

Abstract
AIM: To comprehensively understand the underlying molecular events accounting for aberrant Wnt signaling activation in hepatocellular carcinoma (HCC).
METHODS: This study was retrospective. The HCC tissue specimens used in this research were obtained from patients who underwent liver surgery. The Catalogue of Somatic Mutations in Cancer (COSMIC) database was searched for the mutation statuses of CTNNB1, TP53, and protein degradation regulator genes of CTNNB1. Dual-luciferase reporter assay was performed with TOP/FOP reporters to detect whether TP53 gain-of-function (GOF) mutations could enhance the transcriptional activity of Wnt signaling. Methylation sensitive restriction enzyme-quantitative PCR was used to explore the methylation status of CpG islands located in the promoters of APC, SFRP1, and SFRP5 in HCCs with different risk factors. Finally, nested-reverse transcription PCR was performed to examine the integration of HBx in front of LINE1 element and the existence of HBx-LINE1 chimeric transcript in Hepatitis B virus-related HCC. All results in this article were analyzed with the software SPSS version 19.0 for Windows, and different groups were compared by χ2 test as appropriate.
RESULTS: Based on the data from COSMIC database, compared with other solid tumors, mutation frequency of CTNNB1 was significantly higher in HCC (P < 0.01). The rate of CTNNB1 mutation was significantly less frequent in Hepatitis B virus-related HCC than in other etiologies (P < 0.01). Dual-luciferase reporter system and TOP/FOP reporter assays confirmed that TP53 GOF mutants were able to enhance the transcriptional ability of Wnt signaling. An exclusive relationship between the status of TP53 and CTNNB1 mutations was observed. However, according to the COSMIC database, TP53 GOF mutation is rare in HCC, which indicates that TP53 GOF mutation is not a reason for the aberrant activation of Wnt signaling in HCC. APC and AXIN1 were mutated in HCC. By using methylation sensitive restriction enzyme-quantitative PCR, hypermethylation of APC was detected in HCC with different risk factors, whereas SFRP1 and SFRP5 were not hypermethylated in any of the HCC etiologies, which indicates that the mutation of APC and AXIN1, together with the methylation of APC could take part in the overactivation of Wnt signaling. Nested-reverse transcription PCR failed to detect the integration of HBx before the LINE1 element, or the existence of an HBx-LINE1 chimeric transcript, suggesting that integration could not play a role in the aberrant activation of Wnt signaling in HCC.
CONCLUSION: In HCC, genetic/epigenetic aberration of CTNNB1 and its protein degradation regulators are the major cause of Wnt signaling overactivation.
Keywords: β-catenin, CTNNB1, Hepatitis B virus, Hepatocellular carcinoma, TP53, Wnt signaling
Core tip: Abnormal activation of Wnt/β-catenin signaling can be detected in approximately 50%-70% of hepatocellular carcinoma (HCC). It is necessary to take the analysis about the cause of Wnt/β-catenin signaling pathway aberration with the etiologic differences into consideration. In this review, the suggested genetic/epigenetic aberrations and their involvement in the abnormal Wnt/β-catenin overactivation in HCC were comprehensively analyzed, with focus on the cause of hepatitis B virus-related HCC. We suggest that genetic/epigenetic aberration of CTNNB1 and its protein degradation regulators are the major cause of Wnt signaling overactivation. TP53 gain-of-function mutation is seldom involved, and HBx-LINE1 chimeric transcripts created by viral integration may not be present.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and the third most deathly human malignancy[1]. The leading causative factors of HCC include chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, exposure to aflatoxin-contaminated food and alcohol consumption. Chronic HBV carriers have a 5-15-fold increased risk of HCC compared with the general population[2]. China alone accounts for more than half of the world’s annually diagnosed new HCC cases, predominately due to high prevalence of HBV infection and consequent cirrhosis[3].
The development of HCC is a multistage process, during which numerous genetic/epigenetic abnormalities are involved. The Wnt/β-catenin signaling pathway, which plays key roles in development and adult tissue homeostasis, is highly conserved throughout evolution. Abnormal activation of this pathway could induce different diseases, especially tumors[4]. When Wnt signaling is activated, canonical Wnt signals are transduced through Frizzled family receptors and LRP5/LRP6 co-receptors located on the cell membrane, initiating the β-catenin signaling cascade[5]. β-catenin is the core component of the Wnt/β-catenin pathway and is sequestered in the cytoplasm by the “destruction complex”, which includes Axin, glycogen synthase kinase 3, and adenomatous polyposis coli (APC). This multi-protein destruction complex could target the proto-oncogene β-catenin for ubiquitin-mediated proteolysis[6,7]. Activation of the wnt signaling could prevent glycogen synthase kinase 3β (GSK-3β)-mediated β-catenin degradation, leading to accumulation and nuclear translocation of β-catenin[8]. The nuclear accumulated β-catenin could then combine with T-cell factor/lymphoid enhancer factor, and thereby promote the transcription of downstream target genes, including FGF20, DKK1, WISP1, MYC, CCND1, and so on. It has been shown that 50%-70% of HCC tissues have abnormal β-catenin protein accumulation[9,10]. Furthermore, β-catenin expression, especially in poorly differentiated tumors, is an indicator of poor prognosis, as HCC patients with β-catenin positive grade III tumors have a significantly poorer prognosis[11]. Therefore, β-catenin could play important roles in the development and prognosis of HCC.
Many mechanisms have been shown to be involved in the aberrant activation of Wnt signaling. First, mutation of the β-catenin coding gene CTNNB1 causes aberrant activation of Wnt signaling in many tumors, including sporadic colorectal cancer[12], anaplastic thyroid carcinoma[13], gastric cancer[14], and HCC[15]. Second, aberration of several constitution molecules such as APC, AXIN, secreted Frizzled related protein (SFRP) 1 and SFRP5 in Wnt signaling could also affect the activation of Wnt signaling. In addition, TP53 gain-of-function (GOF) mutations were reported to activate Wnt signaling[16]. However, whether all these mechanisms play important roles in HCC, especially in the HBV-related HCC, have not been fully understood. Furthermore, a recent study reported that HBx could integrate into human genome and form an HBx-LINE1 chimeric transcript, and this transcript could activate Wnt signaling as an long noncoding RNA[17]. Through literature review and experimental detection, the possible presence of HBx-LINE1 in primary live tumor was also addressed. In this study, by integrative analysis of these potential factors, we summarize the known molecular mechanisms of aberrant activation of Wnt signaling in HCC.
The aim of the study was to understand the underlying molecular events accounting for aberrant Wnt signaling activation in HCC, particularly in the HCC with background of chronic HBV infection.
