

Abstract
Inhibition of anti-apoptotic Mcl-1 is a promising anticancer strategy to overcome the survival and chemoresistance of a broad spectrum of human cancers. We previously reported on the identification of a natural product marinopyrrole A (1) that induces apoptosis in Mcl-1-dependent cells through Mcl-1 degradation. Here, we report the design and synthesis of novel marinopyrrole-based analogues and their evaluation as selective inhibitors of Mcl-1 as well as dual Mcl-1/Bcl-xL inhibitors. The most selective Mcl-1 antagonists were 34, 36 and 37 with 16-, 13- and 9-fold more selectivity for disrupting Mcl-1/Bim over Bcl-xL/Bim binding, respectively. Among the most potent dual inhibitors is 42 which inhibited Mcl-1/Bim and Bcl-xL/Bim binding 15-fold (IC50 = 600 nM) and 33-fold (500 nM) more potently than (±)-marinopyrrole A (1), respectively. Fluorescence quenching, NMR analysis and molecular docking indicated binding of marinopyrroles to the BH3 binding site of Mcl-1. Several marinopyrroles potently decreased Mcl-1 cellular levels and induced caspase 3 activation in human breast cancer cells. Our studies provide novel “lead” marinopyrroles for further optimization as selective Mcl-1 inhibitors and dual Mcl-1 and Bcl-xL inhibitors.
1. INTRODUCTION
Programmed cell death, or apoptosis, is a physiological mechanism that removes damaged or unwanted cells during development and maintains tissue homeostasis [1,2]. Deregulation of apoptosis is a hallmark of malignant transformation as well as tumor resistance to chemotherapy [3,4]. The B-cell lymphoma 2 (Bcl-2) family of proteins are critical regulators of the mitochondrial apoptotic pathway, comprising pro- and anti-apoptotic members [5,6]. The c-terminal hydrophobic grooves of anti-apoptotic members such as Bcl-2, Bcl-xL and Mcl-1 neutralize the activity of pro-apoptotic members by binding the BH3 death helix of pro-apoptotic members Bax and Bak and the BH3-only proteins [5,6]. The BH3-only proteins such as Bim, Bid, and Noxa, are a subgroup of proapoptotic Bcl-2 proteins that have a single BH3 death helix that competes the inhibitory activity of anti-apoptotic Bcl-2 grooves and also directly induces activation of Bax and Bak [7]. Several structures of anti-apoptotic Bcl-2 proteins demonstrate how their c-terminal hydrophobic grooves bind the pro-apoptotic BH3 death helices, defining the specific protein interaction surfaces involved in apoptosis regulation. The inhibition of such protein-protein interactions can restore apoptosis in cancer cells and it is a promising therapeutic strategy for cancer therapy [8–10].
Effective targeting of anti-apoptotic Bcl-2 proteins with small molecules poses two challenges. First, the anti-apoptotic/pro-apoptotic protein-protein interactions have large and flexible interfaces that are more difficult to target than enzyme/substrate interactions that involve smaller and more defined active sites [11]. Second, the potency and selectivity of small molecule inhibitors require optimization for each anti-apoptotic Bcl-2 groove despite the similarities among them. Despite these challenges, successful drug discovery campaigns from academic and industry led to anti-apoptotic Bcl-2 inhibitors [12]. For example, efforts from Abbot Laboratories have yielded two inhibitors undergoing clinical evaluation, ABT-263 and ABT-199 [13,14]. ABT-263, the orally available analog of ABT-737 [15], potently inhibits Bcl-xL and Bcl-2 whereas ABT-199 is a selective inhibitor of Bcl-2. Several studies have shown that Bcl-2/Bcl-xL inhibitors have efficacy in select cancers as single agents when Mcl-1 levels are kept low, and demonstrated that resistance to these agents can develop from the presence of overexpressed Mcl-1, therefore, limiting the efficacy of these agents to broader spectrum of cancers [16,17]. Similarly, the significance of Mcl-1 inhibition in cell survival of several hematological and solid tumors has been demonstrated by indirect approaches that downregulate the expression or stability of Mcl-1 [18,19]. Thus, selective and potent Mcl-1 inhibitors will be attractive agents for the treatment of broad human cancers as single agents or in combination with other Bcl-2 family inhibitors [20].
Previously several Mcl-1 small molecules inhibitors and stapled peptides have been discovered through structure-based design, high-throughput and fragment-based screening approaches [21–31]. Most compounds are not highly selective for Mcl-1 or have not been developed sufficiently for potent cellular and in vivo activity. We previously reported the identification of a natural product marinopyrrole A (1) that induces apoptosis in Mcl-1-dependent cells or ABT-737 resistant cells through targeting Mcl-1 degradation [25]. Here, we report the design and synthesis of novel marinopyrrole-based analogues and their evaluation as inhibitors of Mcl-1 and Bcl-xL-selective inhibitors as well as dual Mcl-1/Bcl-xL inhibitors. Fluorescence quenching employed for measurement of direct binding affinity and HSQC NMR analysis provided information about the binding mode of marinopyrrole-based analogues. NMR-guided docking studies informed to rationalize the structure-activity relationship studies and further design of analogues. The marinopyrrole analogues were tested in cellular assays for their ability to decrease Mcl-1 levels, to inhibit tumor cell survival and to induce apoptosis in human breast cancer cells.
2. RESULTS AND DISCUSSION
2.1. Design of marynopyrole A derivatives that disrupt Mcl-1/Bim and Bcl-xL/Bim protein-protein interactions
The unique molecular geometry of (±)-marinopyrrole A (1) offers excellent opportunities to decorate this natural product-based bispyrrole system for desired activity and selectivity. Previous NMR studies suggested that marinopyrrole A (1) most favorably binds to Mcl-1 in a position centered at the p2 and p3 hydrophobic pockets formed by Mcl-1 helices 4, 5 and 3 that bind the conserved hydrophobic residues Leu and Ile of the BIM BH3 death helix [25]. The critical contributions of p2 and p3 pockets to the high affinity and selectivity to Mcl-1 prompted the design of selective small molecule Mcl-1 inhibitors based on these pockets [32–34]. The structural model of docked marinopyrrole A (1) (Supplemental Figure 1) suggests that the marinopyrrole scaffold makes interaction contacts with residues known to interact with Bim such as Phe 254, Val253, Met250, Met231 in p2 and with Phe228, Ala227, N260, Arg263, Thr266 in p3. Therefore, the molecular docking suggested that the marinopyrrole scaffold possesses favorable features that mimic the BIM BH3 binding to the Mcl-1 groove and is amenable to further expansion and optimization for the generation of more potent and selective binding to Mcl-1.
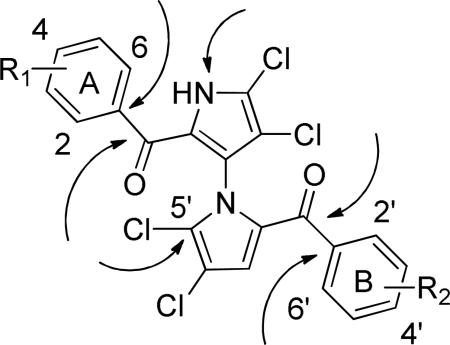
We undertook a structure-based design approach to better understand and explore structure-activity relationships (SARs) using potential sites of marinopyrrole A amenable for optimization as shown in Table 1. Our goal was to expand the interactions towards p1 and p4 pockets to increase binding affinity and selectivity. We aimed to target hydrophobic interactions with Met231, Leu235, Val249 and hydrogen bonds with Lys234 or His252 towards the p1 pocket. We also aimed to target hydrophobic interactions with residues Val216, Val220 and potential hydrogen bonds with residues His224 and Asn223 in the shallower p4 pocket. To this end, we designed a series of novel marinopyrrole derivatives with substitution at the para-positions (4 and 4′) of the two phenyl rings to the carbonyl groups. Di-substitutions with hydrophobic groups on both phenyl rings furnished compounds (23 to 28) while those with hydrophilic groups yielded derivatives 29, 30, 34 and 35. Tri-substitutions of “symmetrical” marinopyrroles on both phenyl rings provided compounds 24, 30 and 36 to 38. Design of “nonsymmetrical” marinopyrroles included compounds 46 to 52. Extension of functional groups in the para-position of the phenyl groups with a sulfide spacer furnished compounds 31 to 34. Marinopyrroles with bistriazole spacer, compounds 36 to 45, were designed to improve solubility and increased binding to pockets p1 to p4. N-methyl analogues 53 and 54 were designed to investigate the importance of the free NH group to Mcl-1 binding and selectivity.
Table 1.
Potential sites amenable for optimization and ELISA results of marinopyrroles.
| ||||
|---|---|---|---|---|
| ID | R1 | R2 | Mcl-1/Bima | Bcl-xL/Bima |
| 1b | 2-OH | 2′-OH | 8.9 ± 1.0 | 16.4 ± 3.3 |
| 23c | 2-OH-4-CF3 | 2′-OH-4′-CF3 | 8.1 ± 0.9 | 9.7 ± 1.3 |
| 24 | 2-OH-5-Cl-4-Me | 2′-OH-5′-Cl-4′-Me | 2.6 ± 0.6 | 2.5 ± 0.4 |
| 25 | 2-OH-4-C=CH | 2′-OH-4′-C=CH | 3.9 ± 0.2 | 5.6 ± 0.5 |
| 26 | 2-OH-4-CH=CH2 | 2′-OH-4′-CH=CH2 | 3.7 ± 0.5 | 3.5 ± 0.7 |
| 27 | 2-OH-4-Et | 2′-OH-4′-Et | 2.1 ± 0.5 | 3.9 ± 1.3 |
| 28c | 2-OH-4-OSO2CF3 | 2′-OH-4′-OSO2CF3 | 1.0 ± 0.3 | 2.1 ± 0.7 |
| 29c | 2-OH-4-OH | 2′-OH-4′-OH | 39.5 ± 6.2 | >50 |
| 30c | 2-OH-5-Cl-4-OH | 2′-OH-5′-Cl-4′-OH | 10.7 ± 0.2 | >50 |
| 31d | 2-OH-4-SCH2CO2Et | 2′-OH-4′-SCH2CO2Et | 1.8 ± 0.3 | 1.2 ± 0.2 |
| 32d | 2-OH-4-SCH2Ph | 2′-OH-4′-SCH2Ph | 0.7 ± 0.2 | 0.6 ± 0.2 |
| 33d | 2-OH-4-SCH2(p-MeOPh) | 2′-OH-4′-SCH2(p-MeOPh) | 0.7 ± 0.1 | 0.6 ± 0.1 |
| 34d | 2-OH-4-SCH2CO2H | 2′-OH-4′-SCH2CO2H | 6.1 ± 1.3 | >100 |
| 35e | 2-OH-4-PO(OH)2 | 2′-OH-4′-PO(OH)2 | 10.9 ± 3.1 | 27.3 ± 7.2 |
| 36 |
|
|
7.8 ± 1.5 | >100 |
| 37 |
|
|
1.6 ± 0.6 | 14.0 ± 4.7 |
| 38 |
|
|
5.2 ± 0.8 | >50 |
| 39 |
|
|
3.3 ± 0.9 | 1.6 ± 0.3 |
| 40 |
|
|
1.5 ± 0.2 | 0.8 ± 0.2 |
| 41 |
|
|
1.4 ± 0.5 | 3.8 ± 1.3 |
| 42 |
|
|
0.6 ± 0.3 | 0.5 ± 0.1 |
| 43 |
|
|
18.4 ± 0.3 | >100 |
| 44 |
|
|
5.1 ± 0.4 | 8.1 ± 2.5 |
| 45 |
|
|
16.5 ± 1.9 | >50 |
| 46f | 2-OMe | 2′-OMe-4′-Cl | 8.0 ± 1.6 | 9.5 ± 2.2 |
| 47f | 2-OH | 2′-OH-3′-Cl | 4.1 ± 1.4 | 10.1 ± 2.2 |
| 48f | 2-OH | 2′-OH-5′-Cl | 3.9 ± 1.1 | 18.3 ± 3.0 |
| 49f | 2-OH | 2′-OH-4′-Cl | 6.5 ± 1.3 | 9.2 ± 2.3 |
| 50g | 2-OH | 2′-OH-5′-F | 8.9 ± 0.9 | 13.3 ± 3.3 |
| 51g | 2-OH | 2′-OH-4′-F | 9.6 ± 0.4 | 21.3 ± 5.6 |
| 52g | 2-OH | 2′-OH-6′-F | 13.1 ± 0.3 | 43.7 ± 10.0 |
| 53 | 2-OMe | 2′-OMe and N-Meh | 15.5 ± 3.3 | 64.9 ± 15.5 |
| 54 | 2-OH | 2′-OH and N-Meh | > 100 | 7.9 ± 1.8 |
| ABT-263 | 4.3 ± 0.4 nM | |||
IC50 in μM (average ± SEM, n ≥ 3) unless specified
Activity as disruptors of Mcl-1 and Bc1-xl reported previously and here for SAR discussion [13]
Chemistry, anti-MRSA activity was reported previously [12]
Chemistry and activity as disruptors of Mcl-1 and Bcl-xL reported previously and here for SAR discussion [15]
Chemistry and activity as disruptors of Mcl-1 and Bcl-xL reported previously and here for SAR discussion [13]
Chemistry and anti-MRSA activity were reported previously [5]
Chemistry and anti-MRSA activity were reported previously [14]
N-Methyl analogue.
