestrol or homoharringtonine, NSCLC cells were electroporated with siRNA control or siRNA against APP (siAPP), plated in RPMI media supplemented with 10% FBS and allowed to attach overnight. After 36 h, the medium was changed to medium without methionine, supplemented with 1% FBS. 12 h later (48 h after transfection), cells were treated with either 0.04 µM or 0.1 µM of silvestrol, 0.1 µM homoharringtonine or vehicle (DMSO). Silvestrol, homoharringtonine or DMSO was added alongside AHA for 20, 30 or 40 min. In experiments performed with Torin‐1, the compound was added at a concentration of 0.250 μM, the medium was replaced with fresh medium containing 0.250 μM Torin‐1 after 24 h, and cells were analyzed 48 h after the original addition of Torin‐1. After incubation, cells were detached, processed, washed, and examined by flow cytometry. In parallel, protein expression levels were determined by immunoblot. Each experiment was performed at least in triplicate.
Gene expression analysis
Total RNA was extracted using the RNeasy Mini kit (Qiagen). RNA concentration was determined using a NanoDrop Spectrophotometer (Thermo Fisher Scientific Inc.) and 1 μg of total RNA was used for cDNA synthesis. cDNA synthesis was performed using iScript Reverse Transcription Supermix RT‐qPCR (Bio‐Rad, Hercules, CA). Quantitative real‐time PCR was done with SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) in an ABI 7300 thermal cycler (Applied Biosystems). Non‐reverse transcription reactions served as negative controls. All measurements were normalized for human ribosomal protein RPL13A, β‐actin, and β‐tubulin mRNAs (reference genes). We used linearity curves calibrations in all experiments.
For the Illumina (San Diego, CA) Micro‐array Gene Expression experiment, total RNA was extracted from A549 and H1299 cells as described above. Samples from three independent transfection experiments along with untransfected cells were used for analysis. Gene expression analysis was performed using the HumanHT12 (48,000 probes, RefSeq plus EST) array. Data was analyzed using Genome Studio software (Illumina). Only changes of ±2‐fold or more in gene expression common to the two cell lines were taken into consideration. These changes were confirmed using Q‐PCR.
Results
γ‐secretase inhibition causes changes in 4E‐BP1 phosphorylation pattern
A hypoxic tumor microenvironment is traditionally considered to be resistant to radio/chemotherapy because cancer cells deprived of oxygen and nutrients are quiescent compared to well‐oxygenated tumor tissue (Kyle et al., 2012). In previous studies, we targeted hypoxic tumor tissue in experimental, orthotopic NSCLC models using γ‐secretase inhibitors in order to inhibit Notch‐1 signaling (Chen et al., 2007; Eliasz et al., 2010; Liang et al., 2012). γ‐secretase treatment proved effective in specifically killing hypoxic NSCLC, reducing its overall volume, increasing median survival, and reducing expression of hypoxia markers in NSCLC biopsies (Liang et al., 2012). In immunofluorescence studies, we used glucose transporter 1 (GLUT‐1) as a marker of hypoxia because this protein's overexpression is a reliable indicator of hypoxic cells and is perfectly co‐expressed alongside hypoxia inducible factor (HIF‐1α) (Eliasz et al., 2010). In vivo BrdU incorporation experiments and Ki67 staining confirmed that hypoxic tumor masses were essentially non‐proliferating (Fig. 1A). mTORC‐1 activation is generally considered to be an indication of active protein synthesis and cell cycle progression; one activation marker is phosphorylation of 4E‐BP1 at T37/46 (Villalonga et al., 2009). Similar to what was obtained with BrdU incorporation or with Ki67 immunofluorescence experiments, hypoxic tumor regions did not show detectable 4E‐BP1 phosphorylation at T37/46, while in tumor regions not expressing the hypoxia marker GLUT‐1, phosphorylation was readily detectable (Fig. 1B). Post‐mortem analyses of γ‐secretase inhibitor (GSI)‐treated mice revealed the presence of 4E‐BP1 phosphorylated at T37/46 in tumor regions also displaying the hypoxia marker GLUT‐1 (Fig. 1C). This data prompted the hypothesis that GSI treatment could have re‐induced mTORC‐1 activity in hypoxic tumors. We sought to test this hypothesis in vitro, but re‐creating in vitro the conditions found in hypoxic tumor tissue is likely impossible, due to its intrinsic heterogeneity and complexity (Palmer et al., 2010). For this reason, we performed tissue culture experiments in complete medium, but preserved HIF‐1α expression in our cells by culturing them under a 0.5% O2, 5.0% CO2, 94.5% N2 atmosphere. In such conditions, cells are not growth or proliferation arrested. Furthermore, at these O2 concentrations, mitochondrial functions are not compromised (Höckel and Vaupel, 2001), hence 0.5% O2 is not necessarily considered deep hypoxia. Higher oxygen concentrations did not reliably stabilize HIF‐1α (not shown).
Figure 1.