MATERIALS AND METHODS
Database resources
Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/) was searched for summarizing the mutation statuses of CTNNB1, APC, AXIN1, AXIN2, and TP53. TP53 GOF mutants were defined including S127Y, P151S, R156P, Y163N, Y163C, V173L, R175H, C176Y, H179R, L194R, Y205C, H214R, Y220C, Y234C, M237I, S241F, G245C, G245S, G245V, G245D, R248W, R248G, R248Q, R273C, R273L, R273H, R273P, C275Y, D281G, and R282W, as suggested[16]. For investigating the link between gene mutation and etiology, we also consulted the articles referred by the COSMIC database, and determined the etiology of each case. The mutation data of CTNNB1, AXIN1, and TP53 is listed in Tables 1, 2, 3, 4, 5, 6, 7, 8 and 9.
Table 1.
CTNNB1 mutation case resources in each article
| PubMed ID | Total cases | HBV infection cases | HCV infection cases | HBV and HCV coinfection cases | Non-viral infection cases |
| 9635572 | 14/75 | NA/NA | NA/NA | NA/NA | NA/NA |
| 9671767 | 8/31 | NA/NA | NA/NA | NA/NA | NA/NA |
| 10487827 | 14/32 | 2/5 | 2/13 | 0/0 | 10/13 |
| 10595907 | 13/22 | 0/0 | 13/22 | 0/0 | 0/0 |
| 10665646 | 9/38 | NA/NA | NA/NA | NA/NA | NA/NA |
| 10700176 | 13/100 | NA/NA | NA/NA | NA/NA | NA/NA |
| 10980116 | 57/421 | 27/265 | 23/95 | 3/44 | 4/17 |
| 11282485 | 6/32 | NA/NA | NA/NA | NA/NA | NA/NA |
| 11375957 | 26/137 | 3/33 | 12/31 | 0/9 | 11/41 |
| 11429783 | 0/22 | 0/0 | 0/0 | 0/0 | 0/22 |
| 11443619 | 7/60 | 5/48 | 0/2 | 0/0 | 2/10 |
| 11477549 | 1/14 | 0/1 | 1/12 | 0/0 | 0/1 |
| 11570580 | 15/34 | NA/NA | NA/NA | NA/NA | NA/NA |
| 12101426 | 14/73 | NA/NA | NA/NA | NA/NA | NA/NA |
| 12375019 | 10/57 | 0/0 | 10/57 | 0/0 | 0/0 |
| 12439747 | 184/1123 | NA/NA | NA/NA | NA/NA | NA/NA |
| 12845670 | 24/100 | 5/26 | 16/51 | 0/0 | 3/23 |
| 14999698 | 3/61 | 0/29 | 0/7 | 0/1 | 0/0 |
| 15067328 | 10/89 | NA/NA | NA/NA | NA/NA | NA/NA |
| 15151624 | 1/61 | NA/NA | NA/NA | NA/NA | NA/NA |
| 15288479 | 2/16 | 0/0 | 0/4 | 0/0 | 2/6 |
| 15305374 | 44/781 | NA/512 | NA/151 | NA/52 | NA/66 |
| 15814635 | 37/265 | NA/161 | NA/53 | NA/26 | NA/25 |
| 17187432 | 34/120 | NA/NA | NA/NA | NA/NA | NA/NA |
| 17393110 | 7/29 | NA/NA | NA/NA | NA/NA | NA/NA |
| 17510384 | 13/81 | 2/13 | 8/44 | 1/2 | 2/18 |
| 17531558 | 4/42 | 0/7 | 4/23 | 0/3 | 0/9 |
| 18171349 | 1/36 | 0/21 | 0/4 | 0/0 | 1/11 |
| 18282277 | 11/52 | NA/NA | NA/NA | NA/NA | NA/NA |
| 18358501 | 4/54 | 3/43 | 0/1 | 0/2 | 1/8 |
| 18467159 | 47/223 | NA/NA | NA/NA | NA/NA | NA/NA |
| 18701503 | 28/81 | 0/0 | 28/81 | 0/0 | 0/0 |
| 19101982 | 10/32 | 0/0 | 3/6 | 0/0 | 7/26 |
| 20347502 | 1/1 | 0/0 | 1/1 | 0/0 | 0/0 |
| 20923573 | 7/15 | 1/4 | 4/10 | 0/0 | 2/2 |
| 20963515 | 2/20 | 2/20 | 0/0 | 0/0 | 0/0 |
| 21457159 | 19/44 | 3/11 | 10/23 | 1/2 | 5/8 |
| 21822264 | 28/139 | 5/50 | 13/43 | 1/2 | 9/44 |
| 22561517 | 42/149 | 3/32 | 7/24 | 1/4 | 31/89 |
| 25021421 | 53/176 | 13/78 | NA/NA | NA/NA | NA/NA |
Data are presented as the number of mutation cases/category total. HBV: Hepatitis B virus; HCV: Hepatitis C virus; NA: Not available.
Table 2.
AXIN1 mutation case resources in each article
| PubMed ID | Total cases | HBV infection cases | HCV infection cases | HBV and HCV coinfection cases | Non-viral infection cases |
| 10700176 | 9/100 | NA/NA | NA/NA | NA/NA | NA/NA |
| 11375957 | 12/112 | 8/32 | 0/31 | 2/9 | 2/40 |
| 12101426 | 4/56 | 0/9 | 0/5 | 0/2 | 4/40 |
| 15067328 | 25/89 | NA/NA | NA/NA | NA/NA | NA/NA |
| 18171349 | 9/36 | 5/21 | 3/4 | 0/0 | 1/11 |
| 21499249 | 1/1 | 0/0 | 1/1 | 0/0 | 0/0 |
| 22561517 | 19/149 | 5/32 | 5/24 | 1/4 | 8/89 |
| 25021421 | 25/176 | 13/78 | NA/NA | NA/NA | NA/NA |
Data are presented as the number of mutation cases/category total. HBV: Hepatitis B virus; HCV: Hepatitis C virus; NA: Not available.
Table 3.
