

Abstract
Mild cognitive impairment (MCI) and Alzheimer’s disease (AD) represent points on a continuum of cognitive performance in aged populations. Cognition may be impaired or preserved in the context of brain aging. One theory to account for memory maintenance in the context of extensive pathology involves ‘cognitive reserve,’ or the ability to compensate for neuropathology through greater recruitment of remaining neurons. In this review, we propose a complementary hypothesis of ‘metabolic reserve’, where a brain with high metabolic reserve is characterized by the presence of neuronal circuits that respond adaptively to perturbations in cellular and somatic energy metabolism and thereby protects against declining cognition. Lifestyle determinants of metabolic reserve, such as exercise, reduced caloric intake, and intake of specific dietary components can promote neuroprotection, while pathological states arising from sedentary lifestyles and excessive caloric intake contribute to neuronal endangerment. This bidirectional relationship between metabolism and cognition may be mediated by alterations in central insulin and neurotrophic factor signaling and glucose metabolism, with downstream consequences for accumulation of amyloid beta and hyperphosphorylated tau. The metabolic reserve hypothesis is supported by epidemiological findings, and the spectrum of individual cognitive trajectories during aging, with additional data from animal models identifying potential mechanisms for this relationship. Identification of biomarkers for metabolic reserve could assist in generating a predictive model for the likelihood of cognitive decline with aging.
Keywords: exercise, BDNF, insulin, caloric restriction, diet, neurodegeneration, diabetes, metabolic syndrome
Introduction
Alzheimer’s disease (AD) is associated with gradual impairment of memory and cognition that occurs late in life. While genetic mechanisms for early-onset familial AD have been identified, little is known about the underlying factors driving the prevalence of late-onset sporadic AD. Mild cognitive impairment (MCI) often presages AD [1], and genetic risk factors such as the apolipoprotein E epsilon 4 allele may also confer some degree of risk [2]. However, genotyping strategies lack specificity and sensitivity, and amnestic MCI may be too late to implement strategies for prevention of further decline.
Lifestyle factors are increasingly recognized for their role in determining cognitive impairment, or the lack thereof, during aging. Participation in recreational physical activity is inversely related with the prevalence of age-related cognitive decline and dementia [3]. Dietary factors, including total caloric intake and diet composition, also correlate with cognitive performance in aging populations, such that reduced caloric intake [4] and lower saturated fat intake [5] have been independently associated with reduced prevalence of dementia. The ability to maintain efficient cognitive performance despite the presence of age-related neuropathology has been defined as ‘cognitive reserve.’ Factors such as education and socioeconomic status are likely contributors to cognitive reserve, as are social networks [6]. We propose a complementary hypothesis of ‘metabolic reserve,’ whereby cognitive preservation in the face of extensive neuropathology, determined in part by the ability of brain cells to regulate energy metabolism in the remaining neuronal circuits , which is in turn influenced by exercise and dietary factors. Although the existence of ‘metabolic reserve’ has been suggested by both longitudinal and cross-sectional studies, the protective mechanisms for the effects of exercise and dietary energy restriction are still being identified.
Diseases arising from lifestyle factors also may increase the risk of developing MCI and AD. Just as normal cognition, MCI, and AD exist along a continuum, glucose and insulin metabolism also represent a metabolic spectrum punctuated by clinical thresholds for prediabetes and type 2 diabetes. Prediabetes is associated with elevated fasting glucose levels and/or impaired glucose tolerance; these parameters may be above the normal range without reaching criteria for type 2 diabetes. Further increases in fasting glucose, with concomitant impairment of glucose and insulin metabolism, are hallmarks of type 2 diabetes. Obesity is a significant risk factor for both prediabetes and diabetes, both of which increase the likelihood of AD [7].
In this review, we will discuss recent findings on the bidirectional relationship between metabolism and cognitive function in the context of aging. The majority of published studies support the existence of ‘metabolic reserve,’ defined as neuroprotection in the context of efficient metabolism, and neuronal endangerment in the context of impaired metabolism. Possible mechanisms for metabolic reserve involve a direct role for insulin [8], insulin-like growth factors [9] and neurotrophic factors [10], as well as pathological changes in glucose metabolism and protein glycosylation [11]. Other components of the hormonal milieu, including the adipocyte cytokine leptin [12], will also be discussed with reference to their participation in MCI and AD.
Epidemiological studies support an association between metabolism and cognition
A cross-sectional analysis of the epidemiological literature supports the metabolic reserve hypothesis. Independent studies have repeatedly shown that participation in recreational physical activity reduces the incidence of cognitive decline in aging populations [3]. Likewise, studies have demonstrated that dietary restraint is associated with maintenance of cognitive function late in life [7]. Cross-cultural studies have further suggested that regular intake of specific dietary components, such as omega-3 fatty acids and antioxidants, preserves cognitive function well into the eighth decade of life [13].