2.2. Chemistry
Starting from our previously reported compound 2 [35], mono-ketone 4 was obtained in 73% yield over two steps by introduction of ortho-methoxy-para-methylphenyl group (3 was not isolated) followed by IBX oxidation (Scheme 1). Removal of TBDMS protecting group with TBAF gave alcohol 5 in 90% yield. Oxidation of 5 by IBX furnished aldehyde 6 in 90% yield. Bisketone 8 was obtained in 54% yield after introduction of second ortho-methoxy-para-methylphenyl group (without isolation of 7) followed by IBX oxidation. Removal of para-toluenesulfonyl group with KOH generated 9 in 98% yield, which was converted to 10 in 65% yield by chlorination with NCS [35]. The final symmetrical marinopyrrole derivative 24 was obtained in 85% yield after demethylation using BBr3/DCM [36]. Using our previously reported intermediate 11 [35] as a starting material, palladium-mediated substitution of the triflate 11 with ethynyltrimethylsilane furnished 12 in 74% yield (Scheme 2). Demethylation of 12 using BBr3/DCM gave 13 in 53% yield, which was converted to the final symmetrical marinopyrrole 1–3 in 78% yield. Reduction of triple bonds in 1–3 with atmospheric H2/Pd/BaSO4 provided para-vinyl substituted marinopyrrole 26 in 60% yield, which was further reduced with atmospheric H2/Pd/BaSO4 to para-ethyl marinopyrrole 27 in 96% yield. Schemes 3–5 showed the chemistry that we developed to synthesize bistriazole marinopyrrole derivatives. Start from a common intermediate 14 [35], palladium-mediated substitution of the triflate 14 with ethynyltrimethylsilane provided 15 in 92% yield, which was converted to 16 in 95% yield after removal of tosyl group by KOH. Bistriazole marinopyrrole 17 in 78% yield was constructed using “Click Chemistry” [37]. Chlorination of 17 with NCS generated 18. The final product 36 was obtained in 50% yield after demethylation of 18 using BBr3/DCM (Scheme 3). In order to improve overall yield, demethylation of 14 [35] using BBr3/DCM was performed first to give 19 in 90% yield as shown in Scheme 4. Palladium-mediated substitution of the triflate 19 with ethynyltrimethylsilane furnished 20 in 98% yield. Removal of tosyl group in 20 provided 21 in 95% yield. Intermediate 22 was obtained in 80% yield using “Click Chemistry” [37], which was subjected to chlorination with NCS to give the final compound 37. The free carboxylic acid 38 was obtained in 65% yield after removal of t-butyl group from 37. Compound 1–3 was used as a common starting material by “Click Chemistry” to produce seven bistriazole marinopyrrole derivatives (39 to 45) as shown in Scheme 5. The final compounds 39 to 44 were obtained in 55%, 70%, 52%, 48%, 52% and 83% yield, respectively. Removal of t-butyl group from 44 furnished 45 in 94% yield. N-methyl analogue of marinopyrrole 53 was synthesized via N-methylation of a precursor that we reported previously followed by O-demethylation using BBr3 to generate 54 [36].
Scheme 1.

Synthesis of marinopyrrole 24.
Scheme 2.

Synthesis of marinopyrroles 25 to 27.
Scheme 3.

Synthesis of marinopyrrole 36.
Scheme 5.

Synthesis of bistriazole marinopyrroles 39 to 45.
Scheme 4.

Synthesis of marinopyrroles 37 and 38.
2.3. Binding and Structural Characterization of Marinopyrroles to Mcl-1
To validate direct binding of marinopyrrole compounds to the BH3-binding groove of Mcl-1 and investigate the protein-ligand interactions, we performed binding by fluorescence-quenching and structural characterization by NMR and molecular docking. We selected representative compound 35 from the list of symmetrical derivatives and compound 38 from the list of compounds containing a bistriazole spacer (Table 1). We first examined the binding affinity of compounds using a fluorescence-quenching (FQ) assay based on the intrinsic Trp fluorescence of Mcl-1. Using this assay we have confirmed direct binding of marinopyrrole analogues to Mcl-1 and calculated binding constants for 35 (Kd = 3.5 μM) and 38 (Kd = 2.5 μM) from their corresponding binding isotherms (Figure 1). Next, we performed NMR analysis of 15N Mcl-1 upon titration of compounds 35 and 38. The HSQC spectra of 15N Mcl-1 demonstrated well dispersed peaks characteristic of a well-folded protein. Upon titration of marinopyrrole compounds Mcl-1 keeps its ordered structure and does not undergo significant conformational changes (Figure 2, Supplemental figure 2). The HSQC analysis confirmed binding to Mcl-1 for both compounds and interaction of the compounds in an intermediate exchange regime, in agreement with the low M binding affinity as suggested by FQ. The titration of compounds up to 2:1 compound-to-protein ratio induced significant chemical shift changes in several residues of Mcl-1 located in the BH3-binding groove and specifically in residues from helices α2, α3, α4, α5 (Figure 3A). Both compounds induced significant chemical changes in the same residues of p1–p4 pockets although compound 38 induced significant broadening in several p1–p4 residues which is consistent with its higher binding affinity compared to 35. Interestingly, some residues in helices α1, α6 and α7 undergo chemical shifts effects possibly from allosteric effects from the binding to the BH3-binding groove. Similar allosteric effects have been also documented with NMR titrations of BH3 helices and Mcl-1 [38]. Mapping of the chemical shift perturbation data onto the structure of Mcl-1 showed the position of residues that are affected by binding of the compounds and conclusively demonstrated that the compounds bind Mcl-1 protein at the same binding site where conserved BIM BH3 residues bind Mcl-1 (Figure 3A). Taken together, NMR analysis and mapping of the chemical shift perturbation data onto the structure of Mcl-1 are consistent with a direct interaction of 35 and 38 with the BH3-binding groove of Mcl-1. Furthermore, the data also show that 35 and 38 engage more residues in the p1 and p4 pockets than compound 1, in agreement with our structure-based design approach (Figure 3B).
Figure 1.
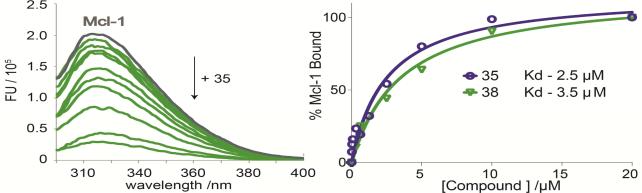
A) Intrinsic fluorescence spectra of Mcl-1 quenched upon titration of 35 B) Binding isotherms of 35 and 38 generated by fluorescence quenching measurements.
Figure 2.

Marinopyrroles 35 and 38 target the hydrophobic BH3-binding groove of Mcl-1. A) Plots of measured chemical shift perturbations Δ(CSP) of 15Mcl-1 upon titration of 35 and 38 as function of Mcl-1 residue. Residues undergoing significant chemical shifts changes (CSP>0.05) upon binding of marinopyrroles correspond to orange bars. Red bars highlight residues with CSP>0.1 and light blue bars correspond to residues undergoing exchange broadening. Light green shading designates the corresponding residues of the BH3-binding site of Mcl-1. B) Structural models of Mcl-1 (grey) with the view of the hydrophobic BH3-binding groove showing compounds 35 and 38 in the BH3-binding groove as derived by NMR-guided docking. Residues undergoing significant chemical shift perturbations are colored in orange and red and residues undergoing exchange broadening are colored in light blue in agreement with (A). The observed NMR chemical shift perturbation data are consistent with the 35 and 38 binding to the BH3-binding pocket of Mcl-1.
Figure 3.

Structural models of NMR-guided docked structures of 35 and 38 at the BH3-binding pocket of Mcl-1. The BH3-binding site residues are colored according to the property of their side chain: hydrophobic (yellow), hydrophilic (green), positive charged (blue) and negatively charged (red).
We next performed molecular docking using constraints from our NMR chemical shift perturbation data analysis to investigate the compounds conformations and interactions with the BH3-binding pocket of Mcl-1. The structural models suggest that compounds 35 and 38 complement the BH3-binding site using an extended conformation and making several hydrophophic and polar contacts (Figure 4). Both compounds bind with the marinopyrrole scaffold positioned to the center of the BH3-binding site occupying p2 and p3 pockets as described above. Furthermore the symmetrical substitutions at either end of the marinopyrrole core extend the interactions of these compounds further to p1 and particularly for the larger 35 compound to p4, consistent with our analogue design approach and NMR characterization (Figure 3). Several hydrophobic residues (Phe228, Met231, L235, Leu246, Val249, Met250, Val253, Phe254, Val258, Gly262 and Leu267) within the p1, p2 and p3 pockets of the BH3-binding site are predicted to interact with 35 and 38. Furthermore, the marinopyrrole scaffold of 35 and 38 is predicted to form hydrogen bond interactions with Arg263, Thr266 and Asn260 of helix α5. Interestingly, the phosphate and carboxylate groups of 35 and 38, respectively, are predicted to form new hydrogen bonds with the positively charged Lys234 in the p1 pocket. Moreover, 38 docking pose suggests that this compound extends its interactions in the p4 pocket with the triazole group having contacts with Gly262 and Val216. The docking model suggests that the aromatic rings of Phe318 and Phe319 are also in close distance with the carboxylate and triazole groups, however chemical shifts are not observed for these phenylalanines in helix α8 (Figure 3). Previous studies suggested that p4 pocket in Mcl-1 is shallower and can accommodate smaller hydrophobic or polar groups in contrast to Bcl-xL [32–33]. Notably, a mutation of the Phe residue in BIM BH3 that binds p4 with a Val residue changed the selectivity preference to Mcl-1 over Bcl-xL. Thus, the presence of the triazole and carboxylate groups provides a possible explanation for the selectivity of 38 for Mcl-1 (Table 1). Taken together, the structural models of 35 and 38 bound to Mcl-1 may explain the interactions of the new marinopyrrole analogues with residues of the BH3-binding groove and suggest how increased affinity or selectivity can be achieved with interactions in p1 and p4 pockets.
Figure 4.

Marinopyrrole derivatives potently decrease Mcl-1 levels and induce caspase 3 activation in human breast cancer MDA-MB-468 cells.
2.4. Structure Activity Relationships of Marinopyrrole Derivatives
To investigate SARs of marinopyrrole derivatives, we selected the six sites amenable for generating analogues as shown in Table 1. The design of analogues included substitutions on the pyrrole nitrogen, two phenyl rings and substitution on the second pyrrole ring. We evaluated potency and selectivity of the new analogues using an ELISA assay that evaluates competitive binding to Bim/Mcl-1 and Bim/Bcl-xL complexes (Table 1). Marinopyrrole 23 [35], the bistrifluoromethyl derivative of 1, exhibited similar potency to that of 1 against Mcl-1/Bim complex and was slightly more potent against Bcl-xL/Bim complex. Nonsymmetrical marinopyrroles with either chlorine [39] or fluorine [40] substitution in ring B (47 to 52) showed that chlorine in either 3′ or 4′ position improves potency against both Mcl-1 and Bcl-xL. Chlorine in position 5′ improves potency against Mcl-1 but not Bcl-xL. The presence of fluorine in either 4′ or 6′ reduced potency against both Mcl-1 and Bcl-xL. Compounds 24 to 27 with hydrophobic substitutions in the para-position are two- to nine-fold more potent than 1 against both Mcl-1 and Bcl-xL, with the ethyl substitution having the best interaction with Mcl-1 (IC50 = 2.1 μM). However the trifluoromethanesulphonate analogue 28 further increased potency against Mcl-1 (IC50 = 1.0 μM) and Bcl-xL (IC50 = 2.1 μM), nine- and eight-fold compared to 1, respectively. Marinopyrroles with hydrophilic substitutions in 4′ or 5′ (29, 30 and 35) are less active than 1 against Bcl-xL/Bim, with 29 being less active against both Bcl-xL and Mcl-1. Interestingly, these hydrophilic substitutions increased solubility of the compounds but significantly reduced the potency against Bcl-xL [41]. Thus, the SAR results suggest that hydrophobic substitutions at either end of the marinopyrrole scaffold lead to increased affinity by reaching to p1 and p4 pockets, and that the trifluoromethanesulphonate moiety may have the best fit compared to other hydrophobic substitutions. The presence of hydrophobic groups in 4′ position results in similar binding for Mcl-1 and Bcl-xL pocket as expected from structural studies with BH3 helices [32,33,42].