TP53 mutation case resources in each article
| PubMed ID | Total cases | HBV infection cases | HCV infection cases | HBV and HCV coinfection cases | Non-viral infection cases |
| 1311638 | 0/21/36 | 0/21/36 | 0/0/0 | 0/0/0 | 0/0/0 |
| 1327523 | 3/20/61 | 3/12/37 | 0/4/17 | 0/3/4 | 0/1/3 |
| 1655254 | 2/9/43 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 1672732 | 0/5/10 | 0/5/8 | 0/0/NA | 0/0/NA | 0/0/NA |
| 1849234 | 0/8/16 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 7903205 | 2/20/35 | 2/15/28 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8093350 | 1/17/53 | 0/3/7 | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 8093978 | 0/3/20 | 0/NA/17 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8100480 | 1/3/20 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 8108145 | 2/6/15 | 2/6/15 | 0/0/0 | 0/0/0 | 0/0/0 |
| 8261444 | 5/12/16 | 1/2/3 | 4/8/10 | 0/1/2 | 0/1/1 |
| 8290606 | 0/19/80 | 0/8/28 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8302580 | 1/6/22 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 8380058 | 0/2/15 | 0/NA/NA | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8382111 | 0/12/38 | 0/NA/NA | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8384081 | 0/4/45 | 0/4/28 | 0/0/11 | 0/0/2 | 0/0/4 |
| 8389246 | 0/11/34 | 0/NA/NA | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8390289 | 0/10/15 | 0/8/13 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8390407 | 0/1/18 | 0/0/3 | 0/0/5 | 0/0/1 | 0/1/9 |
| 8393166 | 1/1/7 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 8407553 | 3/19/63 | 2/10/16 | 1/4/15 | 0/0/0 | 0/5/32 |
| 8565124 | 0/2/12 | 0/0/3 | 0/0/2 | 0/0/1 | 0/2/6 |
| 8655704 | 3/10/38 | NA/NA/19 | NA/NA/10 | NA/NA/5 | NA/NA/4 |
| 8655958 | 2/2/20 | 1/1/4 | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 8672994 | 0/1/18 | 0/NA/NA | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 8895490 | 0/9/12 | 0/0/0 | 0/1/3 | 0/2/3 | 0/3/6 |
| 9012469 | 0/8/16 | 0/1/8 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 9270015 | 2/26/105 | 2/24/78 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 9463584 | 0/2/8 | 0/NA/NA | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 9699537 | 2/6/97 | NA/NA/16 | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 9781942 | 1/2/15 | 0/0/0 | 1/2/15 | 0/0/0 | 0/0/0 |
| 10389750 | 0/17/31 | 0/11/17 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 10389978 | 0/12/24 | 0/10/19 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 10564952 | 2/9/11 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 10662591 | 0/5/27 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 10699891 | 4/11/18 | 0/0/0 | 0/0/0 | 0/0/0 | 4/11/18 |
| 10743047 | 0/35/40 | 0/33/38 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 11051249 | 2/21/83 | 0/14/50 | 1/6/16 | 0/0/0 | 1/1/17 |
| 11191353 | 0/14/55 | 0/NA/45 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 11282486 | 2/6/22 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 11375957 | 1/36/137 | 1/15/33 | 0/10/31 | 0/3/9 | 0/8/41 |
| 11704835 | 8/30/71 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 12483005 | 3/7/34 | 2/5/27 | 0/1/3 | 0/0/0 | 1/1/4 |
| 12640682 | 0/2/18 | 0/1/5 | 0/1/8 | 0/0/4 | 0/0/1 |
| 12759240 | 2/16/33 | 2/16/33 | 0/0/0 | 0/0/0 | 0/0/0 |
| 12845670 | 0/14/100 | 0/4/26 | 0/7/51 | 0/0/0 | 0/3/23 |
| 14499690 | 0/18/68 | 0/NA/52 | 0/NA/2 | 0/0/0 | 0/NA/14 |
| 14675778 | 0/5/22 | 0/0/0 | 0/5/22 | 0/0/0 | 0/0/0 |
| 14687797 | 3/9/80 | 2/7/NA | 0/1/NA | 0/0/NA | 1/1/NA |
| 15017592 | 4/22/50 | NA/NA/13 | NA/NA/33 | NA/NA/3 | NA/NA/1 |
| 15126338 | 3/27/48 | NA/NA/NA | NA/NA/NA | NA/NA/NA | NA/NA/NA |
| 15943041 | 8/8/20 | 8/8/20 | 0/0/0 | 0/0/0 | 0/0/0 |
| 16078640 | 2/7/41 | NA/NA/1 | NA/NA/26 | NA/NA/NA | NA/NA/NA |
| 16570275 | 1/11/40 | 1/4/22 | 0/2/5 | 0/2/5 | 0/3/8 |
| 16685387 | 0/28/55 | 0/NA/49 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 16697535 | 0/7/23 | 0/7/23 | 0/0/0 | 0/0/0 | 0/0/0 |
| 17066440 | 0/5/51 | 0/0/14 | 0/4/30 | 0/0/0 | 0/1/7 |
| 17266182 | 1/28/50 | 1/24/41 | 0/NA/NA | 0/NA/NA | 0/NA/NA |
| 17350822 | 4/16/83 | NA/2/13 | NA/13/64 | NA/0/2 | NA/1/4 |
| 17510384 | 0/25/81 | 0/5/13 | 0/15/44 | 0/2/2 | 0/3/18 |
| 17531558 | 2/7/42 | 1/1/7 | 1/4/24 | 0/1/2 | 0/1/9 |
| 21499249 | 0/1/1 | 0/0/0 | 0/1/1 | 0/0/0 | 0/0/0 |
| 21760996 | 0/9/26 | 0/6/13 | 0/2/4 | 0/0/0 | 0/0/4 |
| 21822264 | 4/39/139 | 2/20/50 | 1/10/43 | 0/2/2 | 1/7/44 |
| 22561517 | 0/26/149 | 0/9/32 | 0/7/24 | 0/2/4 | 0/8/89 |
| 22634756 | 3/14/27 | 3/7/11 | 0/7/14 | 0/0/0 | 0/0/2 |
| 22922871 | 1/1/10 | 1/1/10 | 0/0/0 | 0/0/0 | 0/0/0 |
Data are presented as the number of gain-of-function mutation cases/number of mutations/category total. HBV: Hepatitis B virus; HCV: Hepatitis C virus; NA: Not available.
Table 4.
Mutation case numbers and rates of CTNNB1 in different tissues
| Cancer type | Mutation cases (n) | Total cases (n) | Mutation rate (%) | P value1 |
| HCC | 671 | 3720 | 18.04 | |
| Colon Adenocarcinoma | 126 | 1318 | 9.56 | < 0.001 |
| Ovary carcinoma | 102 | 1599 | 6.38 | < 0.001 |
| Stomach Adenocarcinoma | 54 | 1137 | 4.75 | < 0.001 |
| Lung Adenocarcinoma | 38 | 1219 | 3.12 | < 0.001 |
| ESCC | 5 | 347 | 1.44 | < 0.001 |
The P value vs HCC. ESCC: Esophageal squamous cell carcinoma; HCC: Hepatocellular carcinoma.
Table 5.
Mutation case numbers and rates of CTNNB1 in hepatocellular carcinoma etiologies
| Viral background | Mutation cases (n) | Total cases (n) | Mutation rate (%) | P value1 |
| HBV | 74 | 686 | 10.79 | |
| HCV | 155 | 554 | 27.98 | < 0.001 |
| HBV and HCV | 7 | 69 | 10.14 | 0.999 |
| Non-viral | 90 | 348 | 25.86 | < 0.001 |
The P value vs HBV. HBV: Hepatitis B virus; HCV: Hepatitis C virus.
Table 6.