On the other hand, obesity in midlife has been linked to cognitive impairment at older ages [14]. Glycemic control, defined as the efficiency of the physiological response to a glucose challenge, has also been linked with cognition in middle-aged adults, such that individuals with better glycemic control also perform better on measures of executive function [15]. Type 2 diabetes is defined by poor glycemic control, and this pathological state also compromises intellectual ability, particularly late in life [16–17]. These data support the idea of a metabolic spectrum, defined as the relationship between somatic metabolism and neurological decline with aging (Figure 1). In addition to a long-standing systemic positive energy balance and associated insulin resistance, data from studies of human subjects and animal models suggest that an acute disruption in brain cell energy metabolism (as occurs in traumatic brain injury and stroke) may precipitate or hasten the onset of AD [18–20].
Figure 1. The metabolic reserve hypothesis.
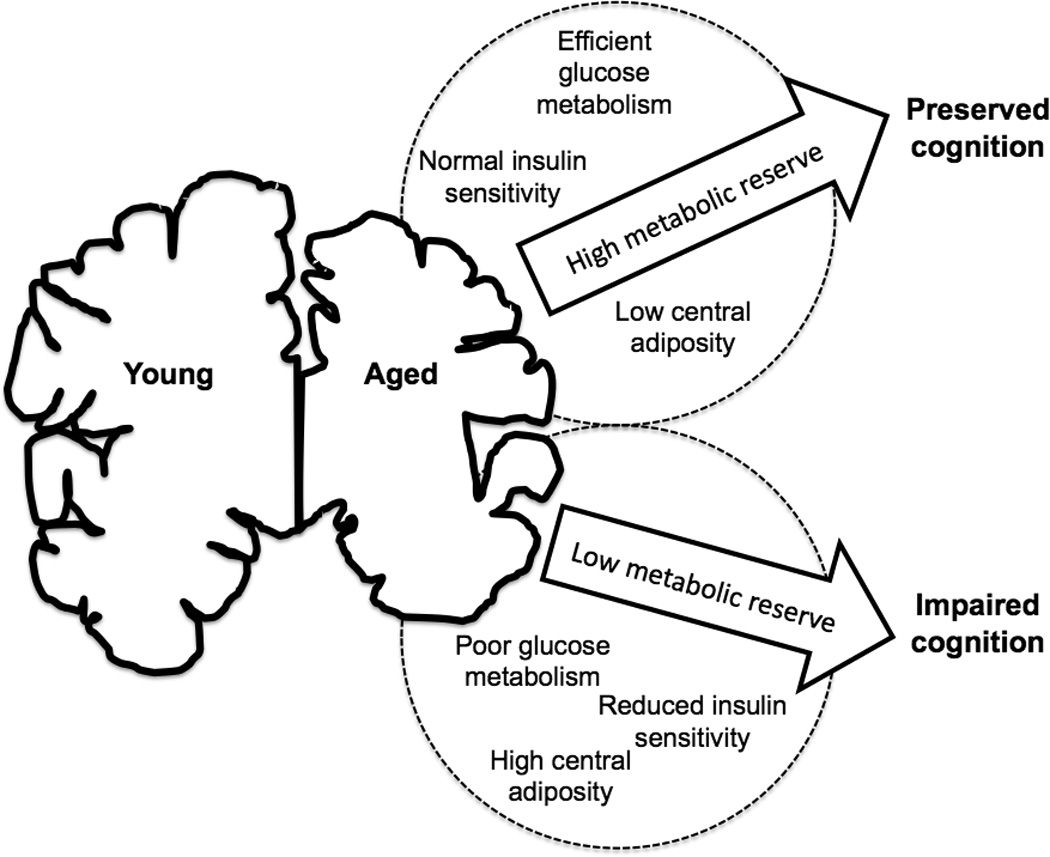
Aging is accompanied by variable degrees of brain atrophy. Having a high metabolic reserve, defined by efficient central and peripheral glucose and insulin metabolism and low levels of visceral body fat, improves the probability that an individual will maintain cognitive function in the face of regional atrophy. By contrast, having low metabolic reserve, defined by inefficient cellular energy metabolism and central adiposity, increases the likelihood of cognitive decline.
Longitudinal associations between metabolic indices and cognition
Both cross-sectional and longitudinal studies have been used to evaluate the relationship between metabolism and cognitive status. In the Health ABC Study, 2,509 participants were followed for 8 years and cognitive outcomes were measured in relationship to physical activity and other lifestyle factors [21]. This longitudinal assessment revealed that exercise reduces the risk of cognitive decline. Similarly, in the Rotterdam study, 5,395 participants were periodically assessed for 6 years, and cognitive function was evaluated in relation to dietary antioxidant intake [22]. Again, greater intake of antioxidants was associated with a lower incidence of dementia. At the other end of the spectrum, when 1,447 participants were assessed over 9 years with regard to cognition, diabetes emerged as a significant predictor of cognitive decline [23]. Central adiposity during midlife was also predictive of incident dementia late in life [14], and in animal models, removal of visceral adipose tissue promotes longevity [24].
The concept of a metabolic set point, determined by central and peripheral factors, has been widely discussed in the literature surrounding food intake and obesity. The metabolic set point represents a particular range of body weight and adiposity that is established and maintained over time [25]. Based on these studies, it is possible that maintenance of a metabolic set point within a specified range also establishes and preserves a cognitive set point during aging. In this regard, enhanced metabolic efficiency may serve as a buffer against age-related cognitive decline.