Next, we tested symmetrical marinopyrroles with a triazole spacers attached to 4′ position of the phenyl rings (36 to 45). These substitutions increased the flexibility of the molecules; however, the hydrophobic nature of some substituents increased significantly the potency for Mcl-1 and Bcl-xL. The most potent compound in this series is 42 which is 15- (IC50 = 0.6 μM) and 33-fold (IC50 = 0.5 μM) more potent than 1 against Mcl-1and Bcl-xL, respectively. Furthermore, substituents like ethylester (36 and 43) or carboxylate (38 and 45) attached to the triazole group has somewhat similar activity to Mcl-1 but reduced significantly potency against Bcl-xL. Compounds 36, 37, 38, 43 and 45 showed three- to 13-fold selectivity for Mcl-1 over Bcl-xL, consistent with the tolerability of Mcl-1 for hydrophilic groups in the p4 pocket and in agreement with our SAR results and molecular docking analysis.
Furthermore, we recently reported on marinopyrrole analogues with sulfide spacers 31–33 found that the most potent 32 and 33 inhibit potently Mcl-1/Bim and Bcl-xL/Bim binding with IC50 values of 700 nM and 600 nM, respectively [41]. Interestingly, analogue 34 with the sulfide-acetic acid functional group in the para- positions lost some activity (IC50 = 6.1 μM) compared to 32 and 33 but exhibited 16.4-fold selectivity for Mcl-1 over Bcl-xL in agreement with our observation in NMR and docking analysis regarding the tolerance of hydrophilic group in p4 pocket.
The SAR results uncovered several selective Mcl-1 inhibitors as well as selective Bcl-xL and dual Mcl-1/Bcl-xL inhibitors. The most selective Mcl-1 antagonist was the above-mentioned 34 with the carboxylate-containing sulfide spacer having over 16-fold more selectivity for disrupting Mcl-1/Bim over Bcl-xL/Bim binding. However, neutralizing the carboxylate negative charge with an ethyl ester as in 31 or replacing the carboxylate with either a phenyl (32) or a methoxyphenyl (1–11) not only greatly increased the potency as discussed above, but also reversed the selectivity resulting in some of the most potent Mcl-1 and Bcl-xL dual inhibitors. In the triazole series, 39 with a benzyl and 40 with a phenyl were 2-fold more selective for Bcl-xL over Mcl-1. However, this selectivity was reversed towards Mcl-1 when the benzyl or phenyl group was replaced with ethyl carboxylate as in 43 (5.4 fold), carboxylate as in 45 (3.0 fold), cyclohexyl as in 41 (2.7 fold) or t-butyl carboxylate as in 44 (1.6 fold). Replacement with an octyl group as in 42 resulted in one of the most potent dual Mcl-1 and Bcl-xL antagonists, inhibiting Mcl-1/Bim and Bcl-xL/Bim binding with IC50 values of 600 nM and 500 nM, respectively. In the triazole series, adding chloro groups to the 5 and 5′ positions increases selectivity for Mcl-1 over Bcl-xL [compare 36 (12.9 fold) to 43 (5.4 fold), 38 (9.6 fold) to 45 (3 fold) and 37 (8.8 fold) to 44 (1.6 fold)]. Moreover, substituting the 4 and 4′ hydrogens in 1 with hydrophobic groups such as vinyl (26), methyl and 5, 5′-Cl (24), ethyne (25), ethyl (27) and CF3 (23) all lead to dual Mcl-1 and Bcl-xL antagonists. Taken together these SAR studies provide further starting points to design selective or dual Mcl-1/Bcl-xL antagonists.
2.5. Marinopyrrole derivatives are highly effective at decreasing the Mcl-1 levels and inducing apoptosis in human breast cancer cells
To assess the ability of the marinopyrroles to reach their target and induce programmed cell death (apoptosis) in human cancer cells, we treated MDA-MB-468 breast cancer cells with the marinopyrrole derivatives at 3 μM and processed the cells for western blotting as described by us previously [42]. Figure 4 shows that several symmetrical marinopyrrole derivatives with hydrophobic substituents such as 23 (CF3), 25 (ethyne), 26 (vinyl), 24 (para-CH3 and meta-Cl), 27 (ethyl) and 28 (trifluoromethanesulfonate) potently decreased Mcl-1 and induced apoptosis as determined by activation of the protease caspase 3. This is consistent with our previous data that shows that 1 binds Mcl-1 and induces its degradation in a proteasome-dependent manner which was associated with apoptosis induction [7] Several non-symmetrical marinopyrrole derivatives were highly potent at decreasing Mcl-1 levels and inducing apoptosis of MDA-MB-468 breast cancer cells. These include 49, 50 and 51. In contrast, 48 and 52 which were active in vitro against Mcl-1were not active in intact cells. Similarly, despite being highly potent in vitro, sulfides 31, 32 and 33 as well as triazoles 37, 40 and 42 had little activity in intact cells (Figure 4). We do not know the reason for these differences between in vitro and intact cells, but lower cellular uptake, higher cellular efflux and/or metabolic inactivation are possible mechanisms. Marinopyrrole derivatives, 29, 30, 34–36, 38, 39, 41, 43–46 were inactive at decreasing Mcl-1 levels and at inducing caspase 3 cleavage at concentrations as high as 10 μM (data not shown). As we have reported previously, none of the sulfides or sulphone derivatives are active at the 3 μM concentration; however, at higher concentrations (40 μM) some of the sulfide and sulphone derivatives are cell-active [40].
3. CONCLUSIONS
This article describes the design, synthesis and SAR studies of novel marinopyrrole derivatives. FQ supports binding of marinopyrroles to Mcl-1 and NMR with molecular docking suggests their binding mode is within the p1–p4 pockets of the Mcl-1 BH3 groove that bind the natural α-helical Bim peptide. Our comprehensive SAR studies clearly demonstrated: i) symmetrical marinopyrroles with hydrophobic, but not hydrophilic, substituents in the para-position to the carbonyl group are desirable for disrupting Mcl-1/Bim and Bcl-xL/Bim protein-protein interactions; ii) substituents with sulfide, but not sulfone, spacers are tolerated and some greatly increase potency; iii) substituents with triazole spacers are allowed in most cases against Mcl-1/Bim but not against Bcl-xL/Bim for 36, 38, 43 and 45; iv) substituents with chlorine or fluorine in B ring of non-symmetrical marinopyrroles are tolerated in most cases; v) N-methylation of marinopyrrole A (54) showed selectivity for Bcl-xL/Bim over Mcl-/Bim. In addition, we have identified several symmetrical and non-symmetrical marinopyrrole derivatives that potently decrease the levels of Mcl-1 and induce tumor cell apoptosis of human breast cancer cells. Our studies generated a series of novel “lead” marinopyrroles for further optimization as highly potent and selective Mcl-1 inhibitors as well as dual Mcl-1 and Bcl-xL inhibitors which can induce apoptosis of human cancer cells.
4. EXPERIMENTAL SECTION
4.1. General
All chemicals were purchased from commercial suppliers and used without further purification. All solvents were dried and distilled before use. Tetrahydrofuran was distilled from sodium/benzophenone. Dichloromethane and acetonitrile were distilled over calcium hydride. Flash column chromatography was performed with silica gel (200–300 mesh). 1H NMR spectra were recorded at either 400 MHz or 600 MHz at ambient temperature. 13C NMR spectra were recorded at either 100 or 150 MHz at ambient temperature. Infrared spectra were recorded on a spectrophotometer (Perkin-Elmer Spectrum 100). Melting points were determined with melting point apparatus (Fukai X-4). High resolution mass spectra were performed by electrospray ionization (ESI) on an Agilent ESI-TOF LC-MS 6200 system. Analytical HPLC was performed on an Agilent 1100 series with diode array detectors and auto samplers. All tested compounds possessed a purity of not less than 95%.
4.1.1. (2-(((tert-Butyldimethylsilyl)oxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-methylphenyl)methanol (3)
To a solution of 1-bromo-2-methoxy-4-methylbenzene (524 mg, 2.62 mmol) in anhydrous THF (5 mL) at –78 °C under N2 was slowly added n-BuLi (1.15 mL, 2.5 M in n-pentane, 2.88 mmol). After being stirred for 30 min, a solution of 2 [39] (600 mg, 1.31 mmol) in anhydrous THF (1 mL) was added slowly via a syringe. The reaction was stirred for about 8 h and quenched by addition of a saturated aqueous NH4Cl (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified quickly by column chromatography (15% EtOAc/petroleum ether, Rf = 0.3) to yield 3 (unstable).
4.1.2. (2-(((tert-Butyldimethylsilyl)oxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-methylphenyl)methanone (4)
To a solution of 3 in anhydrous DMSO (20 mL) was added IBX (618 mg, 2.20 mmol) at room temperature. The reaction was allowed to warm up to 30 °C and stirred additionally for about 6 h. The reaction was quenched with water (30 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (15% EtOAc/petroleum ether, Rf = 0.2) to yield 4 (550 mg, 73% yield in two steps) as a pale brown solid. mp 30.7–33.0 °C; 1H NMR (400 MHz, CDCl3) δ 0.006 (s, 6H), 0.86 (s, 9H), 2.38 (s, 3H), 2.40 (s, 3H), 3.75 (s, 3H), 4.67 (s, 2H), 6.20 (dd, J = 4.0, 2.4 Hz, 1H), 6.32 (d, J = 3.2 Hz, 1H), 6.67 (dd, J = 4.0, 1.6 Hz, 1H), 6.74 (s, 1H), 6.75 (d, J = 7.6 Hz, 1H), 7.06 (dd, J = 2.4, 1.6 Hz, 1H), 7.18 (d, J = 3.6 Hz, 1H), 7.24–7.28 (m, 3H), 7.84 (d, J = 8.8 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ –5.70, –5.70, 18.33, 21.46, 21.71, 25.80, 25.80, 25.80, 53.45, 55.45, 108.90, 111.46, 112.05, 120.34, 121.16, 122.99, 126.70, 126.86, 126.86, 127.20, 129.50, 129.64, 129.64, 129.81, 132.40, 132.62, 136.66, 141.84, 144.60, 157.22, 184.12 ppm; HRMS ESI (M+H+) calcd for C31H39N2O5SSi 579.2349, found 579.2358; IR (KBr) 3434, 3443, 2954, 2930, 2856, 1916, 1708, 1936, 1608, 1498, 1409, 1368, 1256, 1179, 1035, 839, 772, 670, 602 cm–1.
4.1.3. (2-(Hydroxymethyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-methylphenyl)methanone (5)
To a solution of 4 (550 mg, 0.95 mmol) in anhydrous THF (10 mL) was added TBAF (745 mg, 2.85 mmol) at room temperature. The reaction was allowed to stir additionally for about 5 h at room temperature. The reaction was quenched with water (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (20% EtOAc/petroleum ether, Rf = 0.3) to yield 5 (397 mg, 90% yield) as a brown-red solid. mp 29.9–31.7 °C; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H), 2.40 (s, 3H), 2.97 (t, J = 6.8 Hz, 1H), 3.74 (s, 3H), 4.55 (d, J = 6.8 Hz, 2H), 6.23 (dd, J = 4.0, 2.4 Hz, 1H), 6.34 (d, J = 3.6 Hz, 1H), 6.63 (dd, J = 4.0, 1.6 Hz, 1H), 6.73 (s, 1H), 6.76 (d, J = 7.6 Hz, 1H), 7.00 (dd, J = 2.4, 2.0 Hz, 1H), 7.22 (d, J = 7.6 Hz, 1H), 7.28 (d, J = 3.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.50, 21.70, 55.02, 55.48, 109.61, 110.85, 112.06, 120.41, 121.03, 123.68, 126.61, 127.08, 127.08, 128.22, 128.72, 129.27, 129.95, 129.95, 132.60, 132.82, 135.73, 141.93, 145.21, 157.09, 185.16 ppm; HRMS ESI (M+Na+) calcd for C25H24N2NaO5S 487.1304, found 487.1297; IR (KBr) 3445, 3141, 2956, 2930, 1771, 1702, 1631, 1609, 1498, 1461, 1410, 1366, 1177, 1138, 1034, 1014, 931, 720 cm–1.
4.1.4. 2′-(2-Methoxy-4-methylbenzoyl)-1′-tosyl-1′H-1,3′-bipyrrole-2-carbaldehyde (6)
To a solution of 5 (413 mg, 0.89 mmol) in DMSO (20 mL) was added IBX (374 mg, 1.33 mmol) at room temperature. The reaction was allowed to warm up to 50 °C and stirred for about 3 h. The reaction was quenched with water (30 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (15% EtOAc/petroleum ether, Rf = 0.3) to yield 6 (370 mg, 90% yield) as a white solid. mp 116.4–118.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H), 2.42 (s, 3H), 3.73 (s, 3H), 6.27 (s, 1H), 6.45 (d, J = 3.2 Hz, 1H), 6.68 (d, J = 2.0 Hz, 1H), 6.72 (s, 1H), 6.76 (d, J = 7.6 Hz, 1H), 6.99 (s, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.68 (d, J = 3.2 Hz, 1H), 7.89 (d, J = 8.0 Hz, 2H), 9.62 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.53, 21.69, 55.41, 109.99, 111.43, 112.02, 120.41, 123.04, 125.22, 125.99, 127.50, 127.94, 127.94, 129.80, 129.83, 129.83, 131.70, 133.68, 134.72, 139.12, 142.26, 145.71, 157.32, 176.91, 184.11 ppm; HRMS ESI (M+H+) calcd for C25H23N2O5S 463.1328, found 463.1336; IR (KBr) 3449, 3150, 3129, 2957, 2924, 2854, 1690, 1671, 1606, 1565, 1408, 1357, 1263, 1170, 1073, 1009, 773 cm–1.