Mutation case numbers and rates of AXIN1 in different tissues
| Cancer type | Mutation cases (n) | Total cases (n) | Mutation rate (%) |
| Hepatocellular carcinoma | 75 | 872 | 8.60 |
| Colon adenocarcinoma | 20 | 513 | 3.90 |
| Ovary carcinoma | 3 | 749 | 0.40 |
| Stomach adenocarcinoma | 18 | 377 | 4.77 |
| Lung adenocarcinoma | 6 | 697 | 0.86 |
| Esophageal squamous cell carcinoma | 3 | 130 | 2.31 |
Table 7.
Mutation case numbers and rates of AXIN1 in hepatocellular carcinoma etiologies
| Viral background | Mutation cases (n) | Total cases (n) | Mutation rate (%) | P value1 |
| HBV | 31 | 172 | 18.02 | |
| HCV | 9 | 65 | 13.85 | 0.964 |
| HBV and HCV | 3 | 15 | 20.00 | 0.999 |
| non-viral | 15 | 180 | 8.33 | 0.122 |
The P value vs HBV. HBV: Hepatitis B virus; HCV: Hepatitis C virus.
Table 8.
Mutation case numbers and rates of TP53 in different tissues
| Cancer type | GOF mutation cases (n) | Mutation cases (n) | Total cases (n) | GOF mutation rate (%) | Non-GOF mutation rate (%) | Total mutation rate (%) | P value1 |
| HCC | 91 | 825 | 2813 | 3.23 | 26.09 | 29.33 | |
| Colon adenocarcinoma | 331 | 1867 | 3596 | 9.20 | 42.71 | 51.92 | < 0.001 |
| Ovary carcinoma | 392 | 1604 | 3329 | 11.78 | 36.41 | 48.18 | < 0.001 |
| Stomach adenocarcinoma | 270 | 1160 | 3459 | 7.81 | 25.73 | 33.54 | < 0.001 |
| Lung adenocarcinoma | 421 | 2414 | 6394 | 6.58 | 31.17 | 37.75 | < 0.001 |
| ESCC | 224 | 972 | 1858 | 12.06 | 40.26 | 52.31 | < 0.001 |
GOF mutation rate vs HCC. ESCC: Esophageal squamous cell carcinoma; GOF: Gain-of-function; HCC: Hepatocellular carcinoma.
Table 9.
Mutation case numbers and rates of TP53 in hepatocellular carcinoma etiologies
| Viral background | GOF mutation cases (n) | Mutation cases (n) | Total cases (n) | GOF mutation rate (%) | Non-GOF mutation rate (%) | Total mutation rate (%) |
| HBV | 35 | 321 | 819 | 4.27 | 34.92 | 39.19 |
| HCV | 9 | 101 | 398 | 2.26 | 23.12 | 25.38 |
| HBV and HCV | 0 | 18 | 41 | 0.00 | 43.90 | 43.90 |
| non-viral | 7 | 60 | 346 | 2.02 | 15.32 | 17.34 |
GOF: Gain-of-function; HBV: Hepatitis B virus; HCV: Hepatitis C virus.
HCC tissue samples
HCC tissue samples were obtained from patients who underwent routine curative surgery at Henan Oncology Hospital in Zhengzhou, Henan Province of China. This study was retrospective, and all tissues were obtained during surgeries. All patients were HBV-positive, which was indicated with serum HBsAg or HBV-DNA presence. For detecting the methylation status of APC, SFRP1, and SFRP5, seven pairs of tissue specimens from HCC patients with serum anti-HCV-positive and ten pairs of tissue specimens from HCC patients without HBV/HCV viral infection were collected. Six tumor-free tissues from patients with hepatic hemangioma were used as controls. All tissues were snap frozen in liquate nitrogen until use. The study was approved by the Ethics Committee in the university, and the informed consents were obtained from all patients and donors before the start of the study.
DNA methylation detection
DNA was extracted from tissues by digestion of frozen samples with 1% proteinase K, followed by standard phenol/chloroform and ethanol precipitation. The DNA methylation status was detected by DNA methylation-sensitive restriction endonuclease digestion, followed by subsequent quantitative (q)PCR assay as described previously[18]. In brief, 2 μg DNA was treated with HhaI methylation-sensitive enzyme at 37 °C for 16 h. HhaI can digest the GCGC sequence if the cytosine is not methylated. Then, qPCR was performed to amplify the target template with primers between which HhaI cutting sites were located. The reaction was performed in a 96-well plate on Roche Lightcyler 480II Real-Time PCR System (Roche, Basel, Switzerland). Methylation intensity was quantified between 0% and 100% by calculating Ct values of tissues treated either with or without HhaI digestion. Primers used for APC, SFRP1, and SFRP5 methylation detection are: APC, F-CGGACCAGGGCGCTCCCCATTCC and R-TGACACCCTGGCGGGCTGCACCAA; SFRP1, F-TCGCCCCGCCGGGAGCTGATTG and R-GGCTGGAGTGCGCGGGGCTCCT; SFRP5 F-CCAGTGCAGCGCCCCCAGCAGCA and R-CGCGGCGCGCACCTGGAGAG.
Reverse transcription-PCR assay
Total RNA was extracted with TRI-Reagent (Invitrogen of Thermo Fisher Scientific, Waltham, MA, United States) according to the manufacturer’s protocol, and reverse transcription was then performed using Reverse Transcription Kit (Thermo Fish Scientific). Nested-PCR was performed to detect HBx-LINE1 transcript (primers: first round: F-TCCCCGTCTGTGCCTTCTC and R-TAGTGCTGCAATAAACATGGGA; second round: F-ACGCGGTCTCCCCGTCTGT and R-GCTGGATCATATGGAAGCTCTGG). β-actin was used as the calibrator gene (primers: F-CTACAGCTTCACCACCACGG and R-TCAGGCAGCTCGTAGCTCTTC).
Plasmid construction
PcDNA3.1-TP53 expression vector was kept in our lab, which covers the coding sequence of TP53 and contains a myc tag. Using this vector as a template, three different GOF mutation sites were precisely mutated. The primers used: R273C, F-AACAGCTTTGAGGTGTGTGTTTGTGCCTGTCCTGGG and R-GACAGGCACAAACACACACCTCAAAGCTGTTCCGTC; R273H, F-AACAGCTTTGAGGTGCATGTTTGTGCCTGTCCTGGG and R-GACAGGCACAAACATGCACCTCAAAGCTGTTCCGTC; Y220C, F-GTGTGGTGGTGCCCTGTGAGCCGCCTGAGGTTGGCT and R-ACCTCAGGCGGCTCACAGGGCACCACCACACTATGT.
