

Abstract
The Amyloid Hypothesis, which has been the predominant framework for research in Alzheimer’s Disease (AD) over the past two decades, has also been the source of considerable controversy within the field. The Amyloid Hypothesis postulates that amyloid-beta peptide (Aβ) is the causative agent in AD, and is strongly supported by data from rare autosomal dominant forms of AD. However, the evidence that Aβ causes age-associated sporadic AD is more complex and less clear, prompting criticism of the hypothesis. Herein, we provide an overview of the major arguments for and against the Amyloid Hypothesis, and review key data supporting or refuting these arguments. We conclude that Aβ likely is the key initiator of a complex pathogenic cascade which causes AD, thus supporting the Amyloid Hypothesis in general. However, we argue that Aβ acts primarily as a trigger of other downstream processes, in particular tau aggregation, which mediate neurodegeneration. Thus, Aβ appears to be necessary but not sufficient to causes AD, and its major pathogenic effects may occur very early in the disease process. We discuss implications for therapeutic development and future research.
Keywords: amyloid hypothesis, Aβ, tau, neurodegeneration, Alzheimer Disease
Introduction
In their original edition of the amyloid cascade hypothesis in 1992, Hardy and Higgins postulated that “amyloid-beta protein (Aβ)…is the causative agent in Alzheimer’s Disease (AD) pathology and that neurofibrillary tangles, cell loss, vascular damage, and dementia follow as a direct result of this deposition” 1. While two decades of research have revealed many layers of complexity, we will argue that the bulk of data still supports a role for Aβ as the primary initiator of AD pathogenesis. Herein, we will discuss data which supports that amyloid hypothesis, address some key arguments against the amyloid hypothesis, and present an updated framework for AD pathogenesis which attempts to reconcile the existing data. It has become clear that Aβ does not exert its effects in a vacuum, and that a complex network of pathologic processes converge to produce the neuropathologic and clinical syndrome that is AD. However, multiple lines of evidence still support the concept that Aβ aggregation plays a unique and critical role as the key initiator of AD pathology. By our interpretation of the existing data, Aβ appears to be necessary but not sufficient for AD, while it may be necessary and sufficient for cerebral amyloid angiopathy (CAA). This argument over the role of Aβ has important implications regarding the development of therapies, the allocation of research funding and the focus of public and private research efforts.
Genetic factors which increase Aβ aggregation cause AD
The strongest data supporting the role of Aβ as disease initiator come from human genetics. Autosomal dominant familial AD (fAD) is caused by mutations in three genes, APP, PSEN1, and PSEN2, which are all integrally involved in Aβ production 2. APP encodes the Amyloid Precursor Protein, which is the precursor to Aβ, while PSEN1 and PSEN2 encode Presenilin 1 and 2, catalytic subunits of the γ-secretase complex which cleaves APP to generate Aβ. FAD mutations lead to accelerated accumulation of Aβ plaques and cause early-onset dementia, cerebral amyloid angiopathy (CAA), or both diseases 2–6. Duplication of the APP locus on chromosome 21 increases Aβ production through increased gene dosage and causes age-related dementia with brain parenchymal Aβ deposits and CAA 7–9. Down syndrome patients, who have trisomy 21 and thus have an extra copy of APP, develop age-related amyloid plaque deposition and dementia in an APP-dependent manner 10. In general, pathogenic mutations in all three genes are specifically clustered in regions which serve to increase the level of amyloidogenic APP cleavage by β- or γ-secretase or increase the relative production of amyloidogenic Aβ1–42 versus shorter Aβ species 2, 11–13. Importantly, several APP mutations which reside in the middle of the Aβ coding region have been identified which cause fAD or CAA by increasing the aggregation propensity or inhibiting degradation of the Aβ peptide and do not affect Aβ production 5, 6. This suggests that it is Aβ aggregation, rather than some other alteration in APP processing or presenilin function, which instigates fAD. Thus, fAD provides an opportunity to examine a pure Aβ-driven disease in relatively young, often healthy patients. In keeping with the amyloid hypothesis, fAD patients first develop fibrillar amyloid plaques, then subsequently accumulate neurofibrillary tau pathology, neuronal loss, and dementia 14, 15. Thus, it is relatively easy to argue that the amyloid hypothesis, or a variant of it, is generally true for fAD. Interestingly, fAD-linked mutations clearly impact age-of-onset, but do not strongly affect rate of progression of symptomatic disease. While some of these mutations cause large increases in the production of Aβ, they do not significantly accelerate progression, suggesting that Aβ serves as an initiator or pathology, but not a major mediator of downstream neurodegeneration 16.