4.1.5. (2-(Hydroxy(2-methoxy-4-methylphenyl)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-methylphenyl)methanone (7)
To a solution of 1-bromo-2-methoxy-4-methylbenzene (378 mg, 1.89 mmol) in anhydrous THF (5 mL) at –78 °C under N2 was slowly added t-BuLi (1.46 mL, 1.3 M, 1.89 mmol). After being stirred for 30 min, a solution of 6 (350 mg, 0.76 mmol) in anhydrous THF (1 mL) was added slowly via a syringe. The reaction was stirred for about 8 h and quenched by addition of a saturated aqueous NH4Cl (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified quickly by column chromatography (12% EtOAc/petroleum ether, Rf = 0.3) to yield 7 (unstable).
4.1.6. (1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-methoxy-4-methylphenyl)methanone) (8)
To a solution of 7 in anhydrous DMSO (20 mL) was added IBX (275 mg, 0.98 mmol) at room temperature. After being stirred for about 3 h, the reaction was quenched with water (30 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (12% EtOAc/petroleum ether, Rf = 0.2) to yield 8 (240 mg, 54% yield two steps) as a pale brown solid. mp 71.0–72.7 °C; 1H NMR (400 MHz, CDCl3) δ 2.27 (s, 3H), 2.37 (s, 3H), 3.43 (s, 3H), 3.65 (s, 3H), 3.75 (s, 3H), 5.85 (t, J = 3.2 Hz, 1H), 6.29 (dd, J = 4.0, 1.6 Hz, 1H), 6.46–6.48 (m, 2H), 6.53 (d, J = 8.0 Hz, 1H), 6.70 (d, J = 8.0 Hz, 1H), 6.73 (s, 2H), 6.96 (d, J = 7.6 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.49 (d, J = 3.6 Hz, 1H), 7.94 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.50, 21.62, 21.79, 55.39, 55.39, 108.52, 111.61, 111.99, 112.04, 119.88, 120.37, 122.95, 123.41, 124.79, 126.25, 128.07, 128.07, 128.61, 129.41, 129.41, 129.41, 129.73, 131.48, 132.17, 132.29, 135.78, 141.65, 144.24, 144.81, 157.25, 158.29, 183.15, 184.47 ppm; HRMS ESI (M+H+) calcd for C33H31N2O6S, found 583.1903, found 583.1890; IR (KBr) 3356, 3006, 2958, 2851, 1631, 1612, 1463, 1408, 1262, 1157, 1088, 1033, 859, 746 cm–1.
4.1.7. 1′H-1,3′-Bipyrrole-2,2′-diylbis((2-methoxy-4-methylphenyl)methanone) (9)
To a solution of 8 (210 mg, 0.36 mmol) in a mixture of MeOH/THF (1:1, 5 mL) was added KOH (60 mg, 1.08 mmol) at room temperature. After being stirred for 15 min, the reaction was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to yield 9 (151 mg, 98% yield) as a light white solid. mp 171.1–172.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.25 (s, 3H), 2.38 (s, 3H), 3.67 (s, 3H), 3.77 (s, 3H), 5.80–5.82 (m, 1H), 6.30 (t, J = 2.8 Hz, 1H), 6.35 (dd, J = 4.0, 1.6 Hz, 1H), 6.46–6.48 (m, 2H), 6.62 (t, J = 2.8 Hz, 1H), 6.73–6.74 (m, 2H), 7.02 (t, J = 2.8 Hz, 1H), 7.08 (dd, J = 9.6, 7.6 Hz, 2H), 9.40 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.78, 21.81, 55.33, 55.58, 108.32, 110.60, 111.35, 112.20, 119.99, 120.63, 122.76, 122.98, 125.23, 125.92, 126.77, 129.01, 129.89, 131.13, 132.22, 132.34, 141.51, 141.55, 156.69, 157.43, 183.43, 183.90 ppm; HRMS ESI (M+H+) calcd for C26H25N2O4 429.1814, found 429.1811; IR (KBr) 3357, 3006, 2957, 2852, 1631, 1612, 1462, 1408, 1264, 1127, 1033, 859, 747 cm–1.
4.1.8. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((5-chloro-2-methoxy-4-methylphenyl)methanone) (10)
To a solution of 9 (10 mg, 0.02 mmol) in anhydrous MeCN (1 mL) at room temperature was added NCS (18.7 mg, 0.14 mmol) slowly. After being stirred for about 20 min at room temperature,the reaction was quenched with water (5 mL) and extracted with EtOAc (5 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (12% EtOAc/petroleum ether, Rf = 0.2) to yield 10 (9 mg, 65% yield) as a pale brown solid. mp 78.3–80.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.30 (s, 3H), 2.32 (s, 3H), 3.72 (s, 3H), 3.76 (s, 3H), 6.44 (s, 1H), 6.67 (s, 1H), 6.80 (s, 1H), 7.06 (s, 1H), 7.20 (s, 1H), 9.90–10.10 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 20.54, 20.65, 55.80, 55.89, 110.91, 111.82, 113.10, 114.04, 120.46, 120.80, 124.20, 124.75, 124.87, 125.07, 125.31, 126.30, 128.27, 128.66, 129.86, 130.65, 139.95, 140.16, 155.25, 156.06, 180.59, 180.86 ppm; HRMS ESI (M+H+) calcd for C26H19Cl6N2O4 632.9476, found 632.9492; IR (KBr) 3232, 2955, 2918, 2849, 1736, 1644, 1604, 1462, 1428, 1401, 1172, 1039, 871, 678 cm–1.
4.1.9. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((5-chloro-2-hydroxy-4-methylphenyl)methanone) (24)
To a solution of 10 (47 mg, 0.07 mmol) in anhydrous CH2Cl2 (5 mL) was slowly added a solution of BBr3 (75 mg, 0.30 mmol) in anhydrous CH2Cl2 (1 mL) via a syringe under N2 at –78 °C. After being stirred for 0.5 h, the reaction was quenched by addition of water (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (12% EtOAc/petroleum ether, Rf = 0.2) to yield 24 (38 mg, 85% yield) as a pale brown solid. mp 84.7–86.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 2.39 (s, 3H), 6.76 (s, 1H), 6.80 (s, 1H), 6.90 (s, 1H), 7.39 (s, 2H), 9.89 (br s, 1H), 10.29 (s, 1H), 10.98 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 20.72, 20.80, 113.28, 117.53, 117.77, 119.80, 119.82, 120.40, 120.44, 120.74, 120.76, 123.34, 123.43, 124.38, 124.72, 128.60, 129.65, 130.91, 145.51, 145.71, 159.52, 160.76, 184.34, 185.20 ppm; HRMS ESI (M+H+) calcd for C24H15Cl6N2O4 604.9163, found 604.9168; IR (KBr) 3415, 3238, 2955, 2927, 2856, 1628, 1595, 1479, 1430, 1215, 1027, 871, 690 cm–1. HPLC purity, 95.6% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 75 : 25; tR = 9.0 min).
4.1.10. (4,4′,5,5′-Tetrachloro-1′-tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-methoxy-4-((trimethylsilyl)ethynyl)phenyl)methanone) (12)
Under N2, a mixture of 11 [12] (300 mg, 0.30 mmol), ethynyltrimethylsilane (176 mg, 1.80 mmol), Pd(PPh3)4 (70 mg, 0.06 mmol) and Et3N (30 mg, 0.30 mmol) was dissolved in anhydrous DMF (5 mL). The reaction was allowed to stir for about 16 h at room temperature. The reaction was quenched with water (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (10% EtOAc/petroleum ether, Rf = 0.3) to yield 12 (199 mg, 74% yield) as a light yellow solid. mp 118.7–120.0 °C; 1H NMR(400 MHz, acetone-d6) δ 0.25 (s, 18H), 2.54 (s, 3H), 3.56 (s, 3H), 3.63 (s, 3H), 6.52 (s, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.93 (d, J = 2.8 Hz, 2H), 7.04 (dd, J = 8.0, 1.2 Hz, 1H), 7.13 (d, J = 7.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 2H), 7.96 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ −0.18, −0.18, −0.18, −0.14, −0.14, −0.14, 21.72, 56.11, 56.11, 96.70, 97.96, 104.84, 105.05, 114.43, 114.66, 115.16, 116.20, 117.25, 118.20, 120.38, 121.80, 123.55, 124.09, 124.78, 127.79, 127.98, 129.23, 129.23, 129.80, 130.33, 131.02, 131.02, 131.10, 131.64, 134.42, 134.99, 147.58, 158.07, 159.35, 181.79, 183.11 ppm; HRMS ESI (M+H+) calcd for C41H39Cl4N2O6SSi2 883.0821, found 883.0812; IR (KBr) 3445, 2960, 2857, 2159, 1654, 1600, 1556, 1456, 1400, 1268, 1250, 1192, 1034, 951, 851 cm–1.
4.1.11. (4,4′,5,5′-Tetrachloro-1′-tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-((trimethylsilyl)ethynyl)phenyl)methanone) (13)
To a solution of 12 (43 mg, 0.05 mmol) in anhydrous CH2Cl2 (5 mL) was slowly added a solution of BBr3 (61 mg, 0.24 mmol) in anhydrous CH2Cl2 (1 mL) via a syringe under N2 at –78 °C. After being stirred for 30 min, the reaction was quenched by addition of water (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (10% EtOAc/petroleum ether, Rf = 0.2) to yield 13 (22 mg, 53% yield) as a pale brown solid. mp 97.3–99.7 °C; 1H NMR (400 MHz, acetone-d6) δ 0.27 (s, 18H), 2.52 (s, 3H), 6.86 (s, 1H), 6.91-6.93 (m, 2H), 6.96 (d, J = 8.8 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 3H), 7.90 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ −0.23, −0.23, −0.23, −0.23, −0.23, −0.23, 21.84, 99.67, 103.39, 103.72, 106.30, 112.25, 113.64, 113.86, 120.07, 120.83, 121.20, 121.25, 121.35, 122.60, 122.62, 122.80, 124.44, 128.25, 128.25, 128.26, 130.14, 130.14, 131.68, 131.94, 131.98, 133.23, 133.80, 146.95, 146.95, 162.00, 162.00, 180.55, 188.94 ppm; HRMS ESI (M+H+) calcd for C39H35Cl4N2O6SSi2 855.0508, found 855.0502; IR (KBr) 2957, 2923, 2852, 2159, 1728, 1624, 1547, 1382, 1343, 1245, 1191, 973, 850, 662 cm–1.
4.1.12. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((4-ethynyl-2-hydroxyphenyl)methanone) (25)
To a solution of 13 (22 mg, 0.03 mmol) in a mixture of MeOH/THF (1:1, 3 mL) was added KOH (7.2 mg, 0.13 mmol) at room temperature. After being stirred for 15 min, the reaction was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (15% EtOAc/petroleum ether, Rf = 0.2) to yield 25 (13 mg, 91% yield) as a light yellow solid. mp 83.3–84.1 °C; 1H NMR (400 MHz, acetone-d6) δ 3.90 (s, 1H), 4.00 (s, 1H), 6.49 (s, 1H), 6.81 (d, J = 8.0 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 7.05 (s, 1H), 7.54 (t, J = 8.4 Hz, 3H), 10.50 (s, 1H), 10.82 (s, 1H) 12.39 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 78.81, 82.37, 82.99, 83.19, 109.91, 120.99, 121.00, 121.09, 121.19, 121.81, 122.40, 122.75, 123.54, 124.80, 125.28, 129.32, 130.71, 131.35, 134.03, 137.85, 141.50, 142.85, 161.00, 161.22, 185.62, 186.50 ppm; HRMS ESI (M+H+) calcd for C26H13Cl4N2O4 556.9629, found 556.9632; IR (KBr) 3405, 3295, 2969, 2929, 2108, 1701, 1624, 1594, 1551, 1448, 1393, 1332, 1246, 1120, 965, 788, 675 cm–1. HPLC purity, 99.1% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 70 : 30; tR = 5.7 min).