Luciferase reporter assays
The TOP/FOP reporter plasmids were co-transfected with Renilla luciferase vector into HEK 293T, SMMC 7721, and Huh-7 cell lines using Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, cells were washed twice with PBS and lysed in passive lysis buffer. PGL3-basic vector was used as a negative control. Luciferase activity was analyzed using a luminometer and a dual luciferase assay kit according to the manufacturer (Promega Corp., Madison, WI, United States). Luciferase counts were normalized using Tk-Renilla-luciferase (Promega).
Statistical analysis
All analyses were performed with the software SPSS version 19.0 (IBM, Armonk, NY, United States). Different groups were compared by χ2 tests as appropriate. All statistical tests were two-sided, and P < 0.01 was considered as statistically significant.
RESULTS
CTNNB1 mutation is a major causative factor of aberrant Wnt signaling activation in HCC
CTNNB1 mutation, especially mutation at the phosphorylation sites in N-terminal domain, could affect β-catenin protein stability and its combining capability with APC and AXIN. Firstly, we summarized the mutation frequency of CTNNB1 in different tumors, including HCC, stomach, lung, ovary, colon tumor and esophageal squamous cell carcinoma, based on the information collected from COSMIC database. As shown in Figure 1A, the CTNNB1 mutation rate in HCC was 18.04%, which was significantly higher than the mutation rates in other tumors (Table 4), implicating that CTNNB1 mutation could be one of the major reasons for aberrant activation of Wnt signaling commonly seen in HCC. Next, the HCC patients were classified into different groups according to the background of viral infection, and then we compared the rates of CTNNB1 mutation among different HCC groups. As shown in Figure 1B, the CTNNB1 mutation rate in chronic HBV-related HCC was 10.79%, which was similar to that in HBV/HCV coinfection-related HCC, but significantly lower than those with HCV-related HCC or non-viral HCC (Table 5). Nevertheless, the CTNNB1 mutation rate in chronic HBV-related HCC was still higher than that in several other human tumors such as esophageal squamous cell carcinoma, lung cancer, and gastric cancer.
Figure 1.
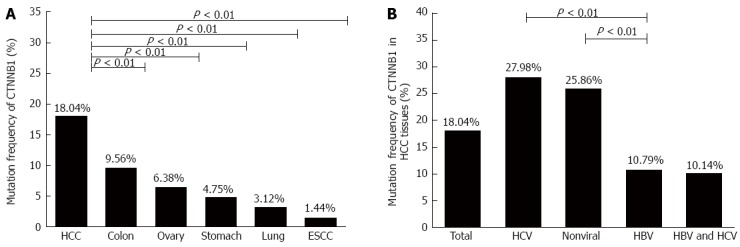
CTNNB1 mutation rate in tumors based on the information collected from COSMIC database. CTNNB1 mutation rates in A: Different human tumors; and B: Hepatocellular carcinoma tissues with different risk factors. ESCC: Esophageal squamous cell carcinoma; HBV: Hepatitis B virus; HCV: Hepatitis C virus; HCC: Hepatocellular carcinoma.
TP53 GOF mutants may not contribute to aberrant Wnt signaling activation in HCC
Tumor suppressor gene TP53 is the most frequently mutated gene in cancer. Among these p53 mutants, some mutations not only lose the tumor suppressive functions, but also gain novel oncogenic activities, including promotion of tumor cell proliferation, survival, metabolic changes, angiogenesis, and metastasis, which were defined as p53 GOF activities[19]. It has been reported that β-catenin expression and the Wnt signaling pathway are highly activated in tumors harboring GOF p53 mutants[16]. To investigate whether TP53 GOF mutant could activate the Wnt signaling pathway in HCC, we performed a TOP/FOP luciferase assay by overexpressing Y220C, R273C, and R273H TP53 GOF mutants in HEK 293T, SMMC7721, and Huh-7 cell lines. Compared with wild-type p53, Y220C enhanced the TOP/FOP value in Huh-7 cells, whereas R273H and R273C enhanced the TOP/FOP value in SMMC 7721 cells. As a positive control, all three GOF mutations enhanced TOP/FOP values in HEK 293T cells (Figure 2A). The effect of p53 GOF mutants in those hepatic origin cells was insignificant and inconsistent. Consistent with the results of the in vitro analysis, searching of the COSMIC database revealed that, though the total TP53 mutation rate in HCC was as high as 29.33%, the rate of TP53 GOF mutation in HCCs was only 4.27%, substantially lower than that observed in other tumors (Figure 2B and Table 8). Furthermore, such a low rate of TP53 GOF mutation was constantly present among HCCs of different etiologies (Figure 2B). Taken together, although TP53 GOF mutants activate Wnt signaling in HCC cell lines in vitro, they seldom occur in HCC and likely do play a major role in aberrant activation of Wnt signaling commonly present in HCC.
Figure 2.
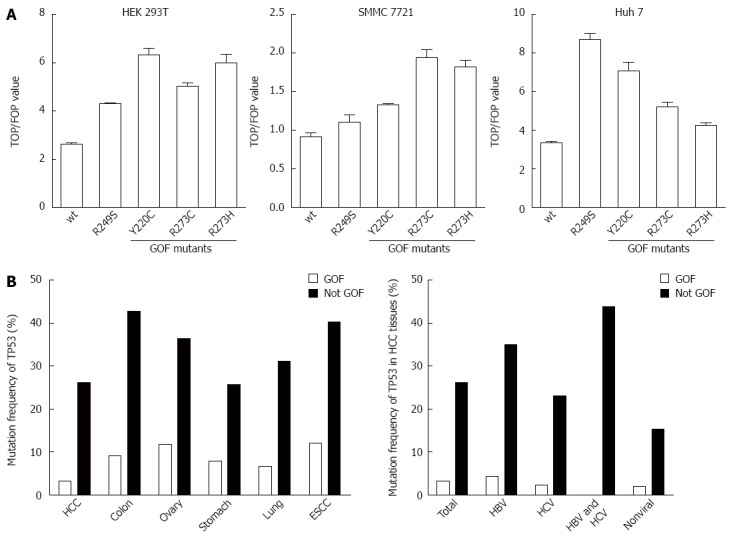
TP53 GOF mutation does not likely contribute to the aberrant activation of Wnt signaling. A: TP53 GOF mutants activate Wnt signaling in vitro; B: TP53 mutation rate in different tumors and different etiologies of HCC. Although TP53 mutation was commonly detected in HCC, GOF mutation was a rare event in HCC with different etiologies. ESCC: Esophageal squamous cell carcinoma; GOF: Gain-of-function; HBV: Hepatitis B virus; HCV: Hepatitis C virus; HCC: Hepatocellular carcinoma.
Frequent genetic/epigenetic aberrations in negative regulators involved in Wnt signaling activation in HCC
Since APC and AXINs can form a degradation complex with GSK-3β to prompt the ubiquitination-dependent degradation of β-catenin protein, aberration of either APC or AXINs might affect the activity of the Wnt/β-catenin signaling pathway. Based on the COSMIC database, the mutation rates of AXIN1, AXIN2, and APC in HCC were 8.60%, 0.42%, and 1.33%, respectively (Figure 3A). The high frequent mutation rate of AXIN1 suggests that it commonly contributes to the aberrant Wnt signaling activation in HCC.