Fortunately, fAD is extremely rare, and the vast majority of AD cases are considered sporadic (sAD). The genetics of sAD are more complex, and the strongest genetic risk factor is APOE. The APOE ε4 allele increases risk for AD and CAA, and the ε2 allele decreases risk for AD relative to the ε3 allele 17, 18. Approximately 20–25% of the population carries at least one copy of ApoE4, which increases the risk of AD by ~4 fold (as compared to those with the more common ApoE3/E3 genotype), while 2% of the population carries two E4 alleles, imparting an ~12 fold increased risk 19. While ApoE4 appears to exert a variety of effects in the brain, it is a strong modulator of Aβ pathology, and knockin mice expressing human ApoE4 along with FAD-linked APP and PSEN1 transgenes have greatly increased Aβ plaque pathology and reduced Aβ clearance 19, 20 while human ApoE2 expressing mice have decreased Aβ plaque pathology 21. Numerous studies have demonstrated accelerated amyloid plaque accumulation in human ApoE4 carriers, while preclinical studies show that ApoE influences Aβ metabolism, with ApoE4 promoting amyloid aggregation and deposition 20–25. Reducing ApoE levels, either genetically or with antibody therapy, also reduces Aβ plaque burden in mice 26–29. Furthermore, human ApoE4 is associated with pathogenic changes in CSF Aβ42 in cognitively-normal subjects, but not changes in tau, again suggesting that ApoE4 exerts is effects in AD by modulating Aβ upstream of tau 23, 30. Thus, the predominant genetic risk factor for sAD clearly exacerbates Aβ aggregation. Recently, a rare protective mutation in the APP gene, which diminishes amyloidogenic Aβ production, has been identified in humans and linked to a decreased risk of developing AD 31, providing another strong genetic link between Aβ and sAD.
Aβ aggregation triggers tau pathology and neurodegeneration in an anatomically discordant manner
One key argument against the amyloid hypothesis is based on the poor correlation, both temporally and anatomically, between Aβ plaque deposition, neuronal death, and clinical symptoms in sAD. Neuroanatomically, fibrillar Aβ deposition occurs first and most severely in regions such as the precuneus and frontal lobes, while neuronal death begins and occurs most readily in the entorhinal cortex and hippocampus, regions with relatively few Aβ plaques 32, 33. Tau pathology correlates much more closely with neuronal loss, both spatially and temporally, than amyloid plaques 33–36. According the work of Braak and others, Aβ pathology appears to begin in the cortex and spread inward, while tau pathology exhibits an opposite progression 32, 37. This has prompted critics to suggest that Aβ must not mediate neurodegeneration in AD and is merely a “bystander”. If this were true, one would expect that in fAD, in which disease pathogenesis is more clearly driven by Aβ, that there would be a stronger anatomical correlation between plaque deposition and neuronal loss. This is not the case, as fAD pathology closely resembles that of sAD, with Aβ plaques anatomically disconnected from areas of severe neuronal loss 14, 15. Thus, genetic data again demonstrates that it is possible for Aβ to drive tau pathology and neuronal loss without inherently obvious anatomic colocalization between plaques and areas of neurodegeneration. This anatomic disconnect between fibrillar Aβ plaques, neurofibrillary tangles, and neuronal loss is still not fully explained, but appears to be consistent between fAD and sAD.