Imaging studies support a link between metabolism and brain structure
Structural imaging analyses have identified a bidirectional relationship between metabolism and the volume of brain regions that are vulnerable to decline with aging. Participation in physical training increases hippocampal volume in humans [26], and reduces hippocampal atrophy in the context of aging [27]. While data on diet and hippocampal structure in human populations are relatively scarce, there is some indication that antioxidant intake reduces the occurrence of brain infarcts with aging [28]. In these respects, exercise and dietary restraint impact not only cognition, but also preserve and maintain brain structure.
A number of studies have shown that diabetes is associated with both peripheral and central neuropathy. This is supported by observations of hippocampal shrinkage in diabetics [29], as well as by increased frequency of white matter abnormalities throughout the brain [30]. Some studies have documented similar structural consequences in obesity [31], and functional alterations in glutamatergic markers have also been implicated in the context of the metabolic syndrome [32]. Hippocampal shrinkage in stress-related psychiatric conditions is well-documented [33], and an imbalance between excitatory and inhibitory neurotransmission is one potential mediator for this relationship, particularly in elderly populations [34]. Even in the absence of drug treatment (which compromises metabolism independently of psychiatric disorders), diseases such as depression impair glycemic control [35]; this further indicates that glucose metabolism is one feature that may determine metabolic reserve, or the lack of reserve in the context of depression. These and other observations align with the metabolic reserve hypothesis in that structural and functional abnormalities, including regional atrophy and changes in excitatory neurotransmission, occur in parts of the brain that are specifically vulnerable to neuropathology during aging.
Direct and indirect contributions of insulin signaling cascades to brain aging
Insulin in the central nervous system can promote or attenuate cognitive decline, depending on the time course for exposure [36]. Acute intranasal insulin improves cognition [37], while chronic exposure to high levels of insulin reduces insulin sensitivity and impairs executive function in humans [38]. One mechanism through which hyperinsulinemia accelerates neuropathology involves the diversion of enzymatic resources that could otherwise be applied to clearance of amyloid beta from the brain. Both insulin and amyloid beta are cleared by insulin-degrading enzyme (IDE), and occupation of IDE by insulin reduces the availability of IDE for amyloid beta clearance [39].
Insulin signaling impacts both amyloid beta accumulation and phosphorylation of tau, the primary component of neurofibrillary tangles in AD. Insulin-induced activation of phosphoinositol-3 kinase (PI3K) reduces phosphorylation of tau by inhibiting glycogen synthase kinase 3 beta (GSK3beta). With hyperinsulinemia, the functional coupling between insulin receptor activation and PI3K is impaired, leading to conditions that are permissive for tau phosphorylation. In animal models, Type 2 diabetes has repeatedly been shown to increase accumulation of phosphorylated tau in the hippocampus, a brain region that is particularly vulnerable during aging and AD [40]. Likewise, in brain tissue from humans with both AD and Type 2 diabetes, immunoreactivity for markers of plaques and tangles is greater than that observed in tissue from nondiabetic humans with AD [41], suggesting that insulin resistance promotes neuropathology.
Multiple studies in animal models have demonstrated that exercise, in the form of voluntary wheel running, attenuates cognitive impairment and neuropathology [42–43]. Caloric restriction also ameliorates cognitive deficits and neuropathological markers in a triple-transgenic mouse model of AD with plaques and tangles [44]. Both exercise and caloric restriction have been associated with reduced insulin levels, indicative of improved insulin sensitivity [42, 45]. This opens the possibility that greater insulin sensitivity in the brain might protect against amyloid beta accumulation and tau phosphorylation. In the case of amyloid beta, reduced levels of insulin with exercise and caloric restriction would increase the capacity for IDE-mediated clearance of amyloid beta from the brain. In terms of tau phosphorylation, increased insulin sensitivity could lead to greater PI3K recruitment, which would suppress GSK3beta-mediated phosphorylation of tau. While these mechanistic pathways have not yet been shown to mediate the neuroprotective effects of exercise and caloric restriction, further studies are warranted, with the goal of developing pharmacological strategies to promote metabolic reserve.
Glucose, glucose transporters, and protein glycosylation in aging and disease
Central glucose utilization is impaired in the hippocampus and associated cortical structures of AD patients [46]. Levels of two glucose transporters expressed in astrocytes (GLUT1) and neurons (GLUT3) are reduced in the AD brain [47–48]. Decreased brain glucose transporter expression was correlated with increased tau phosphorylation in patient tissue [36]. In another study, decreased GLUT1 was associated with increased amyloid beta accumulation in a double-transgenic mouse model of AD [49]. However, the extent to which restoration of glucose transporter expression might attenuate disease progression in AD models has not yet been assessed.
Patterns of tau protein glycosylation are also altered in AD. There seems to be an inverse relationship between O-linked N-acetylglucosamine (O-GlcNAc) attachment to tau and tau phosphorylation, such that sugar residues can occupy sites that would otherwise be phosphorylated. O-GlcNAc glycosylation of tau is reduced in AD brains [50], in support of this competitive hypothesis. Because diabetes is associated with impaired glycemic control, it is possible that a reduction the level of intracellular energy substrates in neurons could be associated with reduced O-GlcNAc glycosylation of tau, thereby contributing to increased tau phosphorylation in the diabetic brain. However, this possibility has yet to be addressed.