Figure 3.
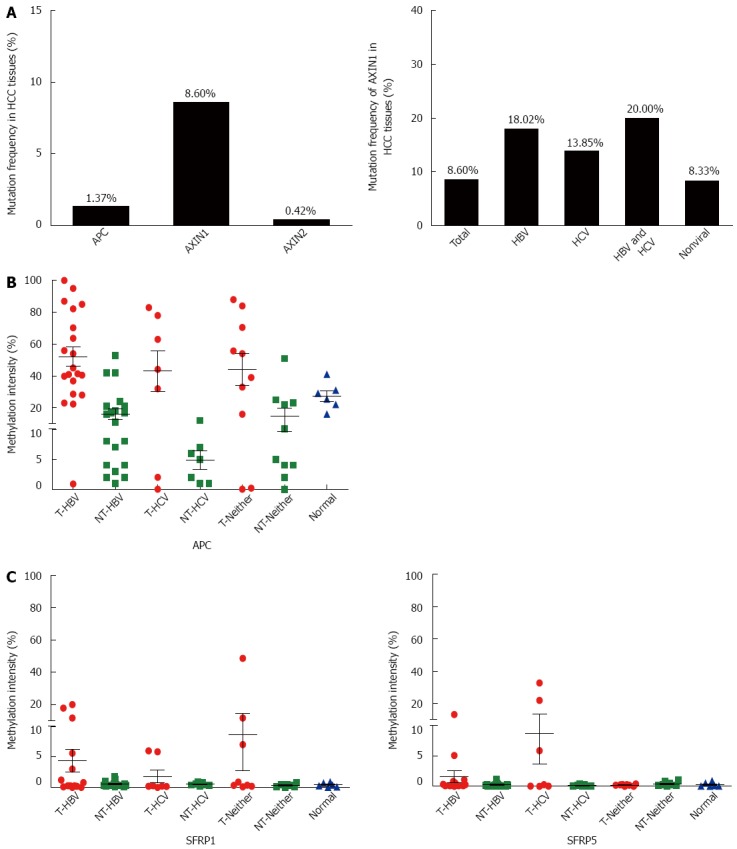
Frequent genetic/epigenetic aberrations of the negative regulators in Wnt signaling in hepatocellular carcinoma. A: AXIN1 was frequently mutated in HCCs with different etiologies; B: APC promoter hypermethylation was frequently found in HCCs with different etiologies; C: Neither SFRP1 nor SFRP5 was frequently hypermethylated in HCCs with different risk factors. HBV: Hepatitis B virus; HCV: Hepatitis C virus; HCC: Hepatocellular carcinoma.
SFRP1 and SFRP5 encode SFRP, which is the antagonist of the Wnt signaling pathway. The epigenetic downregulation of SFRP has been shown to be involved in hepatocarcinogenesis[20]. We previously demonstrated that the promoter CpG island of APC is hypermethylated in HCC with HBV infection[21]. To further demonstrate the involvement of the negative regulators in Wnt/β-catenin overactivation in HCC, methylation sensitive restriction enzyme-qPCR was performed to detect the methylation statuses of APC, SFRP1, and SFRP5 in HCC with different risk factors, including between HCV-related HCC and those without viral infection. The results showed extensive hypermethylation of the APC promoter in all HCC of different etiologies (Figure 3B). As shown in Figure 3C, hypermethylation of SFRP1 was present in 15% (3/20) of HBV-infected tumor tissues, 0% (0/7) of HCV-infected tumor tissues, 20% (2/10) of non-infected tumor tissues, and 0% (0/6) of hepatic hemangioma tissues, whereas hypermethylation of SFRP5 was present in 5% (1/20) of HBV-infected tumor tissues, 28.6% (2/7) of HCV-infected tumor tissues, 0% (0/10) of non-infected tumor tissues, and 0% (0/6) of hepatic hemangioma tissues. These results suggest that SFRP1 and SFRP5 are not primary causes of aberrant Wnt signaling activation in HCCs.
HBx-LINE1 transcripts are not detected in tissues of HBV-related HCC
A recent report described that HBx frequently forms a chimeric transcript (HBx-LINE1) after integrating into the LINE1 element in 8p11.21 of the host genome, and the authors further suggested that HBx-LINE1 could activate the Wnt signaling pathway as a long noncoding RNA[17]. To explore whether HBx-LINE1 could take part in the aberrant activation of Wnt signaling in HBV-related HCC, the expression of this transcript was measured in up to 30 HCC tissues with a chronic HBV infection background. Unfortunately, even by nested-PCR, no HBx-LINE1 chimeric transcript was detected. To exclude the possibility that the absence was due to an experimental failure, PCR primers were designed to detect the HBx-LINE1 viral-host junction sequences at the DNA level. Still, no HBx-LINE1 integration was detected (data not shown). Moreover, we carefully reviewed the available viral integration information from published data. In a total of 1115 HBV integration sites derived from 299 HBV-HCC patients, only one was found mapped at chromosome 8p11.21. However, further precise analysis excluded the possibility to form an HBx-LINE1 transcript[22], because the integration site was away from the LINE1 site. Collectively, HBx was not expected to exactly integrate at such an accurate site to form the HBx-LINE1 chimeric transcript, at least not at the high frequency as described by the report[17]. As a result, HBx-LINE1, the suggested viral-host junction transcript, may not commonly present in HBV-infection related HCCs.
DISCUSSION
Abnormal activation of Wnt/β-catenin signaling is detected in 50%-70% of HCC cases, making it the most common signaling pathway aberration in this cancer[10]. However, it is necessary to analyze the cause of Wnt/β-catenin signaling pathway aberration while considering the different etiologic causes of HCC. In the present study, we summarized all the suggested factors relevant to the aberrant activation of the Wnt signaling pathway in HCC with different causative etiologies. Those genetic/epigenetic events include CTNNB1 gene mutation, TP53 GOF mutation, the presence of an HBx-LINE1 chimeric transcript, and aberrations of other genes within this signaling pathway.
CTNNB1, which encodes β-catenin, has been recognized as one of the most frequently mutated genes in primary HCC. Indeed, according to the COSMIC database, the mutation of CTNNB1 was detected in 671/3720 HCC cases, with detailed mutation information provided for 543 of these cases (http://cancer.sanger.ac.uk/). It is worthwhile to note that among the 543 identified mutations, 477 were point mutations located at N-terminal domain, 309 of which were at β-catenin phosphorylation sites (serines 33, 37, and 45 and threonine 41). This result is consistent with the fact that the stability of β-catenin relies on the phosphorylation of its N-terminal domain.