GSI treatment causes increased 4E‐BP1 phosphorylation at T37/46 in hypoxic NSCLC. A: Hypoxic tumor microenvironment is quiescent. Top: coimmunofluorescence of GLUT‐1 (red) and Ki67 (green). Bottom: in vivo BrdU incorporation (green) and GLUT‐1 (red). Note that at this stage, lungs of mice contain 93 ± 0.8% human cancer cells (Eliasz et al., 2010). B: Control mice. C: GSI‐treated mice. Red: GLUT‐1; green, phosphorylated T37/46 4E‐BP1‐specific antibody. Note color segregation in control animals and co‐localization in GSI‐treated animals. D: Immunoblot of the specified proteins and phosphoproteins in total cell lysates obtained from the indicated cell lines after exposure to either DMSO (c) or GSI. In cell lines A549, H1299 and H1650 the intensity of the total 4E‐BP1 bands seems to increase because the three isoforms (α, β, γ, corresponding to 4E‐BP1 phosphorylated at T37/46, T37/46 plus T70, and T37/46 plus T70 and S65, respectively; Gingras et al., 2001) merge into one or two bands, indicating loss of 4E‐BP1 phosphorylation at S65 and/or T70 after GSI treatment. E: Q‐PCR experiments performed on total RNA extracted from cells exposed to DMSO vehicle (c) or GSI for 48 h. Columns represent averages of three independent experiments, bars represent S.D. Reference genes: RPL13, β‐actin, β‐globin. F: Left, representative immunoblot of the specified proteins and phosphoproteins in total cell lysates obtained from cell line H1299 after transfection with either a control siRNA (c), or cells transfected with siRNA to preselinin‐1 and nicastrin (siPres/Nic); right, immunoblot of the specified proteins. Similar results were obtained in cell lines A549 and H1437.
In vitro treatment of different NSCLC cell lines with GSI produced a marked increase of 4E‐BP1 phosphorylation at T37/46 accompanied by an increase in steady state levels of total 4E‐BP1 in one cell line (Fig. 1D; in other cell lines we observed collapsing of the three 4E‐BP1 phosphorylated isoforms within the fastest migrating one). However, the 4E‐BP1 mRNA expression levels did not significantly change (Fig. 1E). Considering that γ‐secretase affects a plethora of substrates (Hemming et al., 2008) and that some GSIs seem to inhibit the proteasome (Clementz and Osipo, 2009), we sought to better determine the role of γ‐secretase by artificially downregulating presenilin‐1 and nicastrin (two critical components of the γ‐secretase complex). Under these conditions of downregulation, siRNA to presenilin‐1 and nicastrin caused an increase in T37/46 phosphorylation with no effects on the 4E‐BP1 steady‐state expression levels (Fig. 1F). Since artificial downregulation of γ‐secretase complexes reproduced the effects of GSI exposure on 4E‐BP1 phosphorylation at T37/46, we did not perform further experiments with GSIs, which can target multiple cellular phenomena.
APP is the γ‐secretase substrate responsible for altered 4E‐BP1 phosphorylation pattern
Next we focused on the identification of the putative γ‐secretase substrate(s) responsible for the enhanced 4E‐BP1 phosphorylation. We first investigated Notch receptors, since Notch‐1 affects Akt activation in hypoxia (Eliasz et al., 2010). Artificial downregulation or forced Notch‐1 through ‐4 intracellular domain expression yielded marginal and poorly reproducible results (not shown). This suggested that Notch receptors have little or no influence on 4E‐BP1 T37/46 phosphorylation in our system. Next, we investigated another γ‐secretase substrate, APP. Using different siRNAs, we determined that APP downregulation caused increased 4E‐BP1 phosphorylation at T37/46 in different NSCLC cell lines, while overexpression of the APP intracellular domain, or AICD, produced the opposite effect (Fig. 2A; a representative APP down‐regulation experiment is shown in Fig. 2B,C, while the rationale for siRNA selection/usage is reported in Materials and Methods). When we genetically altered APP expression, we did not observe variations in the steady state total 4E‐BP1 expression levels within the same cell line. This reinforces the idea that increased total 4E‐BP1 expression upon GSI treatment in certain cell lines was due to γ‐secretase inhibition (which affects about 91 cellular proteins) and possible off target effects of GSIs. APP appeared to be actively cleaved in NSCLC cells, as GSI treatment led to a dose‐dependent accumulation of total APP (Fig. 2D), similar to what was observed for Notch‐1 in the same cells (Chen et al., 2007). Further examination of the overall 4E‐BP1 phosphorylation status after APP downregulation revealed decreased phosphorylation at S65 and unchanged phosphorylation at T70 (Fig. 2E). Previous studies have suggested that APP can influence ERK and/or Akt activation (Nizzari et al., 2007; Lee et al., 2009). We observed decreased ERK‐1 and ‐2 phosphorylation at 24, 48, and 72 h after siRNA‐mediated knockdown of APP in NSCLC cell lines A549 and H1299 (Fig. 3A, H1299 results). This supports the findings that APP can activate Ras (Russo et al., 2002). During an observation period of 72 h post siRNA transfection, downregulation of APP did not affect Akt phosphorylation at either T308 or S473 in any cell line tested (Fig. 3B,C, H1299 results).
Figure 3.