In sAD, the relationship between Aβ and tau pathology is complex but ultimately supportive of the amyloid hypothesis. Aggregated, phosphorylated tau pathology is present in the brainstem and entorhinal cortex of young, normal, asymptomatic people and precedes the onset of amyloid accumulation 38. With age, hippocampal neurofibrillary tau pathology is nearly ubiquitous but remains confined to limbic regions in cognitively-normal, amyloid-free individuals 39, 40. However, tau appears to spread into neocortical regions almost solely in people with coexistent Aβ pathology 39, 41. These individuals generally go on to develop AD dementia. In several studies examining the cognitively-normal elderly, or patients with mild cognitive impairment or AD widespread cortical tau pathology (Braak stage ≥3) was commonly observed in patients with concomitant Aβ plaques, it was never observed in those without plaques, suggesting that the presence of Aβ aggregation is required for the appearance of high-grade cortical tau pathology 39, 41, 42. Furthermore, while hippocampal tau phosphorylation is commonly observed in cognitively-normal adults, studies show no evidence of neuronal loss in these regions with normal aging 43, 44. However, neurodegeneration does occur prominently in these regions in AD patients with amyloid plaques 43, 44, suggesting that tau-mediated toxicity again requires some trigger from Aβ. Accordingly, “neuritic plaques”, which are amyloid plaques with associated neurofibrillary tau pathology, correlate more closely with neuronal loss and dementia in AD than either plaques or tangles alone 45. These neuropathologic observations support the idea that while fibrillar Aβ and neurofibrillary tau pathologies are anatomically separated, Aβ aggregation appears to somehow result in accelerated neurofibrillary tau pathology and neuronal toxicity. As noted above, fAD mutations, which directly influence Aβ aggregation, cause tau pathology which is similar to that seen in sAD and correlates closely with neuronal death. However, while mutations in the gene encoding tau (Mapt) cause neurodegenerative disease, they do not induce Aβ pathology 46, 47. Accordingly, polymorphisms in Mapt are associated with increased CSF phospho-tau levels, but only in patients with CSF evidence of Aβ pathology 48. Thus, human neuropathological data supports a key tenet of the amyloid hypothesis: that Aβ triggers or exacerbates downstream tau pathology, though it appears to do so without anatomic colocalization.
Preclinical data from cell culture and animal experiments also support Aβ as the initiator of tau pathology and downstream neuronal injury. Exposure of primary mouse neuronal cultures to aggregated Aβ species, derived either from synthetic Aβ or isolated from human brain, induces tau phosphorylation, neuritic damage, and dendritic spine loss 49, 50. Interestingly, tau knockout neurons were resistant to neuritic degeneration induced by synthetic or human-derived Aβ species, while tau overexpression exacerbated Aβ-induced damage 51, 52. It is notable that a developmental effect of tau deletion cannot be excluded in these and other experiments, as experiments using post-natal tau deletion have not been reported. In human neurons grown in a matrigel culture system, expression of fAD mutation-bearing APP elicits extracellular Aβ aggregation and causes subsequent tau pathology 53. This tau pathology could be blocked with β- or γ-secretase inhibitors, illustrating the dependence on APP cleavage. In mice, coexpression of mutant human App and Mapt caused increased severity and spread of neurofibrillary tau pathology, but did not alter Aβ pathology, again demonstrating that Aβ aggregation appears upstream of tau 54, 55. Accordingly, injection of synthetic Aβ fibrils into the brain of mutant human tau-expressing mice leads to markedly increased tau pathology 56. Aβ also impairs axonal transport in cultured neurons in a tau-dependent manner, while behavioral deficits observed in human APP expressing mice were alleviated by tau deletion. Again in these experiments, tau had no impact on Aβ pathology 57, 58. In total, substantial preclinical data demonstrates that Aβ species can directly trigger tau pathology in cells and mice, while human neuropathologic data supports a model in which tau pathology does not spread into the neocortex until after significant Aβ pathology has developed. Aβ aggregation appears to somehow be triggering an exacerbation of tauopathy, which may in turn cause neuronal dysfunction and death. While Aβ and tau pathology are anatomically separate in the AD brain, considerable evidence demonstrates that Aβ can still trigger tau pathology and neurodegeneration in regions with minimal fibrillar Aβ pathology.