The large percentage (18.04%; 671/3720) of CTNNB1 mutations makes it one of the major causative mechanisms contributing to the aberrant activation of Wnt signaling in HCC. However, the mutation rate of CTNNB1 in chronic HBV-related HCC was only 10.79%, much lower than in HCV-related cases or of other etiologic backgrounds. The lower rate of CTNNB1 mutation in HBV-related HCC has been reported previously by other laboratories, in which relatively small patient cohorts were used[1,23]. The other gene with a high frequency of mutation in HCC is AXIN1, another component of the “destruction complex”. Interestingly, in contrast to CTNNB1, the rate of AXIN1 mutation was higher in HBV-related HCC compared to HCC of other etiologic backgrounds.
The above results suggest that mutation of genes comprising the “destruction complex”, together with their target CTNNB1 mutation, are common events contributing to the altered Wnt signaling pathway activation in HCC. In addition to genetic mutations, epigenetic modulation can also cause aberrant gene expression. We previously reported that in HBV-related HCCs, the CpG islands in the promoter regions of APC and AXIN2 were frequently hypermethylated[20]. Frequent APC hypermethylation in HBV- and HCV-related HCC tissues has also been reported by Feng et al[24]. In this study, we compared the methylation status in HCC with different etiologies. However, there was no noticeable difference among HCCs with different etiologic backgrounds. Taken together, these results suggest that APC promoter hypermethylation is a causative factor for aberrant Wnt signaling activation in HCC tissues. In addition, our data and those from other laboratories suggest that SFRP methylation may not be a primary factor for aberrant Wnt signaling activation in HCC[24].
The recent report that HBx-LINE1 chimeric transcripts can activate Wnt signaling has attracted some recognition[17,25,26]. However, we doubted the possibility of such a high frequency of identical integration (present in 23% of primary HCC tissues, as described by the author[17]). As a matter of fact, the integration of HBV into the host cellular genome is generally a random event, and it is hard to imagine that HBV integrates precisely at chr.8p11.21 in close to one-quarter of HCC tumor tissues. Moreover, in order to make an HBx-LINE1 junction, the HBV genome must also be broken exactly at the same site of its genome. Our lab had searched up to 1115 HBV host genome-adjacent sequences from several articles, including our data, and none of them locate at the site of the LINE1 element, which is essential for the formation of the HBx-LINE1 chimeric transcripts[27-31]. In addition, the effort to detect the presence of HBx-LINE1 chimeric transcripts by the powerful nested Reverse transcription-PCR, or PCR at the tumor genome level, failed. In addition, the previous report utilized Sanger sequencing to confirm the formation of HBx-LINE1 transcripts[17]; we noticed that the viral nucleic acid sequence was exactly the same, not even a single nucleotide variant to this high mutation-rate virus. Therefore, we had sufficient cause to doubt that HBx can accurately integrate with LINE1 with such an extremely high frequency.
Besides the above factors, some other mechanisms that can activate Wnt signaling should be further explored, such as the phosphorylation of extracellular signal-regulated kinase and protein kinase B by HBx, which may be the major mechanism for Wnt signaling activation in HBV-related HCC[32,33]. Additionally, it was reported that HBx can competitively combine with APC protein to release GSK-3β[34]. However, further experimental evidences are still needed to confirm this postulation.
Together with other previous reports, we propose that several possible mechanisms account for the aberrant Wnt signaling activation in HCC, including CTNNB1 mutation, as well as hypermethylation of APC and AXIN1 mutation. In contrast, hypermethylation-mediated silencing of SFRP1 and SFRP5 expression was not a common event. Additionally, although TP53 GOF mutations have the potential to activate Wnt signaling, they rarely occur in HCC, and therefore, it should not be counted as a causative factor of aberrant Wnt signaling overactivation in HCC. Unfortunately, our results do not support the hypothesis that HBx-LINE1 chimeric transcripts activate Wnt signaling in HCC, as none of the 1115 known HBV viral-host cellular genome junction sequences involved the LINE1 sequence, and furthermore, the HBx-LINE1 chimeric transcript was not detected at the mRNA or genomic DNA level.
As our understanding about Wnt signaling pathways continues to grow, the potential clinical value of our knowledge on Wnt signaling and HCC should be further studied.
COMMENTS
Background
The development of hepatocellular carcinoma (HCC) is a multistage process, during which numerous genetic/epigenetic factors could be involved. As one of the most important factors, the Wnt/β-catenin signaling pathway is frequently activated, about 50%-70% of HCC tissues show abnormal β-catenin protein accumulation, which predicts poor prognosis. It has become recognized that the Wnt signaling pathway plays an important role in the development and prognosis of HCC.
Research frontiers
Many mechanisms have been reported to be involved in the aberrant activation of Wnt signaling in HCC. Mutation of CTNNB1, which encodes β-catenin, could cause cytoplasmic accumulation of β-catenin. Genetic or epigenetic aberrations of several constitution molecules in the Wnt signaling pathway could also affect its activation. Furthermore, TP53 gain-of-function mutations have the ability to upregulate the expression of CTNNB1. Finally, a recent report suggested a frequent integration of HBx into LINE1 elements of the human genome and formation of HBx-LINE1 chimeric transcripts, which enhance the transcriptional activity of Wnt signaling.
Innovations and breakthroughs
Previous reports have suggested the presence of different underlying mechanisms for the aberrant activation of Wnt signaling in HCC. However, whether all these mechanisms could really take part in this process is not known. In this article, by integrative analysis of the potential factors, the involvement of the following suggested mechanisms in the aberrant activation of Wnt signaling in HCC were investigated: the mutation rate of CTNNB1, TP53, APC, AXIN1, and AXIN2 by searching in COSMIC database; and the epigenetic aberrations of the constituent molecules in Wnt signaling, such as APC, SFRP1, and SFRP5, by determining their CpG island methylation status using a methylation sensitive restriction enzyme-quantitative PCR technique developed in the laboratory. In addition, in order to judge whether HBx integration in LINE1 elements could activate Wnt signaling in HBV-related HCC, the proposed HBx-LINE integration was also examined at both the genome and RNA levels among HBV-related HCC tissue specimens by nested reverse transcription-PCR and PCR.
Applications
This integrative study provides a panoramic view of the underlying mechanisms relevant to the aberrant activation of Wnt signaling in HCC. The discovery will enhance our understanding of hepatocarcinogenesis.
Terminology
The Wnt/β-catenin pathway is highly conserved throughout evolution, and plays key roles in development in adult tissue homeostasis. β-catenin is the core component which is precisely regulated. β-catenin can be degraded by the destruction complex composed of APC, AXIN, and GSK-3β. Wnt signaling activation leads to nuclear translocation of β-catenin, where it promotes the transcription of several downstream target genes.
Peer-review
The authors present a comprehensive study. The methodology is correct. The conclusions are consistent with the results obtained. This study represents a significant contribution to advance our study on the process of hepatocarcinogenesis.