Artificial downregulation of APP causes a reduced ERKs activation but it does not affect Akt‐1 activation. APP modulates 4E‐BP1 phosphorylation through Styx. A‐C: Western blot analyses of the indicated proteins and phosphoproteins at the indicated time‐points in H1299 cells with depleted APP (siAPP) or in control siRNA‐transfected cells (c). Similar results were obtained in cell lines A549 and H1437. D: Immunoblot of the indicated protein and phosphoproteins in cell lysates obtained from cell line H1299 48 h after exposure to vehicle (c) or roscovitine (Rosc.). E: Immunoblot of the indicated protein and phosphoproteins in cell lysates obtained from cell line H1299 after exposure to vehicle (c) or UO126. In this experiment we show the 24 h time point because longer UO126 exposures caused paradoxical ERKs overactivation, as often described in scientific literature. F: Q‐PCR of the Styx mRNA in H1299 cells transfected with a control siRNA (c) or with the indicated siRNAs to Styx. Columns, averages of three independent experiments; bars, S.D. G: Western blot analysis of the indicated proteins and phosphoproteins 48 h after transfection of the indicated siRNAs. H: Q‐PCR of the Styx mRNA in cells transfected with the control plasmid pCAX (c) or with APP 695 cloned in pCAX (APP695). Columns represent averages of independent experiments performed in A549, H1299, and H1650 cells; bars represent S.D. I: Western blot analysis of the indicated protein and phosphoproteins in H1299 48 h after transfection with a variety of nucleic acids. Lanes: 1, cells transfected with siRNA control (c); 2, cells transfected with siRNA to APP (siAPP); 3, cells transfected with siRNA to APP and siRNA to Styx (siAPP + siStyx); 4, cells transfected with siRNA to Styx (siStyx); 5, cells transfected with control plasmid (pc); 6, cells transfected with a plasmid encoding AICD (AICD); 7, cells transfected with the control plasmid and siRNA to Styx (pc + siStyx); 8, cells transfected with a plasmid encoding AICD and with siRNA to Styx (AICD + siStyx). The apparent additive effect of siAPP and siStyx (lane 3) can be explained by a synergistic effect of the two treatments on the Styx mRNA expression level. APP forced expression, on the other hand, elevates the Styx mRNA expression levels (Fig. 3H). The fact that relatively small variations of the Styx mRNA level can produce measurably different effects on the 4E‐BP1 phosphorylation level at T37/46 is supported by the comparisons between siStyx2 and siStyx3 (Fig. 3F). SiStyx3 seems slightly less efficient in downregulating the Styx mRNA compared to siStyx2. This small variation yields measurable differences in 4E‐BP1 phosphorylation at T37/46 as measured using Western blot analysis (Fig. 3G).
Although phosphorylation of 4E‐BP1 at T37/46 is thought to be dependent on mTORC‐1 activity, S65 and T70 residues are targeted by additional kinases, including cyclin‐dependent kinase 1 (CDK‐1) (Heesom et al., 2001) and ERKs (Herbert et al., 2002). We determined that APP downregulation caused reduced ERKs activation (Fig. 3A) and reduced CDK activity (to be published elsewhere). To address a possible role of these kinases, we performed experiments using the CDK‐1 and MAP‐kinase inhibitors roscovitine and UO126, respectively. We determined that neither kinase was exclusively involved in the 4E‐BP1 phosphorylation pattern exhibited upon APP downregulation (Fig. 3D,E). Their inhibition may contribute to the phosphorylation status of S65, but do not explain the enhanced phosphorylation at T37/46. These results, alongside APP regulation of ERKs activity, support the idea that the effects seen on 4E‐BP1 upon APP depletion may be mediated by multiple kinases. This is in keeping with the highly pleiotropic effects described for APP signaling.
APP seems to affect 4E‐BP1 phosphorylation at T37/46 through transcriptional regulation of Styx
A number of results did not support the notion that APP could regulate 4E‐BP1 phosphorylation at T37/46 through modulation of mTORC‐1 (see below). Hence, we started investigating alternative mechanisms, including transcriptional regulation of possible mediators. APP can regulate transcription alongside Fe65 (which is recruited on the plasma membrane by full‐length APP). Upon γ‐secretase cleavage, Fe65 is associated to AICD and seems to protect free AICD from proteolytic degradation. It has been proposed that this complex can translocate to the nucleus where it regulates transcription alongside the factor Tip60 (Cao and Südhof, 2004). To identify putative genes that could possibly mediate APP effects on the 4E‐BP1 phosphorylation pattern, we performed gene expression arrays using the Illumina HumanHT12 (48,000 probes, RefSeq plus EST). Cell lines H1299 and A549 were transfected with either a control siRNA, siRNA to APP or mock‐transfected. Three independent experiments were performed in duplicate, total RNA was extracted from cells, and then labeled and hybridized to the Illumina chips. We found a number of genes, which expression was modified in both cell lines in similar ways, alongside a large number of genes that appeared to be altered in one cell line only. We identified a total of 17 genes whose expression was either downregulated to at least 50% or upregulated two‐folds or more upon APP depletion in both H1299 and A549 cells. These modifications in expression were confirmed by individual Q‐PCR experiments (Supplementary Fig. S1). We artificially downregulated the expression of all genes for which commercial siRNAs were available (boxed in blue in Supplementary Fig. S1A) and tested whether artificial downregulation of these genes affected 4E‐BP1 phosphorylation. Gene silencing of the pseudophosphatase Styx using two independent siRNAs (Fig. 3F) led to an increase in 4E‐BP1 phosphorylation at T37/46 without affecting other critical 4E‐BP1 residues' phosphorylation sites (Fig. 3G). To provide additional evidence supporting a role for APP transcriptional regulation of Styx, we transfected H1299 and A549 cells with APP 695 cloned in pCAX vector (and used the empty vector as the negative control). APP forced expression alone increased the Styx mRNA expression level 1.6 fold (Fig. 3H). Thus, APP forced expression produced opposite effects compared to APP downregulation. To further confirm the link between APP regulation of Styx and the effects operated by Styx on the phosphorylation status of T37/46, we transfected H1299 cells with a variety of nucleic acids (Fig. 3I). Downregulation of either APP or Styx individually increased T37/46 phosphorylation to approximately similar levels. Simultaneous depletion of both APP and Styx produced additive effects on T37/46 phosphorylation (compare lanes 1 through 4). As expected, forced expression of AICD reduced 4E‐BP1 T37/46 phosphorylation level when compared to cells transfected with control plasmid (compare lanes 5 and 6). Co‐transfection of control plasmid and siRNA to Styx enhanced T37/46 phosphorylation, while co‐transfection of AICD alongside siRNA to Styx partially reversed this effect (lanes 7 and 8). Taken altogether, these results reinforced the link between the regulation of Styx by APP and the enhancement of phosphorylation of 4E‐BP1 at the T37/46 residues (upon Styx depletion). We attempted to confirm variation of the Styx protein using Western blot assays; however, available commercial antibodies raised against Styx seemed unable to identify the endogenous protein. Considering all this evidence, we concluded that APP regulates the phosphorylation status of 4E‐BP1 T37/46 through Styx (which is a poorly investigated protein) rather than primarily acting alongside the canonical PI3K/Akt/mTORC‐1 axis (see below for further evidence concerning APP independence from mTORC‐1). 4E‐BP1 is an important node where the activity of different kinases and phosphatases converge to regulate cell growth (She et al., 2010).