Induction of toxic protein misfolding by Aβ is not isolated to tau. Many individuals with either fAD or sAD also develop α-synuclein aggregation in limbic and cortical regions, suggesting that Aβ may also accelerate synuclein pathology 59, 60. Accordingly, preclinical models demonstrate that Aβ can induce synuclein pathology 61, 62. TDP-43 pathology is also commonly seen in AD patients, and may contribute to neurodegeneration 63. Thus, the ability of Aβ to induce misfolding of other toxic protein, such as tau, synuclein, and TDP-43, may play a critical role in AD pathogenesis, though the exact contribution of some of these protein aggregates is not fully understood.
Aβ aggregation triggers tau pathology and neurodegeneration in a temporally discordant manner
Temporal discrepancy between the appearance of amyloid plaques, tau tangles, neuronal loss, and clinical dementia is another point of frequent contention with the amyloid hypothesis. Many clinically asymptomatic older individuals are known to have significant amyloid plaque pathology, either at autopsy or by PET imaging. Critics claim that this shows that Aβ does not cause dementia. Again, it should be noted that fAD patients also develop fibrillar amyloid plaque pathology years before symptom onset, but clearly have Aβ-driven disease 15. Large cross-sectional biomarker studies suggest that amyloid plaque pathology is not asymptomatic, but instead represents preclinical AD, though the pathologic “incubation time” between the appearance of plaques and onset of tau pathology can be several years, and the time to onset of clinical dementia can be over a decade 64–68. Emerging longitudinal evidence from several groups supports this concept, as asymptomatic individuals with biomarker evidence solely of amyloid plaques (either by CSF or PET amyloid imaging markers, with no tau or other cell injury markers) are ~4 times more likely than those without plaques to develop clinical dementia within the ensuing 2–7 years, though the length of followup is likely still too short to fully appreciate this risk 66–71. If amyloid plaques are present and there is biomarker evidence of brain injury (such as decrease glucose uptake, hippocampal atrophy, or elevated CSF tau), risk of conversion to clinical dementia is even higher, with relative risks exceeding 10 66, 67, 71, 72. People who are ostensibly cognitively normal but have amyloid plaques also exhibit very subtle cognitive deficits on detailed neuropsychometric testing and accelerated hippocampal atrophy when compared to plaque-free controls, suggesting that Aβ deposition may cause a mild but progressive pathological state prior to the onset of more rampant neurodegeneration with tauopathy and synucleinopathy 73, 74. While not conclusive evidence of the central role of Aβ in AD, the bulk of human biomarker data supports the amyloid hypothesis, as Aβ-related changes precede tau biomarker changes and cognitive markers by years, are associated with mild but progressive neuropathology, and are predictive of subsequent dementia.
How does Aβ initiate AD pathology?