4.1.13. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-vinylphenyl)methanone) (26)
Under 1 atm H2, 25 (100 mg, 0.18 mmol) and Pd/BaSO4 (5 mg) were dissolved in MeOH (3 mL). The reaction was allowed to cool to 10 °C and stirred for about 30 min. The suspension was filtered and the filtrate was washed with EtOAc (50 mL). The combined organic layers were concentrated in vacuum and the residue was purified by column chromatography (20% EtOAc/petroleum ether, Rf = 0.2) to yield 26 (61 mg, 60% yield) as a yellow solid. mp 76.4–77.7 °C; 1H NMR (400 MHz, acetone-d6) δ 5.41 (d, J = 10.8 Hz, 1H), 5.49 (d, J = 11.2 Hz, 1H), 5.95 (d, J = 17.6 Hz, 1H), 6.04 (d, J = 17.6 Hz, 1H), 6.50 (s, 1H), 6.95-6.79 (m, 2H), 6.81 (dd, J = 8.0, 1.2 Hz, 1H), 6.98 (d, J = 1.2 Hz, 1H), 7.03 (s, 1H), 7.07 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 7.2 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 11.16 (s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 90.24, 107.07, 109.66, 111.20, 115.52, 115.60, 117.12, 118.04, 118.27, 119.04, 119.89, 128.70, 130.80, 131.83, 134.74, 136.58, 136.60, 137.88, 145.64, 147.00, 152.18, 162.22, 171.91, 172.95, 185.92, 186.38 ppm; HRMS ESI (M+H+) calcd for C26H17Cl4N2O4 560.9942, found 560.9952; IR (KBr) 3423, 3275, 2961, 2926, 1920, 1847, 1737, 1626, 1575, 1499, 1450, 1390, 1353, 1216, 887, 797, 723 cm–1. HPLC purity, 99.2% (Flow rate, 1.0 mL/min; Column, Agilent ZORBAX 300SB-C8, 5 μm, 150×4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 90 : 10; tR = 5.1 min).
4.1.14. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((4-ethyl-2-hydroxyphenyl)methanone) (27)
Under 1 atm H2, 26 (50 mg, 0.09 mmol) and Pd/BaSO4 (5 mg) were dissolved in MeOH (3 mL). The reaction was allowed to stir for about 3 h at room temperature. The suspension was filtered and the filtrate was washed with EtOAc (50 mL). The combined organic layers were concentrated in vacuum and the residue was purified by column chromatography (12% EtOAc/petroleum ether, Rf = 0.2) to yield 27 (48 mg, 96% yield) as a yellow solid. mp 90.3–92.0 °C; 1H NMR (400 MHz, acetone-d6) δ 1.17–1.24 (m, 6H), 2.59 (dd, J = 15.2, 7.6 Hz, 2H), 2.65 (dd, J = 15.2, 7.6 Hz, 2H), 6.47 (s, 1H), 6.53 (dd, J = 8.4, 2.8 Hz, 1H), 6.77–6.80 (m, 3H), 7.35 (br s, 1H), 7.47 (d, J = 8.0 Hz, 1H), 10.80 (br s, 1H), 11.24 (s, 1H), 12.27 (br s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 15.01, 15.01, 15.59, 15.59, 109.53, 117.00, 117.16, 118.24, 118.34, 119.10, 120.31, 121.72, 124.79, 126.30, 128.10, 131.56, 131.56, 134.54, 154.47, 154.47, 156.17, 156.17, 163.15, 163.41, 186.65, 188.42 ppm; HRMS ESI (M+H+) calcd for C26H21Cl4N2O4 565.0255, found 565.0261; IR (KBr) 3420, 3251, 2967, 2932, 1628, 1590, 1500, 1450, 1393, 1258, 1124, 944, 792, 531cm–1. HPLC purity, 97.5% (Flow rate: 1.0 mL/min; Column, Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 75 : 25; tR = 9.4 min).
4.1.15. (1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-methoxy-4-((trimethylsilyl)ethynyl)phenyl)methanone) (15)
Under N2, a mixture of 14 [12] (50 mg, 0.06 mmol), ethynyltrimethylsilane (34 mg, 0.35 mmol), Pd(PPh3)4 (15 mg, 0.01 mmol) and Et3N (18 mg, 0.18 mmol) was dissolved in anhydrous DMF (5 mL). The reaction was heated to 60 °C and stirred for 10 h. The reaction was quenched with water (10 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (10% EtOAc/petroleum, Rf = 0.3) to yield 15 (40 mg, 92% yield) as a brown solid. mp 105.3–106.7 °C; 1H NMR (400 MHz, CDCl3) δ 0.07 (s, 6H), 0.26 (s, 6H), 0.27 (s, 6H), 2.44 (s, 3H), 3.65 (s, 3H), 3.77 (s, 3H), 5.90 (t, J = 2.8 Hz, 1H), 6.29 (dd, J = 4.0, 1.2 Hz, 1H), 6.44 (d, J = 3.2 Hz, 1H), 6.71 (br s, 2H), 6.79 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 8.0 Hz, 1H), 6.99 (s, 1H), 7.04 (d, J = 7.6 Hz, 1H), 7.21 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 3.2 Hz, 1H), 7.94 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ –0.13, –0.13, –0.13, –0.13, –0.13, –0.13, –21.71, 55.67, 55.67, 95.75, 96.80, 104.23, 104.31, 109.13, 111.89, 114.16, 114.39, 123.46, 123.55, 123.55, 124.47, 125.81, 127.61, 127.65, 128.31, 128.31, 128.52, 129.06, 129.63, 129.63, 129.90, 130.86, 131.92, 132.76, 133.02, 135.74, 145.16, 156.89, 157.58, 182.54, 184.05 ppm; HRMS ESI (M+H+) calcd for C41H43N2O6SSi2 747.2380, found 747.2382; IR (KBr) 3443, 3145, 2959, 2857, 2158, 1649, 1600, 1556, 1405, 1377, 1272, 1253, 1175, 1135, 1034, 952, 852, 667 cm–1.
4.1.16. 1′H-1,3′-Bipyrrole-2,2′-diylbis((4-ethynyl-2-methoxyphenyl)methanone) (16)
To a solution of 15 (300 mg, 0.40 mmol) in a mixture of MeOH/THF (1:1, 10 mL) was added KOH (113 mg, 2.0 mmol) at room temperature. After being stirred for 2 h, the reaction was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to yield 16 (171 mg, 95% yield) as a brown solid. mp 178.7–180.0 °C; 1H NMR (400 MHz, CDCl3) δ 3.10 (s, 1H), 3.15 (s, 1H), 3.69 (s, 3H), 3.79 (s, 3H), 5.88 (dd, J = 4.0, 2.4 Hz, 1H), 6.31 (t, J = 2.4 Hz, 1H), 6.36 (dd, J = 4.0, 1.6 Hz, 1H), 6.64 (t, J = 2.0 Hz, 1H), 6.78 (s, 1H), 6.79 (d, J = 7.6 Hz, 1H), 7.04 (s, 1H), 7.08 (t, J = 2.8 Hz, 1H), 7.10–7.12 (m, 3H), 9.43 (br s, 1H) ppm; 13C NMR (DMSO-d6+acetone-d6, 100 MHz) δ 55.39, 55.65, 80.75, 80.98, 83.21, 83.38, 108.79, 109.96, 114.08, 114.94, 122.74, 123.51, 123.68, 123.76, 124.20, 124.45, 126.25, 128.65, 129.17, 129.75, 130.70, 131.13, 132.10, 133.06, 156.16, 156.69, 181.92, 182.70 ppm; HRMS ESI (M+H+) calcd for C28H21N2O4 449.1501, found 449.1494; IR (KBr) 3339, 3281, 3259, 3130, 2942, 2855, 1645, 1612, 1559, 1494, 1409, 1263, 1121, 938, 749 cm–1.
4.1.17. Diethyl 2,2′-(4,4′-((1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(3-methoxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (17)
Under N2, a mixture of 16 (50 mg, 0.11 mmol), ethyl 2-azidoacetate (58 mg, 0.44 mmol), and CuCl (10 mg, 0.11 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 10 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to yield 17 (61 mg, 78% yield) as a light yellow solid. mp 136.3–137.7 °C; 1H NMR (400 MHz, CDCl3) δ 1.31–1.41 (m, 6H), 3.78 (s, 3H), 3.87 (s, 3H), 4.27–4.33 (m, 4H), 5.22 (s, 4H), 5.87 (dd, J = 4.0, 2.8 Hz, 1H), 6.33 (t, J = 2.4 Hz, 1H), 6.36 (dd, J = 4.0, 1.6, 1H), 6.71 (t, J = 2.4 Hz, 1H), 7.01 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 3.2 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.22–7.24 (m, 2H), 7.36 (s, 1H), 7.55 (s, 1H), 7.89 (s, 1H), 8.00 (s, 1H), 9.45 (br s, 1H) ppm; 13C NMR (DMSO-d6+CD3OD, 100 MHz) δ 14.51, 14.59, 51.80, 51.80, 56.04, 56.28, 63.15, 63.15, 108.73, 109.47, 110.08, 111.05, 117.76, 118.22, 124.40, 124.50, 124.55, 124.59, 124.59, 127.38, 129.49, 130.13, 130.54, 131.28, 132.62, 133.40, 134.38, 134.48, 134.55, 147.93, 158.25, 158.82, 168.38, 168.43, 184.34, 184.86 ppm; HRMS ESI (M+H+) calcd for C36H35N8O8 707.2578, found 707.2588; IR (KBr) 3420, 3265, 3139, 2986, 2942, 2852, 1745, 1634, 1614, 1562, 1412, 1247, 1226, 1132, 1028, 932, 783 cm–1.
4.1.18. Diethyl 2,2′-(4,4′-((4,4′,5,5′-tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(2-chloro-5-methoxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (18)
To a solution of 17 (40 mg, 0.06mmol) in AcOH (3 mL) at room temperature was added NCS (54 mg, 0.40 mmol) slowly. The reaction was allowed to stir for about 8 h at room temperature. The reaction was quenched with water (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (25% EtOAc/petroleum ether, Rf = 0.2) to yield 18 (5 mg, 10% yield) as a brown solid. mp 118.7–120.3 °C; 1H NMR (400 MHz, CDCl3) δ 1.28-1.36 (m, 6H), 3.84 (s, 3H), 3.87 (s, 3H), 4.25–4.33 (m, 4H), 5.23 (s, 2H), 5.27 (s, 2H), 6.49 (s, 1H), 7.23 (s, 1H), 7.36 (s, 1H), 7.79 (s, 1H), 7.90 (s, 1H), 8.36 (s, 1H), 8.45 (s, 1H), 10.86 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 29.59, 29.63, 50.94, 51.00, 56.11, 56.14, 62.52, 62.58, 111.11, 111.44, 112.20, 112.31, 120.78, 121.14, 121.59, 121.82, 124.20, 125.09, 125.19, 125.24, 125.28, 126.76, 128.03, 129.63, 130.51, 131.09, 131.71, 131.93, 143.50, 143.66, 155.59, 156.17, 166.04, 166.10, 180.07, 180.49 ppm; HRMS ESI (M+H+) calcd for C36H29Cl6N8O8 911.0240, found 911.0265; IR (KBr) 3419, 3162, 2925, 2852, 1750, 1645, 1607, 1463, 1396, 1254, 1218, 1022, 915 cm–1.
4.1.19. Diethyl 2,2′-(4,4′-((4,4′,5,5′-tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(2-chloro-5-hydroxy-4,1- phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (36)
To a solution of 18 (40 mg, 0.04 mmol) in anhydrous CH2Cl2 (5 mL) was slowly added a solution of BBr3 (44 mg, 0.16 mmol) in anhydrous CH2Cl2 (1 mL) via a syringe under N2 at –78 °C. After being stirred for 2 h, the reaction was quenched by addition of water (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (30% EtOAc/petroleum ether, Rf = 0.2) to yield 36 (19 mg, 50% yield) as a brown solid. mp 157.3–169.7 °C; 1H NMR (400 MHz, CDCl3) δ 1.29–1.35 (m, 6H), 4.25–4.34 (m, 4H), 5.21 (s, 2H), 5.25 (s, 2H), 6.81 (s, 1H), 7.65 (br s, 2H), 7.92 (s, 1H), 7.98 (s, 1H), 8.37 (s, 1H), 8.46 (s, 1H), 10.91 (br s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 14.31, 14.31, 51.39, 51.49, 62.40, 62.46, 109.10, 111.18, 118.09, 118.25, 118.87, 118.95, 119.75, 120.46, 120.93, 121.53, 123.79, 126.17, 126.71, 126.92, 127.51, 131.06, 134.03, 134.39, 134.61, 136.25, 143.15, 143.78, 158.99, 160.48, 167.59, 167.67, 181.63, 186.11 ppm; HRMS ESI (M+H+) calcd for C34H25Cl6N8O8 882.9927, found 882.9933; IR (KBr) 3446, 2955, 2923, 2850, 1749, 1627, 1458, 1377, 1218, 1020, 919, 775 cm–1. HPLC purity, 95.2% (Flow rate: 1.0 mL/min; Column: Phenomenex C6-phenyl, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 80 : 20; tR =10.8 min).