Data on the regulation of central O-GlcNAc glycosylation following exercise or caloric restriction is sparse. Twenty-four hours of fasting decreased hippocampal O-GlcNAc glycosylation [51], but the extent to which findings on fasting align with caloric restriction is indeterminate. Moreover, the vast majority of studies demonstrating protective effects of caloric restriction used adult [45] or aged animals [52]. The mice used in the study by Li et al. [51] were eight weeks old, and it is likely that fasting evokes a different physiological response at different developmental stages. More studies are warranted to determine whether O-GlcNAc glycosylation of tau, or any other protein, is altered by exercise or caloric restriction.
Leptin and leptin sensitivity in vulnerable brain regions
The hippocampus is enriched with receptors for the adipocyte cytokine leptin [53]. Together with insulin, leptin participates in the regulation of feeding behavior [54], but its actions are not restricted to circuits traditionally associated with feeding and metabolism. Peripheral leptin injections activate signaling cascades associated with leptin receptor activation in the hippocampus [55]. High levels of circulating leptin reduce leptin sensitivity in multiple organ systems, including the brain [56]. Therefore, it is paradoxical that elevated circulating leptin levels would be associated with reduced AD prevalence in a longitudinal study of humans [57]. This apparent disparity between the animal literature, which suggests that leptin resistance would increase AD pathology, and the human data can be explained by the study population examined. Although subjects in the human study were hyperleptinemic, the proportion of hyperleptinemic subjects with insulin resistance was relatively low (19%; [57]). It is possible that, in the context of diabetes, leptin resistance might accelerate neuropathology, while in nondiabetics, high circulating leptin levels might actually be protective. There may also be a critical window for leptin resistance, with midlife obesity-induced leptin resistance increasing the likelihood of dementia [14], and late-life elevated leptin levels associated with neuroprotection [57].
Leptin inhibits GSK3beta-mediated phosphorylation of tau [58]. Leptin administration also reduces accumulation of amyloid beta, and peripheral leptin injections attenuate cognitive impairment in mouse models of AD [59]. In the hippocampus, leptin promotes synaptic plasticity and increases arborization among hippocampal neurons [12]. Leptin receptor-deficient mice also have fewer dendritic spines, which are the predominant sites for excitatory neurotransmission in the adult brain [45]. Obesity and leptin resistance following deletion of hypothalamic insulin receptors also impairs hippocampal plasticity [60]. Based on these observations, leptin clearly has some role in neuroplasticity, and pathology within this system is a likely contributor to metabolic reserve.
Exercise [61] and caloric restriction [45] are associated with reduced circulating leptin levels, indicative of improved leptin sensitivity. The dynamic interaction between systemic leptin, blood-brain barrier transport of leptin, and functional coupling between leptin receptor activation and downstream effector systems is an ongoing area of research. How can exercise and caloric restriction delay or prevent AD neuropathology, when high levels of leptin have been correlated with reduced AD prevalence [57]? Again, there may be a critical window where exposure to high levels of leptin induces resistance and promotes cognitive decline [14]; during this same window, improved leptin sensitivity with exercise and caloric restriction might prevent cognitive decline later in life. The question of whether exercise and caloric restriction exert protective effects during specific periods over the lifespan has not yet been fully addressed.
Neurotrophic factor signaling as a determinant of metabolic reserve
There is abundant evidence from studies of cultured neurons and animal models that several different neurotrophic factors can promote neuronal plasticity and survival, and so may promote continued cognitive acuity during aging. Neurotrophic factors can protect neurons in culture and in vivo against insults relevant to aging and AD including oxidative, excitotoxic and energetic stress. For example, basic fibroblast growth factor (FGF2) can increase the resistance of hippocampal and cortical neurons to glutamate toxicity, iron (an oxidative insult), glucose deprivation and ischemia [62–64]. FGF2 also protected neurons from being damaged and killed by amyloid beta [65]. Insulin-like growth factor 1 (IGF-1) has also been reported to protect neurons against oxidative and metabolic insults [66] and amyloid beta toxicity [67]. However, it was recently reported that reducing IGF-1 signaling ameliorates behavioral deficits, neuroinflammation and neuronal degeneration in a mouse model of AD [68], suggesting an adverse effect of IGF-1 signaling on the AD process. Brain-derived neurotrophic factor (BDNF) has been the most intensively studied neurotrophic factor with regards to cognition because of its important roles in synaptic plasticity and neurogenesis. BDNF can also protect neurons and improve functional outcome in experimental models relevant to AD [69–71]. Nerve growth factor (NGF) promotes the survival of basal forebrain cholinergic neurons, and there is some evidence from clinical studies that infusion of NGF into the brain can improve cognitive function in AD patients [72].