So how do we reconcile this anatomic and temporal discordance between fibrillar Aβ pathology, tau aggregation, and neurodegeneration in both fAD and sAD? One explanation is that in addition to fibrillar plaques, oligomeric forms of Aβ may mediate further pathology in AD. Indeed, a variety of cytotoxic oligomeric Aβ species have been identified in AD brain lysates which can exert a wide variety of pathogenic effects in vitro and in the mouse brain 75–77. These oligomeric Aβ species do not necessarily correlate temporally or spatially with plaques, and can be purified from brain regions which are subject to intense neuronal loss, such as the hippocampus 78–80. Indeed, there is some evidence that the presence of Aβ oligomers predicts the presence of dementia more accurately than plaque burden 78, 80. In humans, Aβ oligomers appear to accumulate with age, and are correlated with the appearance of tau pathology 81, 82. Mice which accumulate Aβ oligomers but not fibrillar plaques also develop synaptic damage, inflammation, and cognitive impairment 83, 84 However, it is difficult to rectify the oligomer hypothesis with the long prodromal phase of human AD during which plaques are evident but significant neurodegeneration is not, or to explain why oligomers would not lead to more significant neuronal loss in animal models of Aβ deposition. Furthermore, the specific species of Aβ which mediate downstream pathology remain an open question 75, 85. While the direct neurotoxicity of Aβ oligomers in vivo is unclear, these species have been shown to directly initiate tau phosphorylation both in vitro and in vivo 52, 83, 86. At a molecular level, oligomeric Aβ may initiate tauopathy through specific signaling events in neurons, such as activation of the Src kinase Fyn 58, 87, 88, inducing proteases which modify tau 89, or activating specific tau-targeting kinases 90, though none of these have been adequately demonstrated in humans. Given increasing evidence that Aβ, tau and α-synuclein aggregates all have prion-like properties and can potentially seed normal forms of these proteins and spread through the brain, it may also be important to investigate the ability of Aβ species to exacerbate the prion-like spread of tau and synuclein 91–96. Finally, Aβ could cause large-scale alterations in the brain’s ability to maintain proper protein quality control (proteostasis), eventually prompting aggregation of tau, synuclein, and other intracellular proteins and causing toxicity 97–99. Proteostatic failure could explain α-synuclein in the brains of fAD patients, many of whom are too young to have accumulated such deposits as a function of age, or coexistent α-synuclein and TDP-43 aggregates in sAD patients 63. Aβ-induced aggregation and subsequent propagation and cross-templating of other toxic proteins could overwhelm proteostatic mechanisms. As many cytoprotective and proteostatic systems in neurons and glial decline with age, aging might thus set the stage for disseminated Aβ toxicity 100, 101. Other age-related effects or comorbid pathologies (such as diabetes, head trauma, oxidative stress, inflammation, vascular factors, or even sleep deprivation) might influence the rate of Aβ aggregation and accumulation, accelerate the rate at which Aβ triggers downstream pathology, or directly exacerbate downstream pathology, thereby modulating the onset of dementia.
Aβ does not operate in a vacuum
A shortcoming of the initial amyloid hypothesis is the suggestion that Aβ itself could directly cause the panoply of pathologic changes observed in AD. Aβ critics have frequently and correctly pointed out the many other destructive phenomena, from oxidative stress to impaired autophagy, which occur in AD brain. Certainly when one uses the clinical endpoint of cognitive impairment, many factors appear to accelerate AD, cerebrovascular disease being foremost among them. The relationship between these factors, Aβ, and neurodegeneration is unclear. Mice harboring fAD mutations develop neuroinflammation, oxidative stress, mitochondrial dysfunction, and other pathologic alterations which thus appear to be downstream changes which indicate Aβ-induced neuronal or glial injury, though many of these markers appear before significant plaque deposition 102–104. However, preclinical data suggests that neuronal stressors such as oxidative and nitrative stress 105, 106, mitochondrial dysfunction 107, and inflammation 108, can also directly influence APP metabolism and Aβ accumulation, suggesting the existence of a possible feed-forward mechanism by which Aβ-induced injury can facilitate Aβ production, or by which other stressors, such as ischemia or trauma, might exacerbate Aβ production and toxicity.