APP depletion causes modifications of eIF‐4F's composition on the mRNA cap
We then asked whether the changes in the 4E‐BP1 phosphorylation pattern corresponded to changes in the translation initiation complex on the mRNA cap. We tested this using a 7‐methylguanosine conjugated resin pull‐down. After APP downregulation, the most noticeable effect we observed was a more than eightfold increase (as determined by densitometry) in 5′‐cap‐bound eIF‐4A. All other major eIF‐4F components showed no significant changes upon APP depletion, while the amounts of 5′ cap‐associated 4E‐BP1 showed ∼50% reduction (Fig. 4A). Reduced 4E‐BP1 binding to the cap is a common indication of possible increased cap‐dependent protein synthesis initiation (Thoreen et al., 2012).
Figure 4.

APP depletion enhances eIF‐4A recruitment on the cap; cells with depleted APP are partially resistant to silvestrol. A: Representative immunoblot of the specified proteins in total cell lysates (cell lys.) and in 7‐methylguanosine(m7G)‐sepharose pulled‐down proteins from cell extracts obtained from H1299 cells transfected with either a control siRNA (c), or a siRNA to APP. Seph. beads, sepharose beads control. Similar results were obtained in multiple experiments and in cell line A549. B: AHA incorporation at the indicated time‐points of cells transfected with either a control siRNA (c) or a siRNA to APP, exposed to 0.1 μM silvestrol or to 0.1 μM homoharringtonine (homohar.). C: As in (B) using 0.04 μM silvestrol. D: As in (B), but cells were exposed to vehicle only (no antibiotics). The graphs summarize three experiments performed in A549 cells. Similar results were obtained in H1299 cells. Bars represent S.D.
To confirm that APP depletion specifically affected eIF‐4A functions in eIF‐4F complexes, we used silvestrol in incorporation assays of the methionine surrogate L‐azidohomoalanine (AHA) followed by FACS analysis. Silverstrol is an antibiotic that prevents eIF‐4A binding to initiation complexes, hence inhibiting protein synthesis initiation (Bordeleau et al., 2008). Cells with depleted APP showed a certain resistance to both completely and partially inhibiting concentrations of silvestrol (Fig. 4B,C, respectively), thus confirming that APP may affect initiation complexes mainly by altering the binding of eIF‐4A to eIF‐4F complexes. This result was further confirmed by the use of homoharringtonine, an antibiotic that inhibits protein synthesis elongation (Tujebajeva et al., 1989). Homoharringtonine completely inhibited methionine analog (AHA) incorporation irrespective of APP depletion, yielding fluorescence readings virtually identical to no AHA incorporation (Fig. 4B).
APP depletion increases the rate of global protein synthesis
The above results suggested that APP regulates eIF‐4F composition, and that it could potentially affect global protein synthesis. To explore this possibility, we measured the rate of protein synthesis in cells transfected with siRNA targeting APP compared to cells transfected with a control siRNA. Cells with depleted APP showed a 27% to 172% increase in fluorescent AHA incorporation compared to controls in different experiments performed in several NSCLC cell lines (Fig. 5A,B and Table 1). Similar results were obtained in a mesothelioma cell line (ME16), in immortalized human keratinocytes (HaCat) and in primary human lung fibroblasts (WI‐38) (Supplementary Fig. S2). This increased protein synthesis rate was similar to what was obtained after insulin‐treatment of cells maintained in 1% FBS overnight (Supplementary Fig. S3). We wanted to ascertain whether this increase in the rate of global protein synthesis was specific to cap‐dependent translation. We used a bicistronic mRNA in which Renilla luciferase is translated using a cap‐dependent mechanism, while firefly luciferase is translated using the poliovirus internal ribosome entry sequence (IRES), both encoded by a reporter plasmid. Multiple experiments in different cell lines (A549, H1299, and H1650) showed that both cap‐ and IRES‐mediated protein translation was enhanced upon APP downregulation (Fig. 5C). This result is not unexpected if APP mainly regulates eIF‐4A functions, since this factor is also heavily involved in IRES‐dependent translation initiation (Komar and Hatzoglou, 2011).
Figure 5.