4.1.20. (1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene)bis(trifluoromethanesulfonate) (19)
To a solution of 14 [12] (2.5 g, 2.94 mmol) in anhydrous CH2Cl2 (100 mL) was slowly added a solution of BBr3 (3.68 g, 14.70 mmol) in anhydrous CH2Cl2 (5 mL) via a syringe under N2 at –78 °C. After being stirred for 0.5 h, the reaction was quenched by addition of water (100 mL) and extracted with CH2Cl2 (50 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (12% EtOAc/petroleum ether, Rf = 0.3) to yield 19 (2.17 g, 90% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 2.48 (s, 3H), 6.15 (t, J = 2.8 Hz, 1H), 6.40 (d, J = 3.6 Hz, 1H), 6.45 (dd, J = 8.8, 2.4 Hz, 1H), 6.70 (d, J = 3.6 Hz, 1H), 6.75 (d, J = 2.4 Hz, 1H), 6.78 (dd, J = 8.8, 2.4 Hz, 1H), 6.84 (s, 1H), 6.91 (d, J = 2.0 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.56 (s, 1H), 7.57 (d, J = 5.2 Hz, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.89 (d, J = 8.4 Hz, 2H), 11.48 (s, 1H), 11.83 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.70, 110.72, 110.72, 111.03, 111.09, 111.72, 112.06, 119.24, 119.59, 123.57, 123.57, 125.45, 128.16, 128.16, 128.40, 129.93, 129.93, 130.00, 131.77, 131.85, 133.88, 133.88, 134.48, 135.00, 146.15, 153.58, 153.98, 163.57, 164.06, 186.46, 189.44 ppm. HRMS ESI (M+H+) calcd for C31H21F6N2O12S3 823.0161, found 823.0173. IR (KBr) 3159, 2943, 1685, 1671, 1550, 1454, 1368, 1272, 1137, 1027, 899 cm–1.
4.1.21. (1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-((trimethylsilyl)ethynyl)phenyl)methanone) (20)
Under N2, a mixture of 19 (100 mg, 0.12 mmol), ethynyltrimethylsilane (72 mg, 0.73 mmol), Pd(PPh3)4 (30 mg, 0.02 mmol) and Et3N (37 mg, 0.36 mmol) was dissolved in anhydrous DMF (5 mL). The reaction was allowed to warm up to 70 °C and stirred for about 10 h. The reaction was quenched with water (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (10% EtOAc/petroleum ether, Rf = 0.3) to yield 20 (85 mg, 98% yield) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 0.23 (s, 9H), 0.26 (s, 9H), 2.45 (s, 3H), 6.09 (t, J = 2.8 Hz, 1H), 6.39 (d, J = 3.2 Hz, 1H), 6.56 (d, J = 8.0 Hz, 1H), 6.65 (d, J = 3.2 Hz, 1H), 6.77 (s, 1H), 6.90 (s, 1H), 6.92 (d, J = 8.8 Hz, 1H), 7.06 (s, 1H), 7.34–7.38 (m, 3H), 7.48 (d, J = 3.6 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.0 Hz, 2H), 11.34 (s, 1H), 11.55 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ –0.26, –0.26, –0.26, –0.21, –0.21, –0.21, 21.76, 98.47, 99.46, 103.56, 103.78, 110.47, 110.95, 119.48, 119.66, 120.61, 121.14, 122.07, 122.55, 123.15, 123.92, 124.19, 128.14, 128.14, 129.86, 129.86, 130.08, 130.35, 131.10, 131.40, 131.48, 131.98, 132.49, 135.18, 145.91, 161.91, 162.05, 187.11, 190.31 ppm; HRMS ESI (M+H+) calcd for C39H39N2O6SSi2 719.2067, found 719.2062. IR (KBr) 3159, 2997, 2973, 1795, 1681, 1580, 1417, 1272, 1167, 1097, 878 cm–1.
4.1.22. 1′H-1,3′-Bipyrrole-2,2′-diylbis((4-ethynyl-2-hydroxyphenyl)methanone) (21)
To a solution of 20 (85 mg, 0.12 mmol) in a mixture of MeOH/THF (1:1, 5 mL) was added KOH (33 mg, 0.59 mmol) at room temperature. After being stirred for 1.5 h, the reaction was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to yield 21 (47 mg, 95% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 3.20 (s, 1H), 3.24 (s, 1H), 6.23 (t, J = 3.2 Hz, 1H), 6.36 (s, 1H), 6.55 (d, J = 8.0 Hz, 1H), 6.72 (d, J = 3.2 Hz, 1H), 6.90 (d, J = 6.8 Hz, 1H), 6.93 (d, J = 4.0 Hz, 2H), 7.07 (s, 1H), 7.14 (t, J = 2.8 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 9.48 (br s, 1H), 10.97 (s, 1H), 11.48 (s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 82.18, 82.40, 83.10, 109.73, 109.79, 110.96, 120.91, 121.00, 121.06, 121.15, 121.24, 122.49, 122.57, 123.83, 124.92, 125.09, 128.81, 129.37, 131.01, 131.37, 133.14, 161.25, 162.14, 162.42, 187.26, 187.82 ppm; HRMS ESI (M+Na+) calcd for C26H16N2NaO4 443.1008, found 443.1003. IR (KBr) 3435, 3239, 2980, 1785, 1691, 1590, 1424, 1127, 1017, 886 cm–1.
4.1.23. Di-tert-butyl 2,2′-(4,4′-((1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (22)
Under N2, a mixture of 21 (200 mg, 0.48 mmol), tert-butyl 2-azidoacetate (300 mg, 1.90 mmol), and CuCl (47 mg, 0.48 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 8 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (40% EtOAc/petroleum ether, Rf = 0.3) to yield 22 (280 mg, 80% yield) as a yellow solid. mp 96.0–97.0 °C; 1H NMR (400 MHz, acetone-d6) δ 1.48 (s, 9H), 1.49 (s, 9H), 5.31 (s, 2H), 5.33 (s, 2H), 6.28 (dd, J = 3.6, 2.8 Hz, 1H), 6.44 (t, J = 2.4 Hz, 1H), 6.77 (dd, J = 4.0, 1.6 Hz, 1H), 7.10 (dd, J = 8.4, 1.6 Hz, 1H), 7.26–7.32 (m, 4H), 7.44 (d, J = 1.6 Hz, 1H), 7.48–7.51 (m, 2H), 8.49 (s, 1H), 8.52 (s, 1H), 11.29 (br s, 1H), 11.36 (s, 1H), 11.83 (s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 28.03, 28.03, 28.03, 28.03, 28.03, 28.03, 52.10, 52.10, 83.40, 83.40, 109.66, 110.87, 114.19, 114.38, 116.28, 116.29, 119.66, 119.89, 123.34, 123.35, 124.09, 124.44, 124.48, 124.67, 130.89, 131.29, 132.42, 132.74, 134.03, 138.29, 138.58, 146.62, 162.93, 163.74, 166.68, 166.71, 188.03, 188.48 ppm; HRMS ESI (M+H+) calcd for C38H39N8O8 735.2891, found 735.2894; IR (KBr) 3405, 3139, 2977, 2933, 1745, 1631, 1590, 1414, 1368, 1242, 1157, 1047, 898, 793 cm–1.
4.1.24. Di-tert-butyl 2,2′-(4,4′-((4,4′,5,5′-tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(2-chloro-5-hydroxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (37)
To a solution of 22 (10 mg, 0.01 mmol) in MeCN (1 mL) at room temperature was added NCS (10 mg, 0.07 mmol) slowly. The reaction was allowed to stir for about 2 h at room temperature. The reaction was quenched with water (5 mL) and extracted with EtOAc (5 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by column chromatography (40% EtOAc/petroleum ether, Rf = 0.2) to yield 37 (2 mg, 14% yield) as a pale brown solid. mp 103.7–105.0 °C; 1H NMR (400 MHz, acetone-d6) δ 1.46 (s, 18H), 5.31 (s, 2H), 5.35 (s, 2H), 7.05 (s, 1H), 7.73 (s, 1H), 7.80 (s, 1H), 8.01 (s, 1H), 8.35 (s, 1H), 8.60 (s, 1H), 8.74 (s, 1H), 10.94 (s, 1H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ 28.00, 28.00, 28.00, 28.00, 28.04, 28.04, 51.50, 56.12, 69.00, 83.00, 108.20, 110.04, 113.40, 115.40, 116.83, 117.40, 119.00, 119.20, 119.58, 119.95, 121.80, 123.34, 124.04, 125.83, 126.50, 126.59, 131.14, 131.62, 132.43, 133.16, 142.33, 142.41, 155.48, 157.51, 166.53, 166.53, 179.98, 181.85 ppm; HRMS ESI (M+Na+) calcd for C38H32Cl6N8NaO8 961.0372, found 961.0388; IR (KBr) 3442, 2979, 2920, 2851, 1744, 1629, 1460, 1427, 1383, 1243, 1155, 1025, 752, 666 cm–1. HPLC purity, 96.2% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 75 : 25; tR = 5.9 min).
4.1.25. 2,2′-(4,4′-((4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(2-chloro-5-hydroxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetic acid (38)
To a solution of 37 (30 mg, 0.03 mmol) in anhydrous CH2Cl2 (2 mL) was slowly added CF3COOH (2 mL) via a syringe at 0 °C. The reaction was allowed to warm up to room temperature and stirred for about 4 h. The reaction was concentrated in vacuum. The residue was purified by reverse-phase column chromatography (C18 reverse silica gel, 6% AcOH, 30% H2O, 64% MeOH, Rf = 0.2) to yield 38 (17 mg, 65% yield) as a pale brown solid. mp 261.4–262.7 °C; 1H NMR (400 MHz, CD3OD) δ 5.18 (s, 2H), 5.21 (s, 2H), 6.73 (s, 1H), 7.23 (s, 1H), 7.35 (s, 1H), 7.48(s, 1H), 8.59 (s, 1H), 8.87 (s, 1H), 8.48 (s, 1H), 8.57 (s, 1H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ 52.98, 53.62, 108.07, 109.84, 116.57, 118.82, 119.32, 119.62, 121.71, 123.24, 123.81, 125.44, 125.90, 126.23, 129.42, 131.17, 131.66, 131.68, 132.88, 133.50, 141.85, 141.97, 155.58, 157.52, 161.00, 162.77, 169.56, 172.54, 179.98, 182.01 ppm; HRMS ESI (M+H+) calcd for C30H17Cl6N8O8 826.9301, found 826.9308; IR (KBr) 3392, 2956, 2921, 2851, 1753, 1626, 1462, 1432, 1380, 1246, 1188, 1081, 1025, 770 cm–1. HPLC purity, 98.8% (Flow rate: 1.0 mL/min; Column: Waters C18, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 65 : 35; tR = 4.2 min).
4.1.26. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((4-(1-benzyl-1H-1,2,3-triazol-5-yl)-2-hydroxyphenyl)methanone) (39)
Under N2, a mixture of 25 (20 mg, 0.04 mmol), (azidomethyl)benzene (29 mg, 0.22 mmol), and CuCl (4 mg, 0.04 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 8 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (5% acetone, 31% EtOAc, 64% petroleum ether, Rf = 0.3) to yield 39 (17 mg, 55% yield) as a yellow solid. mp 116.5–118.0 °C; 1H NMR (400 MHz, CDCl3) δ 5.56 (s, 2H), 5.57 (s, 2H), 6.16 (s, 1H), 7.01 (d, J = 8.4 Hz, 1H), 7.28–7.31 (m, 7H), 7.32–7.40 (m, 7H), 7.51 (br s, 1H), 7.69 (s, 1H), 7.75 (s, 1H), 10.35 (br s, 1H), 10.83 (s, 1H), 11.39 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 54.36, 54.36, 112.01, 114.28, 114.28, 115.89, 116.54, 117.18, 118.47, 118.65, 121.17, 121.32, 121.74, 121.94, 124.33, 124.84, 128.12, 128.12, 128.12, 128.19, 128.19, 128.19, 128.95, 128.95, 128.95, 129.22, 129.22, 129.22, 129.22, 131.25, 134.16, 134.16, 134.16, 137.75, 138.85, 146.47, 162.06, 163.05, 185.56, 187.37 ppm; HRMS ESI (M+H+) calcd for C40H27Cl4N8O4 823.0909, found 823.0903; IR (KBr) 2955, 2919, 2850, 1717, 1628, 1592, 1457, 1228, 1047, 935, 912, 790, 699 cm–1. HPLC purity, 98.5% (Flow rate: 1.0 mL/min; Column, Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 85 : 15; tR = 12.5 min).