Metabolic reserve is implicated in the ability of nerve cell circuits to adjust their structure adaptively in response to functional demands and environmental conditions. Neurotrophic factors produced in an activity-dependent and stress-responsive manner are believed to play a major role in such adaptive plasticity. BDNF has emerged as key mediator of the structural and functional changes in neurons responsible for learning and memory processes. BDNF is critical for long-term potentiation at hippocampal synapses [73], and also promotes neurogenesis by enhancing the differentiation of neural progenitor cells [74]. BDNF production is responsive to changes in energy metabolism at both the whole-body and nerve cell levels. Thus, both exercise and dietary energy restriction increase BDNF production [45, 75–76], while a sedentary lifestyle and overeating may decrease BDNF production [77]. BDNF promotes neuronal plasticity by activating a receptor (trkB) coupled to PI3 kinase, Akt kinase, mitogen-activated protein kinases and the transcription factors cyclic AMP response element-binding protein (CREB) and NF-kB [78].
Since the initial observation that mice with reduced levels of BDNF (BDNF+/− mice) overeat and become obese, and that infusion of BDNF can ameliorate the hyperphagia and obesity [79], several novel and interesting roles for BDNF in the regulation of energy metabolism have been discovered. BDNF acts on neurons in the ventromedial nucleus of the hypothlamus to suppress appetite, and this action of BDNF occurs downstream of melanocortin signaling [80]. However, BDNF’s role in energy metabolism goes beyond suppression of appetite as it has been shown that BDNF infusion into the lateral ventricles improves glucose regulation independently of its effects on food intake, possibly by increasing energy expenditure [81]. Systemic administration of BDNF improved glucose regulation in leptin receptor mutant diabetic mice [82]. In addition, the obese, insulin-resistant phenotype of BDNF+/− mice can be reversed by alternate day fasting, a manipulation that also elevates BDNF production in the CNS [83]. Moreover, BDNF signaling can modify cellular energy metabolism in neurons by enhancing glucose transport and by increasing Na+-dependent amino acid transport and protein synthesis in cultured cerebral cortical neurons [84]. Thus, BDNF is involved in regulating energy metabolism at multiple levels including cellular energy metabolism in individual neurons, food intake and peripheral glucose metabolism.
The question then arises as to whether reductions in the expression of neurotrophic factors, and/or the ability of neurons to respond to the trophic factors, may be one factor that contributes to insufficient metabolic reserve during aging and AD. Studies in which levels of BDNF mRNA and protein have been measured in brain tissue samples from AD patients compared to age-matched control subjects suggest that BDNF expression is reduced in vulnerable brain regions in AD [85–86]. Studies using experimental models suggest that amyloid beta impairs CREB signaling resulting in suppression of BDNF expression, which may contribute to adverse effects on synaptic plasticity and neuronal viability [87]. Basal forebrain cholinergic neurons play an important role in memory and degenerate in AD, and cholinesterase inhibitors are the most widely prescribed drugs for AD patients. NGF promotes the survival of cholinergic neurons and their production of acetylcholine, and expression of the high-affinity NGF receptor trkA is reduced in cholinergic neurons in AD [88]. Data from studies of human subjects and animal models have led not only to clinical trials of neurotrophin delivered directly into the brain, but also to the preclinical development of low molecular weight agents that induce the expression of neurotrophins or activate neurotrophin receptors and exhibit beneficial effects in experimental models relevant to AD [89–92]. Collectively, the findings described in this section suggest that neurotrophic factor signaling-based approaches hold considerable potential to counteract the metabolic perturbations involved in age-related cognitive decline and AD.
Summary and conclusion
The metabolic reserve hypothesis states that there is a relationship between global energy metabolism and brain function that can provoke or protect against neuropathology with aging. This relationship has multiple potential mediators, including changes in cerebral insulin and glucose metabolism, alterations in glucocorticoid levels and rhythmicity, and fluctuations in leptin levels and sensitivity. Metabolic reserve contributes to cognitive reserve in that the likelihood of neuronal recruitment in the aging brain is related to bioenergetic efficiency among neurons and astrocytes. By virtue of their beneficial effects on energy balance, neuronal survival, synaptic plasticity and neurogenesis, neurotrophic factors likely play pivotal roles in the development and maintenance of cognitive reserve during aging.
Acknowledgments
A.M.S. is currently supported by start-up funds from Georgia Health Sciences University and M.P.M. is supported by the Intramural Research Program of the National Institute on Aging.