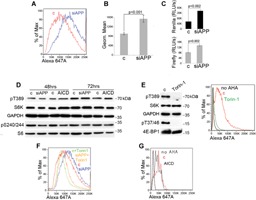
APP depletion increases the rate of global protein synthesis independently of mTOR. A: AHA (L‐azidohomoalanine) incorporation followed by FACS analysis of A549 cells after transfection with either control siRNA (red) or with a siRNA to APP (blue). B: Summary of the results of AHA incorporation in NSCLC cell lines A549, H1299, and H1650. Columns represent averages; bars represent S.D. Three experiments for each cell line are shown. C: Summary of nine dual luciferase experiments performed in three NSCLC cell lines (columns represent averages, bars represent S.D.). Luciferase light units were normalized for plasmid DNA content measured using Q‐PCR. D: Western blot analysis of the indicated proteins and phosphoproteins in H1299 cells transfected with the indicated nucleic acids. E: Torin‐1 completely suppresses mTORC‐1 markers of activation and protein synthesis; left, Western blot analysis of the indicated proteins and phosphoproteins in total cell lysates obtained from H1299 cells treated with DMSO (c), or with Torin‐1; right, AHA incorporation in H1299 exposed to DMSO control or Torin‐1. F: AHA incorporation in H1299 cells transfected with a control siRNA and exposed to DMSO (red), transfected with siRNA to APP and exposed to DMSO (blue), transfected with a control siRNA and exposed to Torin‐1 (light green), and transfected with siRNA to APP and exposed to Torin‐1 (light brown). G: AHA incorporation in H1299 cells transfected with pCDNA3 (c) or with AICD cloned in pCDNA3 (AICD). All nucleic acids were transfected by electroporation. Note that cells electroporated with plasmid DNA appear to incorporate substantially less AHA compared to cells electroporated with siRNAs only.
Table 1.
% increase of the rate of global protein synthesis upon APP artificial downregulation
| Cells | % increase siAPP/cont. | % increase siAPP/cont. |
|---|---|---|
| A549 | 44.0 ± 5.1 | 28.6 ± 3.3 |
| H1299 | 27.0 ± 3.4 | 18.6 ± 2.0 |
| H1437 | 164.5 ± 38.6 | 188.0 ± 32.8 |
| H1650 | 42.7 ± 3.6 | 28.1 ± 1.9 |
| ME16 | 38.7 ± 2.6 | 22.4 ± 2.4 |
| HaCat | 32.6 ± 2.4 | 18.3 ± 1.6 |
| WI38 | 62.4 ± 7.2 | 37.4 ± 4.2 |
Summary of three experiments performed in each of the indicated cell lines/cultures. Results are shown as percentage increase (average) ± S.D.
APP affects protein synthesis independently of mTORC‐1
The rate of global protein translation is primarily regulated by mTORC‐1 activity. APP depletion caused increased phosphorylation of 4E‐BP1 at T37/46, two mTORC‐1 targets. However, increased APP‐dependent phosphorylation at T37/46 appeared to be mediated by Styx, a pseudophosphatase not linked in any way to mTORC‐1 function. To clarify whether APP effects on protein synthesis were dependent or related to mTORC‐1 activity, we further explored the mTORC‐1 signaling pathway. Surprisingly, APP downregulation did not affect other key markers of mTORC‐1 activation, those being phosphorylation of p70S6 kinase or its target small ribosomal subunit protein S6 (Fig. 5D). This observation caused us to doubt the existence of a link between APP effects and mTORC‐1 activity. To further elucidate whether mTOR played a role in the observed increase in the global protein synthesis rate upon APP downregulation, we used Torin‐1, a potent and selective mTOR inhibitor (Thoreen et al., 2012). A number of experiments performed on different NSCLC cell lines confirmed that Torin‐1 is a very potent suppressor of global protein translation (Fig. 5E). Forty‐eight hours of Torin‐1 treatment suppressed 4E‐BP1 and S6 kinase phosphorylation, while AHA incorporation was virtually abolished (Fig. 5E). However, the effects of APP downregulation on protein synthesis did not appear to be dependent on mTOR activity (Fig. 5F, Table 1, and Supplementary Fig. S4), since Torin‐1‐treated, APP‐depleted NSCLC cells still displayed a conspicuous increase in methionine‐analog incorporation compared to the relevant controls.
Overexpression of AICD caused a decrease in AHA incorporation (Fig. 5G), opposite to the effect of APP depletion. These opposing results indicate that the levels of APP control the global rate of protein synthesis. Moreover, this effect appears to be independent of mTOR functions. Increased IRES‐driven translation further supports a main role for eIF‐4A in this process over regulation of eIF‐4E binding to the cap (the central step in protein synthesis initiation regulated by mTORC‐1).
A recent study suggested that the APP fragment Aβ stimulates mTORC‐1 activity via PRAS40 phosphorylation (Caccamo et al., 2011). We observed a modest increase in PRAS40 phosphorylation upon APP depletion (Supplementary Fig. S5A). To further explore this phenomenon in coimmunofluorescence experiments, we also observed APP and PRAS40's co‐localization in the Golgi apparatus (Supplementary Fig. S5B). mTOR appeared to be uniformly localized through the cytoplasm, which does not mirror the cellular distribution of APP and PRAS40 (Supplementary Fig. S5C). When we transfected cells with siRNA to APP, PRAS40 localization to the Golgi was lost and became similar to that of mTOR (Supplementary Fig. S5D). To investigate whether APP interacted with mTORC‐1, we performed a number of co‐immunoprecipitation experiments and consistently failed to detect APP or its fragments in association with RAPTOR (not shown). Furthermore, APP depletion did not affect the expression levels of either RAPTOR or mTOR (Supplementary Fig. S5A). This evidence led us to conclude that APP likely does not affect mTORC‐1 functions through PRAS40 phosphorylation. This potential contribution seemed tenuous and mechanistically problematic in order to address the phenotype observed on global protein synthesis, which seems independent of mTORC‐1.
Finally, in parallel control experiments, we performed siRNA to the APP family protein APLP‐2, which shares the highest degree of sequence homology and tissue distribution compared to APP. Although siRNA to APLP‐2 was effective and specific, APLP‐2 depletion did not affect the rate of protein synthesis of tested cells (Supplementary Fig. S6). Hence, it appears that what we describe here is specific to APP and not to APLP‐2.