While it may be tempting to implicate these other processes as the causative factors in AD, it is important to consider that Aβ plaque pathology is uniquely indicative of the clinical syndrome of AD, while oxidative damage, inflammation, mitochondrial dysfunction, and other pathogenic markers are observed in many neurologic diseases. Furthermore, there is no strong evidence from humans that genetic mutations which increase oxidative stress, inflammation, mitochondrial dysfunction, or inhibit autophagy lead to Aβ pathology in the brain. Risk factors such as smoking can have Aβ-dependent and independent effects on AD pathogenesis, as smoking causes systemic oxidative stress, vascular injury, and may increase Aβ accumulation 109, 110. Similarly, polymorphisms in TREM2 affect microglial activation and increase risk of AD, yet TREM2 hemizygous mice have no increase in Aβ plaques, suggesting that microglial dysfunction could influence AD pathogenesis independently of the amount of Aβ accumulation 111. In both cases, it is likely that these risk factors influence pathogenic mechanism downstream of Aβ, and are not unique to AD. Accordingly, smoking increases the risk of many disease of the brain, including vascular dementia, while TREM2 likely does the same (it has recently been linked to increased risk of ALS, FTD, and Parkinson disease) 112, 113. It would be foolish to argue that these many processes are not important in AD, as they clearly contribute to disease pathogenesis and might provide excellent therapeutic targets. But the existence of these other mechanisms of neural injury in AD does not mean they are causative, nor does it negate the central importance of Aβ.
Another alternative concept of AD pathogenesis suggests that APP cleavage is indeed critical to AD, but that APP fragments other than Aβ are the pathogenic species. Mice with inducible expression of APP develop Aβ plaques and cognitive impairment. The cognitive impairment resolved in part when the APP transgene expression was turned off, despite persisting Aβ plaques 114. Conversely, mice which express Aβ from a truncated transgene (and not full-length APP) accumulate multiple Aβ species as well as plaques, but do not develop cognitive impairment 115. Several studies have implicated the APP β-C-terminal fragment (β-CTF) as a possible mediator of cognitive impairment in APP-overexpressing mice 116, 117. However, it should be noted that antibodies targeting Aβ reduce plaque burden and rescue cognition in APP transgenic mice without eliminating plaques 118. Moreover, as mentioned earlier, APP mutations within the Aβ coding region which enhance Aβ aggregation without influence APP cleavage still cause human fAD 5, 6. Thus, the data implicating alternative APP fragments in AD is intriguing, it is still largely restricted to mouse models, and does not discount the contribution of Aβ.
APP is a complex molecule with a variety of neurobiological functions 119. Thus, it is important to note that cognitive/behavioral changes in APP-overexpressing mice are not fully correlated with Aβ pathology, and are likely influenced greatly by both the APP transgene and overexpression promoter. Cognitive impairment in APP mice can appear before, after, or without significant Aβ deposition, and are generally reversible 85, 118. In human AD, however, cognitive decline is notable years or even decades after extensive plaque pathology accumulates, and is almost always associated with the onset of tau pathology and neurodegeneration, and has not been shown to be reversible. Therefore, the nature of cognitive impairment in mouse APP models and humans may be fundamentally different, and mouse results should be interpreted carefully.”
What initiates Aβ aggregation?
If Aβ is the initiator of AD pathology, what initially triggers the aggregation and accumulation of Aβ in sAD? How is this related to age? Currently, we have no iron-clad explanation for why Aβ initially accumulates and forms plaques in sAD, or why this pathology occurs late in life. Aβ might accumulate in a concentration-dependent manner throughout life, with increased neuronal activity in certain brain regions leading to excessive Aβ production, ultimately causing aggregation and seed formation which then propagates91, 120. Factors which enhance Aβ production or aggregation, or which suppress Aβ clearance could contribute to this over a lifetime. Impaired sleep-wake cycle, for example, could contribute to gradual Aβ accumulation by promoting a higher level of neuronal Aβ release, and by disrupting normal diurnal oscillations in extracellular Aβ levels 121. Sleep deprivation could also impair the clearance of Aβ from the brain by bulk fluid movement, or “glymphatic” flow 122. As another example, age-related oxidative and nitrative stress could also exert gradual influence on Aβ production by altering secretase function 105, 106, and by promoting Aβ aggregation through oxidative modification of the peptide 123, 124. Another possibility is that age-related decline in the clearance of Aβ causes initial Aβ accumulation. Glial uptake of Aβ, as well as the aforementioned “glymphatic” bulk-flow removal of proteins form the brain, both decline with age 125, 126. Studies in humans have shown diminished Aβ clearance in AD patients 127, though this has not yet been demonstrated to precede plaque deposition. Age-related disturbances in proteostasis and neuronal stress response signaling might also shift the balance of Aβ metabolism toward aggregation 100, 101, 128.