References
- 1.Albert MS, Blacker D. Mild cognitive impairment and dementia. Annu Rev Clin Psychol. 2008;2:379–388. doi: 10.1146/annurev.clinpsy.1.102803.144039. [DOI] [PubMed] [Google Scholar]
- 2.Evans DA, Beckett LA, Field TS, Feng L, Albert MS, Bennett DA, Tycko B, Mayeux R. Apolipoprotein E epsilon4 and incidence of Alzheimer disease in a community population of older persons. JAMA. 1997;277:822–824. [PubMed] [Google Scholar]
- 3.Hillman CH, Erickson KI, Kramer AF. Be smart, exercise your heart: exercise effects on brain and cognition. Nat Rev Neurosci. 2008;9:58–65. doi: 10.1038/nrn2298. [DOI] [PubMed] [Google Scholar]
- 4.Luchsinger JA, Tang MX, Shea S, Mayeux R. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59:1258–1263. doi: 10.1001/archneur.59.8.1258. [DOI] [PubMed] [Google Scholar]
- 5.Scarmeas N, Luchsinger JA, Schupf N, Brickman AM, Cosentino S, Tang MX, Stern Y. Physical activity, diet, and risk of Alzheimer disease. JAMA. 2009;302:627–637. doi: 10.1001/jama.2009.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stern Y. Cognitive reserve. Neuropsychologia. 2009;47:2015–2028. doi: 10.1016/j.neuropsychologia.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 2011;10:187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llorens-Martín M, Torres-Alemán I, Trejo JL. Mechanisms mediating brain plasticity: IGF1 and adult hippocampal neurogenesis. Neuroscientist. 2009;15:134–148. doi: 10.1177/1073858408331371. [DOI] [PubMed] [Google Scholar]
- 10.Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- 11.Suji G, Sivakami S. Glucose, glycation and aging. Biogerontology. 2004;5:365–373. doi: 10.1007/s10522-004-3189-0. [DOI] [PubMed] [Google Scholar]
- 12.Harvey J. Leptin regulation of neuronal excitability and cognitive function. Curr Opin Pharmacol. 2007;7:643–647. doi: 10.1016/j.coph.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scarmeas N, Stern Y, Mayeux R, Manly JJ, Schupf N, Luchsinger JA. Mediterranean diet and mild cognitive impairment. Arch Neurol. 2009;66:216–225. doi: 10.1001/archneurol.2008.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008;71:1057–1064. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- 15.Rolandsson O, Backeström A, Eriksson S, Hallmans G, Nilsson LG. Increased glucose levels are associated with episodic memory in nondiabetic women. Diabetes. 2008;57:440–443. doi: 10.2337/db07-1215. [DOI] [PubMed] [Google Scholar]
- 16.Velayudhan L, Poppe M, Archer N, Proitsi P, Brown RG, Lovestone S. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br J Psychiatry. 2010;196:36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- 17.Reaven GM, Thompson LW, Nahum D, Haskins E. Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care. 1990;13:16–21. doi: 10.2337/diacare.13.1.16. [DOI] [PubMed] [Google Scholar]
- 18.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- 19.Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- 20.Uryu K, Laurer H, McIntosh T, Praticò D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yaffe K, Fiocco AJ, Lindquist K, Vittinghoff E, Simonsick EM, Newman AB, Satterfield S, Rosano C, Rubin SM, Ayonayon HN, Harris TB Health ABC Study. Predictors of maintaining cognitive function in older adults: the Health ABC study. Neurology. 2009;72:2029–2035. doi: 10.1212/WNL.0b013e3181a92c36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 23.Xu W, Caracciolo B, Wang HX, Winblad B, Bäckman L, Qiu C, Fratiglioni L. Accelerated progression from mild cognitive impairment to dementia in people with diabetes. Diabetes. 2010;59:2928–2935. doi: 10.2337/db10-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muzumdar R, Allison DB, Huffman DM, Ma X, Atzmon G, Einstein FH, Fishman S, Poduval AD, McVei T, Keith SW, Barzilai N. Visceral adipose tissue modulates mammalian longevity. Aging Cell. 2008;7:438–440. doi: 10.1111/j.1474-9726.2008.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin BE. Factors promoting and ameliorating the development of obesity. Physiol Behav. 2005;86:633–639. doi: 10.1016/j.physbeh.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 26.Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, Kim JS, Heo S, Alves H, White SM, Wojcicki TR, Mailey E, Vieira VJ, Martin SA, Pence BD, Woods JA, McAuley E, Kramer AF. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108:3017–3022. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erickson KI, Prakash RS, Voss MW, Chaddock L, Hu L, Morris KS, White SM, Wójcicki TR, McAuley E, Kramer AF. Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus. 2009;19:1030–1039. doi: 10.1002/hipo.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scarmeas N, Luchsinger JA, Stern Y, Gu Y, He J, Decarli C, Brown T, Brickman AM. Mediterranean diet and magnetic resonance imaging-assessed cerebrovascular disease. Ann Neurol. 2011;69:257–268. doi: 10.1002/ana.22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruehl H, Wolf OT, Sweat V, Tirsi A, Richardson S, Convit A. Modifiers of cognitive function and brain structure in middle-aged and elderly individuals with type 2 diabetes mellitus. Brain Res. 2009;1280:186–194. doi: 10.1016/j.brainres.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reijmer YD, van den Berg E, de Bresser J, Kessels RP, Kappelle LJ, Algra A, Biessels GJ Utrecht Diabetic Encephalopathy Study Group. Accelerated cognitive decline in patients with type 2 diabetes: MRI correlates and risk factors. Diabetes Metab Res Rev. 2011;27:195–202. doi: 10.1002/dmrr.1163. [DOI] [PubMed] [Google Scholar]