Discussion
We described that APP depletion causes a rearrangement in the 4E‐BP1 phosphorylation pattern and enhances eIF‐4A recruitment within eIF‐4F complexes on the cap. This is paralleled by a substantial increase in the rate of global protein synthesis in a variety of human dividing cells, cancerous and normal.
We presented data using GSI because they provide additional confirmatory evidence in support of the experiments involving APP downregulation. However, results obtained with GSIs can only be indicative, since γ‐secretase complexes cleave a plethora of different substrates (to date, 91 proteins have been identified as substrates for γ‐secretase cleavage; Hemming et al., 2008; McCarthy et al., 2009).
APP seems to affect 4E‐BP1 phosphorylation primarily through its AICD, due to its transcriptional regulation of Styx. This pseudophosphatase has the potential to affect different proteins/pathways, including ERKs (Reiterer et al., 2013), PTPMP1 (Niemi et al., 2014) and Ras signaling through interaction with G3BP‐1 (Barr et al., 2013). Downregulation of Styx caused an increase in 4E‐BP1 phosphorylation at T37/46, similarly to what was observed after APP depletion. We propose that the results observed concerning 4E‐BP1 phosphorylation are likely the result of modified activities of multiple kinases/phosphatases, which include ERKs in concert with Styx (Fig. 6).
Figure 6.
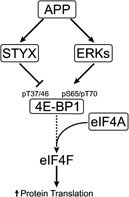
Simplified schematics summarizing key findings in this study. APP seems to positively regulate Styx expression at the transcriptional level. Styx appears to moderate phosphorylation at residues T37/46 of 4E‐BP1. On the other hand, APP sustains activation of ERKs, which support, directly and indirectly, phosphorylation of 4E‐BP1 S65 and T70. Upon APP depletion, the rearrangement of 4E‐BP1 phosphorylation pattern favors increased recruitment of eIF‐4A in eIF‐4F initiation complexes. This event leads to augmented rate of global protein synthesis.
Albeit apparently non‐canonical, the changes in the 4E‐BP1 phosphorylation pattern were paralleled by a 50% decreased recruitment of this protein on the cap. This event is an indication of increased cap‐dependent translation (She et al., 2010 Thoreen et al., 2012). Our data support the idea that APP influences translation primarily by affecting the efficiency of eIF‐4A association with initiation complexes. This is supported by a number of observations including: (i) increased eIF‐4A binding to the cap; (ii) cells with depleted APP show resistance to silvestrol; and (iii) the measured increase of both cap‐ and IRES‐dependent protein translation. eIF‐4A is a central initiation factor involved in the unwinding of secondary structures of probably all mRNAs (Schütz et al., 2008); therefore, eIF‐4A appears to be the main eIF‐4F subunit that can enhance both cap‐ and IRES‐driven translation.
APP depletion caused an increase in the global rate of protein synthesis quantitatively similar to what was elicited by insulin stimulation of serum‐starved cells. We hence propose that the effects mediated by APP on protein synthesis are mTORC‐1 independent. We used Torin‐1 instead of rapamycin, since it is well known that prolonged rapamycin exposure can cause compensatory responses that are mechanistically difficult to explain (Sarbassov et al., 2006; Choo et al., 2008; Thoreen et al., 2012). Torin‐1 completely inhibited mTORC‐1 activity, virtually abolished AHA incorporation, and led to the death of about one third of exposed cells within 48 h (not shown). Despite the harsh inhibitory conditions, APP downregulation caused increased AHA incorporation. Evidence suggesting that APP operates independently of mTORC‐1 are as follows: (i) APP downregulation did not modify the phosphorylation state of p70S6 kinase or its downstream target S6; (ii) APP depletion did not yield any modification in the mTOR/RAPTOR ratio; (iii) APP depletion had no effect on Akt phosphorylation at either T308 or S473; and (iv) we failed to find any association of APP to mTORC‐1 complexes (not shown). In support of previous studies (Caccamo et al., 2011), we found that APP depletion caused a modest increase of PRAS40 phosphorylation. PRAS40 has been proposed as an mTORC‐1 inhibitor, which, upon Akt phosphorylation at T246, activates mTORC‐1 (Sancak et al., 2007). However, recent studies have revealed that the PRAS40 effects are highly context‐dependent (Pallares‐Cartes et al., 2012). The data obtained here do not suggest a major role for PRAS40 for two reasons: (i) the increase in phosphorylation at T246 appears to be very modest; and (ii) APP, before depletion, seems to co‐localize with PRAS40 in the Golgi apparatus, while mTOR seems uniformly distributed throughout the cytoplasm. Upon APP depletion, the expression levels of PRAS40 are unchanged, but PRAS40 distribution is more similar to that of mTOR. Therefore, it is quite problematic to reconcile the release of a putative inhibitor with mTORC‐1 activation, unless PRAS40 is an mTORC‐1 stimulator in our system. This needs to be clarified in further studies. In any case, if PRAS40 contributes to mTORC‐1 activation, it should be confined only to its ability to phosphorylate 4E‐BP1 at T37/46 and not p70S6K.
The phenotypes described here are dependent on the APP's AICD, since forced expression of it reverses the effects of APP depletion. APP has been extensively studied in recent decades. Overall, a complex network of interactions with both extracellular and intracellular proteins has emerged (Müller et al., 2013). Several roles for AICD have been described, including its interaction with more than 20 cellular proteins. AICD instability has hampered a unifying interpretation of its function(s). Understanding the AICD binding partner(s) responsible for the phenomena presented here goes beyond the scope of this investigation and represents a future direction of our research. Nonetheless, our data suggest an active role for cleaved AICD. γ‐secretase cleavage inhibition phenotypes seemed to be mirrored by APP depletion, while APP depletion‐derived phenotypes were reversed by AICD overexpression. Overexpression of AICD can mimic excessive APP cleavage. Promoting endogenous APP cleavage using assays similar to those developed for Notch activation would test this hypothesis (Varnum‐Finney et al., 2000; Yvon et al., 2003;). Although TAG‐1 has been shown to interact with APP and trigger AICD signaling in mice (Ma et al., 2008), a standardized test to induce APP cleavage in human cells has not yet been developed.