We propose a model in which Aβ serves as the primary initiator of AD pathogenesis (see Fig. 1). In our model, Aβ levels exist in a carefully orchestrated homeostasis throughout life, and factors during middle age which disturb that balance facilitate Aβ aggregation. These factors include increased synaptic activity and sleep disruption, which lead to increase Aβ release, psychological stress (which can increase Aβ production via different mechanisms), the function or genotype of proteins (e.g. ApoE, Lrp1, and SorL1) which regulate Aβ trafficking and clearance, age-related declines in Aβ clearance mechanisms, and cellular stressors such as reactive oxygen species and ischemia which might facilitate Aβ production or aggregation. As Aβ homeostasis is lost, oligomeric and fibrillar Aβ species begin to accumulate, and the first plaques appear, providing the first biomarker evidence of AD in humans, though the patient remains clinically asymptomatic. After several years of Aβ aggregation, Aβ somehow triggers the acceleration of AD-type tau pathology, as neurofibrillary tangles begin to spread outside the limbic system and into the neocortex, prompting increases in neurodegeneration reflected as an increase in CSF tau and p-tau levels as well as other neuronal markers (e.g. VILIP-1) 72 . This “tau trigger” event is still poorly understood, and may be mediated by the appearance of particular toxic Aβ species, the activation an intermediary signaling process or kinase cascade, changes in the innate immune system, or represent a breaking point at which the proteostatic capacity of the brain has been overwhelmed, facilitating tau aggregation and spreading. Aggregation of other toxic proteins such as synuclein and TDP-43 may also begin at this stage. At this point, the cascade is set in full motion, as neuronal loss, oxidative damage, inflammation, and clinical symptoms become evident, and neutralizing or removing Aβ is unlikely to have a major effect. Age-related factors and comorbid pathologies likely contribute to the occurrence of AD by injuring neurons in parallel with Aβ and tau, thereby accelerating the appearance of symptoms, though some such factors may directly regulate Aβ levels or impact the Aβ-tau interaction. Thus, preventative strategies for AD would focus on treatment of conditions which promote Aβ accumulation in middle age, such as sleep disorders. Anti-Aβ therapies would need to be delivered as early in the process as possible, while the appearance of positive Aβ biomarkers would be most helpful for timing the initiation of therapies targeting downstream pathologies, such as anti-tau agents.
Figure 1.
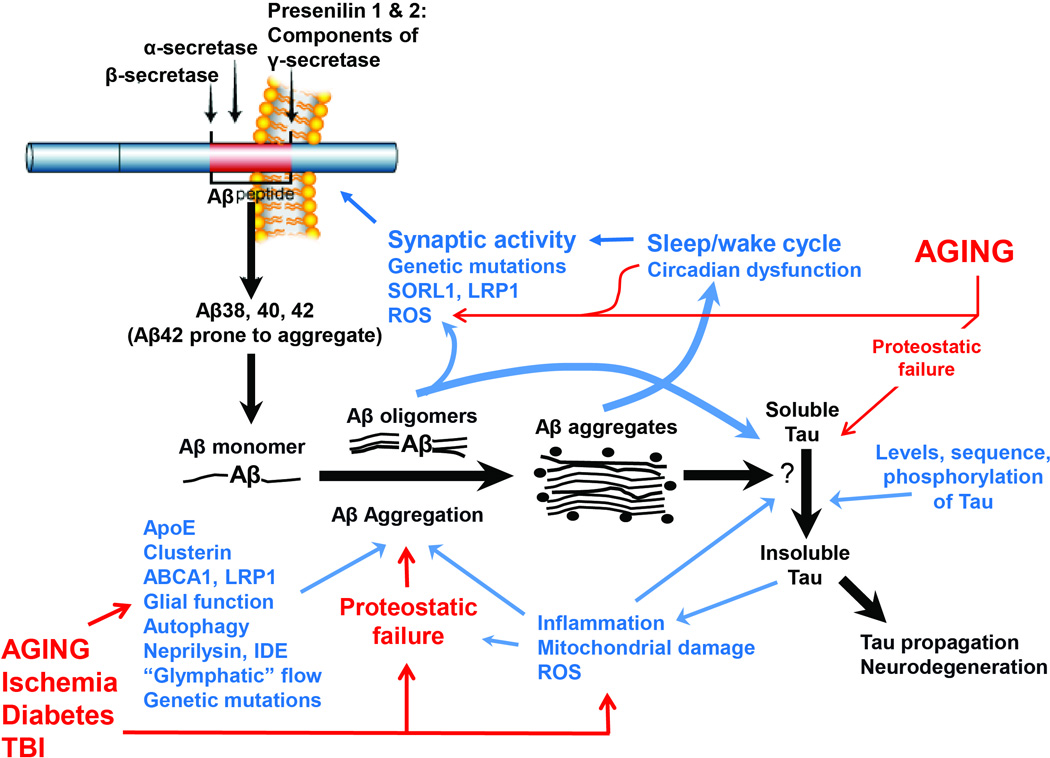