- 31.Raji CA, Ho AJ, Parikshak NN, Becker JT, Lopez OL, Kuller LH, Hua X, Leow AD, Toga AW, Thompson PM. Brain structure and obesity. Hum Brain Mapp. 2010;31:353–364. doi: 10.1002/hbm.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haley AP, Gonzales MM, Tarumi T, Miles SC, Goudarzi K, Tanaka H. Elevated cerebral glutamate and myo-inositol levels in cognitively normal middle-aged adults with metabolic syndrome. Metab Brain Dis. 2010;25:397–405. doi: 10.1007/s11011-010-9221-y. [DOI] [PubMed] [Google Scholar]
- 33.Gerritsen L, Comijs HC, van der Graaf Y, Knoops AJ, Penninx BW, Geerlings MI. Depression, Hypothalamic Pituitary Adrenal Axis, and Hippocampal and Entorhinal Cortex Volumes-The SMART Medea Study. Biol Psychiatry. 2011;70:373–380. doi: 10.1016/j.biopsych.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 34.Frisardi V, Panza F, Farooqui AA. Late-life depression and Alzheimer's disease: The glutamatergic system inside of this mirror relationship. Brain Res Rev. 2011;67:344–355. doi: 10.1016/j.brainresrev.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 35.Arroyo C, Hu FB, Ryan LM, Kawachi I, Colditz GA, Speizer FE, Manson J. Depressive symptoms and risk of type 2 diabetes in women. Diabetes Care. 2004;27:129–133. doi: 10.2337/diacare.27.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frisardi V, Solfrizzi V, Capurso C, Imbimbo BP, Vendemiale G, Seripa D, Pilotto A, Panza F. Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J Alzheimers Dis. 2010;21:57–63. doi: 10.3233/JAD-2010-100015. [DOI] [PubMed] [Google Scholar]
- 37.Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey WH, 2nd, Craft S. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis. 2008;13:323–331. doi: 10.3233/jad-2008-13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuur M, Henneman P, van Swieten JC, Zillikens MC, de Koning I, Janssens AC, Witteman JC, Aulchenko YS, Frants RR, Oostra BA, van Dijk KW, van Duijn CM. Insulin-resistance and metabolic syndrome are related to executive function in women in a large family-based study. Eur J Epidemiol. 2010;25:561–568. doi: 10.1007/s10654-010-9476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messier C, Teutenberg K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer's disease. Neural Plasticity. 2005;12:311–328. doi: 10.1155/NP.2005.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009;150:5294–5301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valente T, Gella A, Fernàndez-Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer's disease and diabetes mellitus. Neurobiol Dis. 2010;37:67–76. doi: 10.1016/j.nbd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 42.Stranahan AM, Lee K, Becker KG, Zhang Y, Maudsley S, Martin B, Cutler RG, Mattson MP. Hippocampal gene expression patterns underlying the enhancement of memory by running in aged mice. Neurobiol Aging. 2010;31:1937–1949. doi: 10.1016/j.neurobiolaging.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:4217–4221. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 45.Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rinne JO, Någren K. Positron emission tomography in at risk patients and in the progression of mild cognitive impairment to Alzheimer's disease. J Alzheimers Dis. 2010;19:291–300. doi: 10.3233/JAD-2010-1224. [DOI] [PubMed] [Google Scholar]
- 47.Mooradian AD, Chung HC, Shah GN. GLUT-1 expression in the cerebra of patients with Alzheimer's disease. Neurobiol Aging. 1997;18:469–474. doi: 10.1016/s0197-4580(97)00111-5. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582:359–364. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hooijmans CR, Graven C, Dederen PJ, Tanila H, van Groen T, Kiliaan AJ. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007;1181:93–103. doi: 10.1016/j.brainres.2007.08.063. [DOI] [PubMed] [Google Scholar]
- 50.Robertson LA, Moya KL, Breen KC. The potential role of tau protein O-glycosylation in Alzheimer's disease. J Alzheimers Dis. 2004;6:489–495. doi: 10.3233/jad-2004-6505. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Lu F, Wang JZ, Gong CX. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci. 2006;23:2078–2086. doi: 10.1111/j.1460-9568.2006.04735.x. [DOI] [PubMed] [Google Scholar]
- 52.Xu X, Zhan M, Duan W, Prabhu V, Brenneman R, Wood W, Firman J, Li H, Zhang P, Ibe C, Zonderman AB, Longo DL, Poosala S, Becker KG, Mattson MP. Gene expression atlas of the mouse central nervous system: impact and interactions of age, energy intake and gender. Genome Biol. 2007;8:R234. doi: 10.1186/gb-2007-8-11-r234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hâkansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci. 1998;18:559–572. doi: 10.1523/JNEUROSCI.18-01-00559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91:389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caron E, Sachot C, Prevot V, Bouret SG. Distribution of leptin-sensitive cells in the postnatal and adult mouse brain. J Comp Neurol. 2010;518:459–476. doi: 10.1002/cne.22219. [DOI] [PubMed] [Google Scholar]