4.1.27. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-(1-phenyl-1H-1,2,3-triazol-5-yl)phenyl)methanone) (40)
Under N2, a mixture of 25 (20 mg, 0.04 mmol), azidobenzene (26 mg, 0.22 mmol), and CuCl (4 mg, 0.04 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 8 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (5% acetone, 31% EtOAc, 64% petroleum ether, Rf = 0.3) to yield 40 (20 mg, 70% yield) as a yellow solid. mp 128.7–130.3 °C; 1H NMR (400 MHz, CDCl3) δ 6.24 (s, 1H), 7.16 (d, J = 8.0 Hz, 1H), 7.41–7.48 (m, 6H), 7.52–7.56 (m, 5H), 7.77 (d, J = 8.0 Hz, 4H), 8.24 (s, 1H), 8.30 (s, 1H), 10.35 (br s, 1H), 10.91 (s, 1H), 11.47 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 109.13, 112.09, 114.48, 114.52, 116.00, 116.69, 117.17, 118.68, 118.83, 119.23, 119.40, 120.51, 120.51, 120.51, 120.51, 120.51, 120.51, 121.76, 122.02, 124.43, 124.86, 129.05, 129.09, 129.83, 129.83, 129.83, 129.83, 129.83, 131.29, 134.25, 136.64, 137.44, 138.55, 146.68, 162.15, 163.12, 185.54, 187.50 ppm; HRMS ESI (M+H+) calcd for C38H23Cl4N8O4 795.0596, found 795.0590; IR (KBr) 2955, 2918, 2849, 1701, 1630, 1594, 1503, 1459, 1238, 1036, 935, 912, 757, 686 cm–1. HPLC purity, 97.4% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 85 : 15; tR = 10.8 min).
4.1.28. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((4-(1-cyclohexyl-1H-1,2,3-triazol-5-yl)-2-hydroxyphenyl)methanone) (41)
Under N2, a mixture of 25 (20 mg, 0.04 mmol), azidocyclohexane (27 mg, 0.22 mmol), and CuCl (4 mg, 0.04 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 8 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (5% acetone, 31% EtOAc, 64% petroleum ether, Rf = 0.3) to yield 41 (15 mg, 52% yield) as a yellow solid. mp 143.8–145.0 °C; 1H NMR (400 MHz, CDCl3) δ 1.25–1.39 (m, 2H), 1.44–1.59 (m, 4H), 1.78–1.87 (m, 6H), 1.94–1.97 (m, 4H), 2.25–2.27 (m, 4H), 4.49 (t, J = 10.0 Hz, 2H), 6.21 (s, 1H), 7.06 (d, J = 8.4 Hz, 1H), 7.31–7.37 (m, 4H), 7.51 (br s, 1H), 7.82 (s, 1H), 7.87 (s, 1H), 10.47 (br s, 1H), 10.91 (s, 1H), 11.44 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 25.04, 25.04, 25.11, 25.11, 29.20, 29.65, 33.52, 33.52, 33.52, 33.52, 60.36, 60.36, 109.00, 112.00, 114.12, 114.22, 115.88, 116.54, 117.09, 118.32, 118.54, 119.10, 119.20, 121.64, 121.86, 124.26, 124.86, 131.25, 134.18, 134.18, 138.22, 139.29, 145.60, 145.60, 162.23, 163.12, 185.59, 187.39 ppm; HRMS ESI (M+H+) calcd for C38H35Cl4N8O4 807.1535, found 807.1532; IR (KBr) 3129, 2923, 2853, 1718, 1628, 1593, 1448, 1413, 1392, 1334, 1297, 1228, 1052, 914, 790 cm–1. HPLC purity, 95.0% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature : 25 °C; Mobile phase: MeOH : H2O = 85 : 15; tR = 20.9 min).
4.1.29. (4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-(1-octyl-1H-1,2,3-triazol-5-yl)phenyl)methanone) (42)
Under N2, a mixture of 25 (20 mg, 0.04 mmol), 1-azidooctane (27 mg, 0.22 mmol), and CuCl (4 mg, 0.04 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 8 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (5% acetone, 31% EtOAc, 64% petroleum ether, Rf = 0.3) to yield 42 (15 mg, 48% yield) as a yellow solid. mp 85.2–86.7 °C; 1H NMR (400 MHz, CDCl3) δ 0.84–0.91 (m, 6H), 1.14–1.35 (m, 20H), 1.93–1.95 (m, 4H), 4.40 (t, J = 6.4 Hz, 4H), 6.20 (s, 1H), 7.06 (d, J = 8.4 Hz, 1H), 7.27–7.37 (m, 4H), 7.52 (br s, 1H), 7.80 (s, 1H), 7.85 (s, 1H), 10.48 (br s, 1H), 10.87 (s, 1H), 11.44 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 22.55, 22.55, 26.44, 26.44, 28.91, 28.91, 28.91, 29.00, 29.00, 29.00, 30.23, 30.26, 31.65, 31.65, 50.60, 50.60, 109.00, 112.02, 114.22, 114.26, 115.89, 116.56, 117.13, 118.41, 118.61, 121.12, 121.25, 121.70, 122.00, 124.34, 124.87, 126.85, 131.27, 134.20, 137.99, 139.09, 146.00, 162.13, 163.10, 163.10, 185.59, 187.40 ppm; HRMS ESI (M+H+) calcd for C42H47Cl4N8O4 867.2474, found 867.2482; IR (KBr) 3392, 3080, 2955, 2922, 2852, 1629, 1587, 1458, 1439, 1336, 1217, 1031, 914, 753, 703 cm–1. HPLC purity, 99.7% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 90 : 10; tR = 10.1 min).
4.1.30. Diethyl 2,2′-(4,4′-((4,4′,5,5′-tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (43)
Under N2, a mixture of 25 (20 mg, 0.04 mmol), ethyl 2-azidoacetate (28 mg, 0.22 mmol), and CuCl (4 mg, 0.04 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 4 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (5% acetone, 31% EtOAc, 64% petroleum ether, Rf = 0.3) to yield 43 (15 mg, 52% yield) as a yellow solid. mp 101.9–103.7 °C; 1H NMR (400 MHz, acetone-d6) δ 1.21-1.29 (m, 6H), 4.19-1.26 (m, 4H), 5.39 (s, 4H), 6.16 (s, 1H), 7.23 (dd, J = 8.4, 2.0 Hz, 1H), 7.33 (d, J = 1.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.42 (d, J = 1.6 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 8.39 (s, 1H), 8.49 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 14.04, 14.04, 50.97, 50.97, 62.62, 62.62, 109.05, 112.01, 114.38, 114.38, 114.38, 115.96, 116.62, 117.08, 118.61, 118.73, 121.67, 121.90, 122.73, 122.87, 124.37, 124.83, 131.25, 134.22, 137.46, 138.60, 146.48, 162.05, 163.03, 166.12, 166.18, 185.58, 187.49, 187.49 ppm; HRMS ESI (M+H+) calcd for C34H27Cl4N8O8 815.0706, found 815.0707; IR (KBr) 3670, 3389, 3080, 2959, 2920, 2850, 1752, 1631, 1584, 1481, 1439, 1216, 1030, 753 cm–1. HPLC purity, 98.4% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 75 : 25; tR = 7.7 min).
4.1.31. Di-tert-butyl 2,2′-(4,4′-((4,4′,5,5′-tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetate (44)
Under N2, a mixture of 25 (10 mg, 0.02 mmol), tert-butyl 2-azidoacetate (14 mg, 0.11 mmol), and CuCl (2 mg, 0.02 mmol) was dissolved in THF (5 mL). The reaction was allowed to warm up to reflux and stirred for about 8 h. The suspension was filtered and the filtrate was concentrated in vacuum. The residue was purified by column chromatography (5% acetone, 31% EtOAc, 64% petroleum ether, Rf = 0.3) to yield 44 (13 mg, 83% yield) as a yellow solid. mp 138.3–140.0 °C; 1H NMR (400 MHz, acetone-d6) δ 1.46 (s, 9H), 1.47 (s, 9H), 5.28 (s, 4H), 6.17 (s, 1H), 7.23 (dd, J = 8.4, 1.6 Hz, 1H), 7.34 (d, J = 1.6 Hz, 1H), 7.40 (dd, J = 8.4, 1.6 Hz, 1H), 7.43 (d, J = 0.8 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.38 (s, 1H), 8.48 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 27.89, 27.89, 27.89, 27.89, 27.89, 27.89, 51.57, 51.57, 84.09, 84.09, 108.98, 111.88, 114.23, 114.30, 115.90, 116.59, 116.98, 118.59, 118.69, 121.66, 121.92, 122.74, 122.90, 122.90, 124.39, 124.82, 126.85, 131.32, 134.19, 137.45, 138.65, 146.32, 161.95, 162.98, 165.10, 165.16, 185.65, 187.45 ppm; HRMS ESI (M+H+) calcd for C38H35Cl4N8O8 871.1332, found 871.1325; IR (KBr) 3425, 3141, 2980, 2931, 1745, 1630, 1599, 1454, 1369, 1239, 1156, 1048, 937, 852, 192 cm–1. HPLC purity, 99.5% (Flow rate: 1.0 mL/min; Column: Agilent ZORBAX 300SB-C8, 5 μm, 150×4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 80 : 20; tR = 7.3 min).
4.1.32. 2,2′-(4,4′-((4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-dicarbonyl)bis(3-hydroxy-4,1-phenylene))bis(1H-1,2,3-triazole-4,1-diyl))diacetic acid (45)
To a solution of 44 (17 mg, 0.02 mmol) in anhydrous CH2Cl2 (2 mL) was slowly added CF3COOH (2 mL) via a syringe at 0 °C. The reaction was allowed to warm up to room temperature and stirred for about 4 h and concentrated in vacuum. The residue was purified by reverse-phase column chromatography (C18 reverse silica gel, 6% AcOH, 30% H2O, 64% MeOH, Rf = 0.2) to yield 45 (14 mg, 94% yield) as a yellow solid. mp 100.3–102.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 4.80 (s, 2H), 4.83 (s, 2H), 6.09 (s, 1H), 7.15 (d, J = 8.4 Hz, 1H), 7.22 (s, 2H), 7.32 (s, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 8.4 Hz, 1H), 8.38 (s, 1H), 8.41 (s, 1H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ 53.17, 53.22, 109.12, 109.58, 112.93, 114.34, 114.99, 116.28, 121.49, 122.71, 123.87, 124.51, 124.67, 129.03, 130.14, 132.35, 133.28, 135.63, 137.40, 145.28, 145.79, 158.86, 159.46, 169.77, 169.80, 172.88, 177.84, 182.18, 185.38, 190.74 ppm; HRMS ESI (M+H+) calcd for C30H19Cl4N8O8 759.0080, found 759.0054; IR (KBr) 3417, 3268, 3136, 1627, 1457, 1431, 1393, 1307, 1234, 1025, 1002, 936, 799, 688 cm–1. HPLC purity, 99.7% (Flow rate: 1.0 mL/min; Column: Waters C18, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 65 : 35; tR = 4.1 min).
4.1.33. 4,4′,5,5′-Tetrachloro-1′-methyl-1′H-[1,3′-bipyrrole]-2,2′-diyl)bis((2-methoxyphenyl)methanone) (53)
To a mixture of 4,4′,5,5′-tetrachloro-1′H-[1,3′-bipyrrole]-2,2′-diyl)bis((2-methoxyphenyl)methanone (107 mg, 2.0 mmol) [2] and NaH (132 mg of 60% oil dispersion, 3.3 mmol) was added anhydrous DMF (24 mL) under argon at 0 °C. After the mixture was stirred for 10 min, methyl iodine (137 μL, 2.2 mmol) was added dropwise via a syringe. The reaction was quenched with water (20 mL) after stirred at 0 °C for 1 h. The mixture was neutralized with 0.1N HCl and extracted with EtOAc (3 × 20 mL). The organic layer was dried over MgSO4 and evaporated under vacuum. The residue was purified by chromatography (10% EtOAc, 90% hexane, Rf = 0.2) to give 53 as a white solid (100 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.81 (s, 3H), 4.06 (s, 3H), 6.26 (s, 1H), 6.67 (t, J = 7.5 Hz, 1H), 6.76 (d, J = 8.3 Hz, 1H), 6.92–6.99 (m, 2H), 7.17–7.25 (m, 3H), 7.38–7.44 (m, 1H). HRMS ESI (M+H+) calcd for C25H19Cl4N2O4 551.0099, found 551.0101. HPLC purity, 99.7% (Flow rate: 1.0 mL/min; Column: Waters C18, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 70 : 30; tR = 8.1 min).
4.1.34. (4,4′,5,5′-tetrachloro-1′-methyl-1′H-[1,3′-bipyrrole]-2,2′-diyl)bis((2-hydroxyphenyl)methanone) (54)
To a solution of 53 (45 mg, 0.08 mmol) in anhydrous DCM (3 mL) was added BBr3 (327 μL of 1.0 M solution in DCM, 0.33 mmol) dropwise under argon at 0 °C. The reaction was quenched with saturated NaHCO3 and the mixture was extracted with DCM. Yellow solid 54 (25 mg, 58%) was obtained after chromatography (5% EtOAc, 95% hexane, Rf = 0.3). 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 3H), 6.56 (dd, J = 8.3 and 8.1 Hz, 2H), 6.86–6.93 (m, 2H), 7.01 (d, J = 8.4 Hz, 1H), 7.30–7.38 (m, 1H), 7.46–7.56 (m, 2H), 7.64 (d, J = 7.8 Hz, 1H), 10.96 (s, 1H), 11.19 (s, 1H). HRMS ESI (M+H+) calcd for C23H15Cl4N2O4 522.9686, found 522.9690. HPLC purity, 99.2% (Flow rate: 1.0 mL/min; Column: Waters C18, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH : H2O = 70 : 30; tR = 7.2 min).