Finally, it must be underscored that APP effects on the rate of global protein synthesis have consequences on the cell cycle and on cell viability. However, this topic has revealed to be complex enough to require being addressed in an independent manuscript.
In summary, we propose a model suggesting two phenomena: (i) AICD regulates global translation through modulation of eIF‐4A and possibly 4E‐BP1 phosphorylation; and (ii) loss of function of AICD results in uncontrolled increase in global protein synthesis. Whether AICD regulates protein synthesis in post‐mitotic cells (such as mature neurons) remains to be established.
Supporting information
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Figure S4.
Supplementary Figure S5.
Supplementary Figure S6.
Supporting Information.
Acknowledgments
We thank Patricia Simms for invaluable help with FACS experiments. We thank Dr. Clodia Osipo and Dr. Lucio Miele for critical reading of the manuscript.
Anna Sobol and Paolo Galluzzo contributed equally to this work.
Conflict of interest: None.
Literature Cited
- Bachmair A, Finley D, Varshavsky A. 1986. In vivo half‐life of a protein is a function of its amino‐terminal residue. Science 234:179–186. [DOI] [PubMed] [Google Scholar]
- Barr JE, Munyikwa MR, Frazier EA, Hinton SD. 2013. The pseudophosphatase MK‐STYX inhibits stress granule assembly independently of Ser149 phosphorylation of G3BP‐1. FEBS J 280:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett C, Nalivaeva NN, Belyaev ND, Turner AJ. 2012. Nuclear signalling by membrane protein intracellular domains: The AICD enigma. Cell Signal 24:402–409. [DOI] [PubMed] [Google Scholar]
- Böhni R, Riesgo‐Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. 1996. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell 97:865–875. [DOI] [PubMed] [Google Scholar]
- Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, Brem B, Greger H, Lowe SW, Porco JA, Jr , Pelletier J. 2008. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest 118:2651–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook‐Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, Nellist M, Gamble V, Harris PC, Sampson JR. 1994. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nat Genet 8:328–332. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magrí A, Oddo S. 2011. Naturally secreted amyloid‐beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40‐mediated mechanism. J Biol Chem 286:8924–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Südhof TC. 2004. Dissection of amyloid‐beta precursor protein‐dependent transcriptional transactivation. J Biol Chem 279:24601–24611. [DOI] [PubMed] [Google Scholar]
- Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, Bocchetta M. 2007. Oxygen concentration determines the biological effects of NOTCH‐1 signaling in adenocarcinoma of the lung. Cancer Res 67:7954–7959. [DOI] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. 2008. Rapamycin differentially inhibits S6Ks and 4E‐BP1 to mediate cell‐type‐specific repression of mRNA translation. Proc Natl Acad Sci USA 105:17414–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementz AG, Osipo C. 2009. Notch versus the proteasome: What is the target of gamma‐secretase inhibitor‐I? Breast Cancer Res 11:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummler B, Tschopp O, Hynx D, Yang ZZ, Dirnhofer S, Hemmings BA. 2006. Life with a single isoform of Akt: Mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 26:8042–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S, Miele L, Bocchetta M. 2010. Notch‐1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF‐1R pathway. Oncogene 29:2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW. 2005. Growth control under stress: mTOR regulation through the REDD1‐TSC pathway. Cell Cycle 4:1500–1502. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch‐Cieszynska A, Aebersold R, Sonenberg N. 2001. Hierarchical phosphorylation of the translation inhibitor 4E‐BP1. Genes Dev 15:2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C. 1999. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3‐kinase signaling pathway. Genes Dev 13:3244–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesom KJ, Gampel A, Mellor H, Denton RM. 2001. Cell cycle‐dependent phosphorylation of the translational repressor eIF‐4E binding protein‐1 (4E‐BP1). Curr Biol 11:1374–1379. [DOI] [PubMed] [Google Scholar]
- Hemming ML, Elias JE, Gygi SP, Selkoe DJ. 2008. Proteomic profiling of gamma‐secretase substrates and mapping of substrate requirements. PLoS Biol 6:e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert TP, Tee AR, Proud CG. 2002. The extracellular signal‐regulated kinase pathway regulates the phosphorylation of 4E‐BP1 at multiple sites. J Biol Chem 277:11591–11596. [DOI] [PubMed] [Google Scholar]
- Höckel M, Vaupel P. 2001. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93:266–276. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. 2009. Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M. 2011. Cellular IRES‐mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 10:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle AH, Baker JH, Minchinton AI. 2012. Targeting quiescent tumor cells via oxygen and IGF‐I supplementation. Cancer Res 72:801–809. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. 2009. The insulin/Akt signaling pathway is targeted by intracellular beta‐amyloid. Mol Biol Cell 20:1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. 1996. The Drosophila phosphoinositide 3‐kinase Dp110 promotes cell growth. EMBO J 15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Liang S, Galluzzo P, Sobol A, Skucha S, Rambo B, Bocchetta M. 2012. Multimodality approaches to treat hypoxic Non‐Small Cell Lung Cancer (NSCLC) microenvironment. Genes Cancer 3:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R. 1997. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16:64–67. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. 1993. Mice carrying null mutations of the genes encoding insulin‐like growth factor I (Igf‐1) and type 1 IGF receptor (Igf1r). Cell 75:59–72. [PubMed] [Google Scholar]
- Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, Xu RX, Bagnard D, Schachner M, Furley AJ, Karagogeos D, Watanabe K, Dawe GS, Xiao ZC. 2008. A TAG1‐APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol 10:283–294. [DOI] [PubMed] [Google Scholar]
- McCarthy JV, Twomey C, Wujek P. 2009. Presenilin‐dependent regulated intramembrane proteolysis and gamma‐secretase activity. Cell Mol Life Sci 66:1534–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller UC, Zheng H. 2012. Physiological functions of APP family proteins. Cold Spring Harb Perspect Med 2:a006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Schrötter A, Loosse C, Pfeiffer K, Theiss C, Kauth M, Meyer HE, Marcus K. 2013. A ternary complex consisting of AICD, FE65, and TIP60 down‐regulates Stathmin1. Biochim Biophys Acta 1834:387–394. [DOI] [PubMed] [Google Scholar]
- Niemi NM, Sacoman JL, Westrate LM, Gaither LA, Lanning NJ, Martin KR, Mackeigan JP. 2014. The pseudophosphatase MK‐STYX physically and genetically interacts with the mitochondrial phosphatase PTPMT1. PLoS One 9:e93896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizzari M, Venezia V, Repetto E, Caorsi V, Magrassi R, Gagliani MC, Carlo P, Florio T, Schettini G, Tacchetti C, Russo T, Diaspro A, Russo C. 2007. Amyloid precursor protein and Presenilin1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling. J Biol Chem 282:13833–13844. [DOI] [PubMed] [Google Scholar]
- Pallares‐Cartes C, Cakan‐Akdogan G, Teleman AA. 2012. Tissue‐specific coupling between insulin/IGF and TORC1 signaling via PRAS40 in Drosophila. Dev Cell 22:172–182. [DOI] [PubMed] [Google Scholar]
- Palmer GM, Fontanella AN, Zhang G, Hanna G, Fraser CL, Dewhirst MW. 2010. Optical imaging of tumor hypoxia dynamics. J Biomed Opt 15:066021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiterer V, Fey D, Kolch W, Kholodenko BN, Farhan H. 2013. Pseudophosphatase STYX modulates cell‐fate decisions and cell migration by spatiotemporal regulation of ERK1/2. Proc Natl Acad Sci USA 110:E2934–E2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo C, Dolcini V, Salis S, Venezia V, Zambrano N, Russo T, Schettini G. 2002. Signal transduction through tyrosine‐phosphorylated C‐terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. J Biol Chem 277:35282–35288. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. 2007. PRAS40 is an insulin‐regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25:903–915. [DOI] [PubMed] [Google Scholar]
- Saqcena M, Menon D, Patel D, Mukhopadhyay S, Chow V, Foster DA. 2013. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS One 8:e74157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168. [DOI] [PubMed] [Google Scholar]
- Schütz P, Bumann M, Oberholzer AE, Bieniossek C, Trachsel H, Altmann M, Baumann U. 2008. Crystal structure of the yeast eIF4A‐eIF4G complex: An RNA‐helicase controlled by protein‐protein interactions. Proc Natl Acad Sci USA 105:9564–9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, Solit DB, Rosen N. 2010. 4E‐BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 18:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, Willis AE. 2010. Translational regulation of gene expression during conditions of cell stress. Mol Cell 40:228–237. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1‐mediated regulation of mRNA translation. Nature 485:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tujebajeva RM, Graifer DM, Karpova GG, Ajtkhozhina NA. 1989. Alkaloid homoharringtonine inhibits polypeptide chain elongation on human ribosomes on the step of peptide bond formation. FEBS Lett 257:254–256. [DOI] [PubMed] [Google Scholar]
- Varnum‐Finney B, Wu L, Yu M, Brashem‐Stein C, Staats S, Flowers D, Griffin JD, Bernstein ID. 2000. Immobilization of Notch ligand, Delta‐1, is required for induction of notch signaling. J Cell Sci 113:4313–4318. [DOI] [PubMed] [Google Scholar]
- Villalonga P, Fernández de Mattos, Ridley S. 2009. RhoE inhibits 4E‐BP1 phosphorylation and eIF4E function impairing cap‐dependent translation. J Biol Chem 284:35287–35296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Okuda H, Watabe M, Kobayashi A, Pai SK, Liu W, Xing F, Okuda H, Watabe M, Kobayashi A, Pai SK, Liu W, Pandey PR, Fukuda K, Hirota S, Sugai T, Wakabayshi G, Koeda K, Kashiwaba M, Suzuki K, Chiba T, Endo M, Mo YY, Watabe K. 2011. Hypoxia‐induced Jagged2 promotes breast cancer metastasis and self‐renewal of cancer stem‐like cells. Oncogene 30:4075–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon ES, Vigouroux S, Rousseau RF, Biagi E, Amrolia P, Dotti G, Wagner HJ, Brenner MK. 2003. Overexpression of the Notch ligand, Jagged‐1, induces alloantigen‐specific human regulatory T cells. Blood 102:3815–3821. [DOI] [PubMed] [Google Scholar]
- Zou J, Li P, Lu F, Liu N, Dai J, Ye J, Qu X, Sun X, Ma D, Park J, Ji C. 2013. Notch1 is required for hypoxia‐induced proliferation, invasion and chemoresistance of T‐cell acute lymphoblastic leukemia cells. J Hematol Oncol 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Figure S4.
Supplementary Figure S5.
Supplementary Figure S6.
Supporting Information.