- 56.Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PLoS One. 2010;5:e11376. doi: 10.1371/journal.pone.0011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, Roubenoff R, Auerbach S, DeCarli C, Wolf PA, Seshadri S. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA. 2009;302:2565–2572. doi: 10.1001/jama.2009.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greco SJ, Sarkar S, Casadesus G, Zhu X, Smith MA, Ashford JW, Johnston JM, Tezapsidis N. Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci Lett. 2009;455:191–194. doi: 10.1016/j.neulet.2009.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greco SJ, Bryan KJ, Sarkar S, Zhu X, Smith MA, Ashford JW, Johnston JM, Tezapsidis N, Casadesus G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis. 2010;19:1155–1167. doi: 10.3233/JAD-2010-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grillo CA, Piroli GG, Evans AN, Macht VA, Wilson SP, Scott KA, Sakai RR, Mott DD, Reagan LP. Obesity/hyperleptinemic phenotype adversely affects hippocampal plasticity: effects of dietary restriction. Physiol Behav. 2011;104:235–241. doi: 10.1016/j.physbeh.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dyck DJ. Leptin sensitivity in skeletal muscle is modulated by diet and exercise. Exerc Sport Sci Rev. 2005;33:189–194. doi: 10.1097/00003677-200510000-00007. [DOI] [PubMed] [Google Scholar]
- 62.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3740. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Tatsuno T, Carney JM, Mattson MP. Basic FGF, NGF, and IGFs protect hippocampal and cortical neurons against iron-induced degeneration. J Cereb Blood Flow Metab. 1993;13:378–388. doi: 10.1038/jcbfm.1993.51. [DOI] [PubMed] [Google Scholar]
- 64.Kawamata T, Dietrich WD, Schallert T, Gotts JE, Cocke RR, Benowitz LI, Finklestein SP. Intracisternal basic fibroblast growth factor enhances functional recovery and up-regulates the expression of a molecular marker of neuronal sprouting following focal cerebral infarction. Proc Natl Acad Sci USA. 1997;94:8179–8184. doi: 10.1073/pnas.94.15.8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mark RJ, Keller JN, Kruman I, Mattson MP. Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997;756:205–214. doi: 10.1016/s0006-8993(97)00196-0. [DOI] [PubMed] [Google Scholar]
- 66.Cheng B, Mattson MP. IGF-I and IGF-II protect cultured hippocampal and septal neurons against calcium-mediated hypoglycemic damage. J Neurosci. 1992;12:1558–1566. doi: 10.1523/JNEUROSCI.12-04-01558.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doré S, Kar S, Quirion R. Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc Natl Acad Sci USA. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng B, Mattson MP. NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res. 1994;640:56–67. doi: 10.1016/0006-8993(94)91857-0. [DOI] [PubMed] [Google Scholar]
- 70.Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Williams BJ, Eriksdotter-Jonhagen M, Granholm AC. Nerve growth factor in treatment and pathogenesis of Alzheimer's disease. Prog Neurobiol. 2006;80:114–128. doi: 10.1016/j.pneurobio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 73.Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng A, Wang S, Cai J, Rao MS, Mattson MP. Nitric oxide acts in a positive feedback loop with BDNF to regulate neural progenitor cell proliferation and differentiation in the mammalian brain. Dev Biol. 2003;258:319–333. doi: 10.1016/s0012-1606(03)00120-9. [DOI] [PubMed] [Google Scholar]
- 75.Neeper SA, Gómez-Pinilla F, Choi J, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- 76.Arumugam TV, Phillips TM, Cheng A, Morrell CH, Mattson MP, Wan R. Age and energy intake interact to modify cell stress pathways and stroke outcome. Ann Neurol. 2010;67:41–52. doi: 10.1002/ana.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nonomura T, Tsuchida A, Ono-Kishino M, Nakagawa T, Taiji M, Noguchi H. Brain-derived neurotrophic factor regulates energy expenditure through the central nervous system in obese diabetic mice. Int J Exp Diabetes Res. 2001;2:201–209. doi: 10.1155/EDR.2001.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tonra JR, Ono M, Liu X, Garcia K, Jackson C, Yancopoulos GD, Wiegand SJ, Wong V. Brain-derived neurotrophic factor improves blood glucose control and alleviates fasting hyperglycemia in C57BLKS-LepRdb)/lepRdb) mice. Diabetes. 1999;48:588–594. doi: 10.2337/diabetes.48.3.588. [DOI] [PubMed] [Google Scholar]
- 83.Duan W, Guo Z, Jiang H, Ware M, Mattson MP. Reversal of behavioral and metabolic abnormalities, and insulin resistance syndrome, by dietary restriction in mice deficient in brain-derived neurotrophic factor. Endocrinology. 2003;144:2446–2453. doi: 10.1210/en.2002-0113. [DOI] [PubMed] [Google Scholar]
- 84.Burkhalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J Neurosci. 2003;23:8212–8220. doi: 10.1523/JNEUROSCI.23-23-08212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 86.Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M. Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Mol Brain Res. 1997;49:71–81. doi: 10.1016/s0169-328x(97)00125-3. [DOI] [PubMed] [Google Scholar]
- 87.Tong L, Thornton PL, Balazs R, Cotman CW. Beta -amyloid-(1–42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival Is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- 88.Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Reduction in p140-TrkA receptor protein within the nucleus basalis and cortex in Alzheimer's disease. Exp Neurol. 1997;146:91–103. doi: 10.1006/exnr.1997.6504. [DOI] [PubMed] [Google Scholar]
- 89.Culmsee C, Stumm RK, Schäfer MK, Weihe E, Krieglstein J. Clenbuterol induces growth factor mRNA, activates astrocytes, and protects rat brain tissue against ischemic damage. Eur J Pharmacol. 1999;379:33–45. doi: 10.1016/s0014-2999(99)00452-5. [DOI] [PubMed] [Google Scholar]
- 90.Jang SW, Liu X, Chan CB, Weinshenker D, Hall RA, Xiao G, Ye K. Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem Biol. 2009;16:644–656. doi: 10.1016/j.chembiol.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]