Footnotes
Supported by National Natural Science Foundation of China, No. 81372603; 973 Program, No. 2015CB554000; National S T Major Project for Infectious Diseases, No. 2012ZX10004-904; and The 111 Project, No. B07001.
Conflict-of-interest: The authors declare no conflicts of interest related to this manuscript.
Data sharing: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 22, 2014
First decision: January 8, 2015
Article in press: March 12, 2015
P- Reviewer: Carrillo MC, Montalti R S- Editor: Qi Y L- Editor: AmEditor E- Editor: Zhang DN
References
- 1.Tornesello ML, Buonaguro L, Tatangelo F, Botti G, Izzo F, Buonaguro FM. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics. 2013;102:74–83. doi: 10.1016/j.ygeno.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Wang HQ, Yang J, Yan LN, Zhang XW, Yang JY. Liver resection in hepatitis B related-hepatocellular carcinoma: clinical outcomes and safety in elderly patients. World J Gastroenterol. 2014;20:6620–6625. doi: 10.3748/wjg.v20.i21.6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 5.Katoh M, Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 2007;13:4042–4045. doi: 10.1158/1078-0432.CCR-06-2316. [DOI] [PubMed] [Google Scholar]
- 6.Yamulla RJ, Kane EG, Moody AE, Politi KA, Lock NE, Foley AV, Roberts DM. Testing models of the APC tumor suppressor/β-catenin interaction reshapes our view of the destruction complex in Wnt signaling. Genetics. 2014;197:1285–1302. doi: 10.1534/genetics.114.166496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song X, Wang S, Li L. New insights into the regulation of Axin function in canonical Wnt signaling pathway. Protein Cell. 2014;5:186–193. doi: 10.1007/s13238-014-0019-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reuter S, Martin H, Beckert H, Bros M, Montermann E, Belz C, Heinz A, Ohngemach S, Sahin U, Stassen M, et al. The Wnt/β-catenin pathway attenuates experimental allergic airway disease. J Immunol. 2014;193:485–495. doi: 10.4049/jimmunol.1400013. [DOI] [PubMed] [Google Scholar]
- 9.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki T, Yano H, Nakashima Y, Nakashima O, Kojiro M. Beta-catenin expression in hepatocellular carcinoma: a possible participation of beta-catenin in the dedifferentiation process. J Gastroenterol Hepatol. 2002;17:994–1000. doi: 10.1046/j.1440-1746.2002.02774.x. [DOI] [PubMed] [Google Scholar]
- 11.Inagawa S, Itabashi M, Adachi S, Kawamoto T, Hori M, Shimazaki J, Yoshimi F, Fukao K. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Clin Cancer Res. 2002;8:450–456. [PubMed] [Google Scholar]
- 12.Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999;59:1811–1815. [PubMed] [Google Scholar]
- 14.Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C, Groden J, Lowy AM. beta-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 2002;62:3503–3506. [PubMed] [Google Scholar]
- 15.Wong CM, Fan ST, Ng IO. beta-Catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136–145. doi: 10.1002/1097-0142(20010701)92:1<136::aid-cncr1301>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 16.Kang HJ, Chun SM, Kim KR, Sohn I, Sung CO. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS One. 2013;8:e72609. doi: 10.1371/journal.pone.0072609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, Co NN, Chan AW, Li PS, Lung RW, et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell. 2014;25:335–349. doi: 10.1016/j.ccr.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Cheng J, Xu C, Liu S, Jiang S, Xu Q, Chen X, Zhuang H, Lu F. Quantitative methylation analysis reveals gender and age differences in p16INK4a hypermethylation in hepatitis B virus-related hepatocellular carcinoma. Liver Int. 2012;32:420–428. doi: 10.1111/j.1478-3231.2011.02696.x. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Zhang C, Feng Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim Biophys Sin (Shanghai) 2014;46:170–179. doi: 10.1093/abbs/gmt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takagi H, Sasaki S, Suzuki H, Toyota M, Maruyama R, Nojima M, Yamamoto H, Omata M, Tokino T, Imai K, et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J Gastroenterol. 2008;43:378–389. doi: 10.1007/s00535-008-2170-0. [DOI] [PubMed] [Google Scholar]
- 21.Liu S, Cheng J, Zhang XL, Jiang SZ, Liu X, Li M, Zhang JB, Li XJ, Xu CH, Chen XM, et al. Quantificational methylation of APC and AXIN2 in HBV-related hepatocellular carcinoma. Curr Cancer Ther Rev. 2013;9:137–146. [Google Scholar]
- 22.Li X, Zhang J, Yang Z, Kang J, Jiang S, Zhang T, Chen T, Li M, Lv Q, Chen X, et al. The function of targeted host genes determines the oncogenicity of HBV integration in hepatocellular carcinoma. J Hepatol. 2014;60:975–984. doi: 10.1016/j.jhep.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, Gaston Mathe Y, Laurent C, Laurent A, Bioulac-Sage P, et al. Integration of tumour and viral genomic characterisations in HBV-related hepatocellular carcinomas. Gut. 2015;64:820–829. doi: 10.1136/gutjnl-2013-306228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Q, Stern JE, Hawes SE, Lu H, Jiang M, Kiviat NB. DNA methylation changes in normal liver tissues and hepatocellular carcinoma with different viral infection. Exp Mol Pathol. 2010;88:287–292. doi: 10.1016/j.yexmp.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heikenwalder M, Protzer U. LINE(1)s of evidence in HBV-driven liver cancer. Cell Host Microbe. 2014;15:249–250. doi: 10.1016/j.chom.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Niknafs YS, Chinnaiyan AM. RNA identity crisis: hepatitis B walks the LINE. Cancer Cell. 2014;25:259–260. doi: 10.1016/j.ccr.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 27.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 28.Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34:787–798. doi: 10.1093/carcin/bgs406. [DOI] [PubMed] [Google Scholar]
- 29.Murakami Y, Saigo K, Takashima H, Minami M, Okanoue T, Bréchot C, Paterlini-Bréchot P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut. 2005;54:1162–1168. doi: 10.1136/gut.2004.054452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012;22:593–601. doi: 10.1101/gr.133926.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li W, Zeng X, Lee NP, Liu X, Chen S, Guo B, Yi S, Zhuang X, Chen F, Wang G, et al. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013;102:338–344. doi: 10.1016/j.ygeno.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–170. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Wang HY, Yang SL, Liang HF, Li CH. HBx protein promotes oval cell proliferation by up-regulation of cyclin D1 via activation of the MEK/ERK and PI3K/Akt pathways. Int J Mol Sci. 2014;15:3507–3518. doi: 10.3390/ijms15033507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett. 2011;300:162–172. doi: 10.1016/j.canlet.2010.09.018. [DOI] [PubMed] [Google Scholar]