4.2. Enzyme-linked Immunosorbent Assay (ELISA)
ELISA assays were performed using the exact same procedure as we have recently described [42].
4.3. Direct Binding Measurement by Fluorescence Quenching
To investigate direct binding of compounds to Mcl-1, we have established a fluorescence-quenching assay based on the intrinsic Trp fluorescence of Mcl-1. Intrinsic fluorescence of Mcl-1 was measured with a TECAN M1000 plate reader, at excitation wavelength of 285 nm and emission wavelengths of 310-400 nm. Serial dilutions of compounds starting from 20 μM in 20 mM K-phosphate pH 7.6, 150 mM KCl were used to assay compound binding using 2.5 μM of Mcl-1. Each compound was tested at triplicate and binding constants were calculated by nonlinear regression analysis of dose response curves using Prism software 6.0 (Graphpad). Control experiments included titration of DMSO and use of denatured Mcl-1.
4.4. Activity in Intact Human Breast Cancer Cells
MDA-MB-468 breast cancer cells were treated and processed for western blotting exactly as we have recently described [42].
4.5. Protein Expression and Purification
Mcl-1 (residues 172-327) was expressed as maltose binding protein (MBP) fusions from the pSV282 vector (provided by Dr. Amy Keating from MIT). Mcl-1 protein was expressed in BL21(DE3) Codon+ at 18 °C for 17 h. Cells were lysed using a high pressure homogenizer in 100 mM Tris-HCl pH 8, 250 mM NaCl, 5 mM 2-mercaptoethanol, 25 mM imidazole, and Complete EDTA-free protease inhibitor cocktail, and purified by Ni-affinity chromatography. Purified Mcl-1-MBP fusion protein was cleaved with TEV protease in 100 mM Tris-HCl pH 8, 60 mM Citrate, 5 mM BME at 4 °C for 17 h. Cleaved Mcl-1 was separated from MBP by subtractive Ni-affinity chromatography followed by gel filtration of the flow through using a Superdex75 10/300 gel filtration column. The uniformly 15N-labelled protein samples were prepared by growing the bacteria in minimal medium containing 15N-labeled NH4Cl followed by the same purification procedure.
4.6. NMR spectroscopy
Correlation 1H-15N-HSQC spectra were recorded on 15N-labelled Mcl-1 at 50 μM prepared in 50 mM potassium phosphate solution at pH 6.7, 50 mM NaCl and 1 mM DTT. Titrations up to 100 μM of 35 and 38 compounds were performed. NMR spectra were acquired at 25 °C on an Inova 600 MHz spectrometer equipped with a cryoprobe and analyzed with CCPNMR43. Mcl-1 chemical shift assignments for 1H, 15N, were transferred from previous assignments and confirmed using 15N-HSQC-NOESY. The weighted average chemical shift difference Δ(CSP) was calculated as √(ΔδH1)2+(ΔδN15/5)2 in ppm. The absence of a bar indicates no chemical shift difference, or the presence of a proline or a residue that is overlapped and not used in the analysis. The significance threshold for backbone amide chemical shift changes of 0.05 ppm was used in accordance with standard methods.
4.7. Molecular Docking
NMR-guided docking of 35 and 38 into the X-ray crystal structure of Mcl-1 (PDB ID: 2PQK) was performed using Glide (Schrodinger) [44-46]. Compounds were converted to 3D all atom structures with Ligprep (Schrodinger) and assigned partial charges with Epik (Schrodinger) [47,48]. The compounds were docked in a binding site that included the BH3-binding groove of Mcl-1 using the extra precision (XP) docking mode. To optimize side chain geometries, the lowest energy possess from XP docking were subjected to a short 120 ps molecular dynamics (MD) simulation using Desmond (Schrodinger) [48]. MD runs were performed in truncated octahedron SPC water box using OPLS_2005 force field, 300K and constant pressure of 1.0325 bar. Clustering and analysis was performed with Maestro [49] tools (Schrodinger). The lowest-energy docking pose is consistent with the observed NMR-chemical shift perturbation data. Pymol is used for preparing the highlighted poses [50].
Supplementary Material
Highlights.
Synthesis of novel marinopyrrole derivatives
Identified marinopyrroles as selective Mcl-1 or dual Mcl-1/Bcl-xL inhibitors
Structure activity relationships of novel marinopyrroles bound to Mcl-1
Marinopyrroles bind to the Mcl-1 BH3 pocket
Marinopyrroles downregulate Mcl-1 levels and induce apoptosis
ACKNOWLEDGMENT
This work was supported by grants from the National Natural Science Foundation of China (21321061 and 21132006), the National Science and Technology Major Project of China (2011ZX09401-304) and the National Basic Research Program of China (973 program, 2010CB833200) to Y.Q., NIH grant R01CA178394 and developmental award P30CA013330-39S1 to E.G., and start-up funds (partial support) from the Moffitt Cancer Center and the University of Nebraska Medical Center to R.L. E.G is a member of the New York Structural Biology Center (NYSBC) and the data collected at NYSBC was made possible by a grant from NYSTAR. We also thank members of the Chemical Biology Core Facility at the Moffitt Cancer Center and Research Institute for producing the GST-Mcl-1 protein used in the ELISA assays. The core facilities at the Moffitt Cancer Center are supported partially by the Cancer Center Support Grant.
ABBREVIATIONS
- Bcl-2
B-cell lymphoma 2
- Bcl-xL
B-cell lymphoma extra large
- BH3
Bcl-2 homology domain 3
- Mcl-1
Myeloid cell leukemia 1
- MDA-MB-468
breast cancer cell line
- SAR
structure activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmch.2014. (publisher to update)
REFERENCES
- 1.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Kelly GL, Strasser A. The essential role of evasion from cell death in cancer. Adv. Canc. Res. 2011;111:39–96. doi: 10.1016/B978-0-12-385524-4.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 6.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol. Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Bioch. Sci. 2014;39:101–111. doi: 10.1016/j.tibs.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr. Opin. Gen. & Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Diff. 2011;18:1414–1424. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 12.Billard C. BH3 mimetics: status of the field and new developments. Mol. Cancer Ther. 2013;12:1691–1700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 13.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 14.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 15.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 16.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 17.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, Fesik SW, Shen Y. 'Seed' analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-XL inhibitor ABT-737. Oncogene. 2007;26:3972–3979. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, Trichet V, Robillard N, Philippe M, Bataille R, Amiot M. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res. 2007;67:5418–5424. doi: 10.1158/0008-5472.CAN-06-4322. [DOI] [PubMed] [Google Scholar]
- 20.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cellular and molecular life sciences: CMLS. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernardo PH, Sivaraman T, Wan KF, Xu J, Krishnamoorthy J, Song CM, Tian L, Chin JS, Lim DS, Mok HY, Yu VC, Tong JC, Chai CL. Structural insights into the design of small molecule inhibitors that selectively antagonize Mcl-1. J. Med. Chem. 2010;53:2314–2318. doi: 10.1021/jm901469p. [DOI] [PubMed] [Google Scholar]
- 23.Dash R, Azab B, Quinn BA, Shen X, Wang XY, Das SK, Rahmani M, Wei J, Hedvat M, Dent P, Dmitriev IP, Curiel DT, Grant S, Wu B, Stebbins JL, Pellecchia M, Reed JC, Sarkar D, Fisher PB. Apogossypol derivative BI-97C1 (Sabutoclax) targeting Mcl-1 sensitizes prostate cancer cells to mda-7/IL-24-mediated toxicity. PNAS. 2011;108:8785–8790. doi: 10.1073/pnas.1100769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, Lin Q. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J. Am. Chem. Soc. 2012;134:14734–14737. doi: 10.1021/ja306864v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doi K, Li R, Sung SS, Wu H, Liu Y, Manieri W, Krishnegowda G, Awwad A, Dewey A, Liu X, Amin S, Cheng C, Qin Y, Schonbrunn E, Daughdrill G, Loughran TP, Jr., Sebti S, Wang HG. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Boil. Chem. 2012;287:10224–10235. doi: 10.1074/jbc.M111.334532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, Opferman JT, Walensky LD. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem. & Biol. 2012;19:1175–1186. doi: 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka Y, Aikawa K, Nishida G, Homma M, Sogabe S, Igaki S, Hayano Y, Sameshima T, Miyahisa I, Kawamoto T, Tawada M, Imai Y, Inazuka M, Cho N, Imaeda Y, Ishikawa T. Discovery of potent Mcl-1/Bcl-xL dual inhibitors by using a hybridization strategy based on structural analysis of target proteins. J. Med. Chem. 2013;56:9635–9645. doi: 10.1021/jm401170c. [DOI] [PubMed] [Google Scholar]
- 28.Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abulwerdi FA, Liao C, Mady AS, Gavin J, Shen C, Cierpicki T, Stuckey JA, Showalter HD, Nikolovska-Coleska Z. 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J. Med. Chem. 2014;57:4111–4133. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, Gulappa T, Cierpicki T, Owens S, Zhang T, Sun D, Stuckey JA, Mohammad RM, Nikolovska-Coleska Z. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2014;13:565–575. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng C, Liu Y, Balasis ME, Garner TP, Li J, Simmons NL, Berndt N, Song H, Pan L, Qin Y, Nicolaou KC, Gavathiotis E, Sebti SM, Li R. Marinopyrrole derivatives with sulfide spacers as selective disruptors of mcl-1 binding to proapoptotic protein bim. Mar. Drugs. 2014;12:4311–4325. doi: 10.3390/md12084311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fire E, Gulla SV, Grant RA, Keating AE. Mcl-1-Bim complexes accommodate surprising point mutations via minor structural changes. Protein Science. 2010;19:507–519. doi: 10.1002/pro.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dutta S, Gulla S, Chen TS, Fire E, Grant RA, Keating AE. Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 2010;398:747–762. doi: 10.1016/j.jmb.2010.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foight GW, Ryan JA, Gulla SV, Letai A, Keating AE. Designed BH3 Peptides with High Affinity and Specificity for Targeting Mcl-1 in Cells. ACS Chem. Biol. 2014 doi: 10.1021/cb500340w. 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng C, Liu Y, Song H, Pan L, Li J, Qin Y, Li R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (II) Mar. Drugs. 2013;11:2927–2948. doi: 10.3390/md11082927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng C, Pan L, Chen Y, Song H, Qin Y, Li R. Total synthesis of (±)-marinopyrrole A and its library as potential antibiotic and anticancer agents. J. Comb. Chem. 2010;12:541–547. doi: 10.1021/cc100052j. [DOI] [PubMed] [Google Scholar]
- 37.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chemie. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Moldoveanu T, Sprules T, Matta-Camacho E, Mansur-Azzam N, Gehring K. Apoptotic regulation by MCL-1 through heterodimerization. J. Biol. Chem. 2010;285:19615–19624. doi: 10.1074/jbc.M110.105452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Haste NM, Thienphrapa W, Nizet V, Hensler M, Li R. Marinopyrrole derivatives as potential antibiotic agents against methicillin-resistant Staphylococcus aureus (I) Mar. Drugs. 2012;10:953–962. doi: 10.3390/md10040953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng C, Liu Y, Balasis ME, Garner TP, Li J, Simmons NL, Berndt N, Song H, Pan L, Qin Y, Nicolaou KC, Gavathiotis E, Sebti SM, Li R. Marinopyrrole derivatives with sulfide spacers as selective disruptors of mcl-1 binding to proapoptotic protein bim. Mar. Drugs. 2014;12:4311–4325. doi: 10.3390/md12084311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng C, Liu Y, Balasis ME, Simmons NL, Li J, Song H, Pan L, Qin Y, Nicolaou KC, Sebti SM, Li R. Cyclic marinopyrrole derivatives as disruptors of Mcl-1 and Bcl-x(L) binding to Bim. Mar. Drugs. 2014;12:1335–1348. doi: 10.3390/md12031335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen TS, Palacios H, Keating AE. Structure-based redesign of the binding specificity of anti-apoptotic Bcl-x(L) J. Mol. Biol. 2013;425:171–185. doi: 10.1016/j.jmb.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 44.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, P Repasky M, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 45.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 46.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 47.Gavathiotis E, Reyna DE, Bellairs JA, Leshchiner ES, Walensky LD. Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol. 2012;8:639–645. doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comp. Mol. Des. 2010;24:591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 47.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. J. Comp. Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 48.Abel R, Salam NK, Shelley J, Farid R, Friesner RA, Sherman W. Contribution of explicit solvent effects to the binding affinity of small-molecule inhibitors in blood coagulation factor serine proteases. ChemMedChem. 2011;6:1049–1066. doi: 10.1002/cmdc.201000533. [DOI] [PubMed] [Google Scholar]
- 49.Maestro, version 9.7. Schrödinger, LLC; New York, NY: 2014. [Google Scholar]
- 50.The PyMOL Molecular Graphics System. Version 1.4. Schrödinger, LLC; New York: 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.