An updated framework of the amyloid hypothesis. The black arrows illustrate the processing of APP by β- and γ-secretases to yield Aβ species, which subsequently aggregate, ultimately triggering tau aggregation and downstream toxicity. Blue text and arrows illustrate proposed modifiers of the Aβ cascade, while red text and arrows show the influence of aging and comorbid pathologies. Note that several feed-forward cycles are hypothesized, including one involving disturbed sleep promoting Aβ production (and perhaps Aβ clearance, though not depicted), while Aβ aggregation in turn disrupts sleep cycles. Multiple factors, from aging to oxidative stress, contribute to proteostatic failure, which in turn promotes aggregation of Aβ, tau, and likely other toxic proteins. Many of the Aβ modifying factors interact with each other (such as ApoE modulating inflammation) though this is not depicted.
Has the amyloid hypothesis been tested pharmacologically in humans?
Mention of AD therapeutics elicits one of the more recent arguments against the amyloid hypothesis: the failure of clinical trials targeting Aβ. The possibility that Aβ initiates pathology very early in the disease suggests that only early anti-Aβ therapy is likely to be effective, and to date, no completed trials have attempted therapy early in the Aβ cascade. Further, Aβ target engagement has not yet been adequate to truly test the amyloid hypothesis. The Elan AN1792 active Aβ immunization study (which was halted due to several cases of meningoencephalitis) is often cited as an indictment of the amyloid hypothesis, as several remaining patients were followed after immunization and failed to show any clinical benefit despite evidence of reduced plaque burden at autopsy 129. However, these patients were immunized late in the course of disease, well after dementia was clinically apparent, and the small sample size in a phase I trial primarily assessing safety which was aborted after only a few vaccinations at most makes it difficult to draw any firm conclusions from this work. If one insists on considering this data, it should be noted that analysis of this same cohort did show a modest decrease in tau pathology, while surviving immunized participants had a slight improvement in functional outcomes, despite late treatment initiation 130–132. Ongoing studies, however, may put the amyloid hypothesis to a much more rigorous test. The Dominantly Inherited Alzheimer Network (DIAN) treatment trial and Alzheimer’s Prevention Initiative (API) should address the role of Aβ in fAD most directly, as both are designed to test if anti-amyloid therapy in primarily presymptomatic fAD patients can prevent pathologic changes and dementia 133, 134. This represents the earliest therapy that is currently possible, and employs a patient population with more “pure” Aβ-driven disease. However, one concerning possibility which arises from the “Aβ as initiator” hypothesis is that the appearance of fibrillar Aβ pathology could represent a point in the pathogenic cascade which is already too late for effective anti-Aβ therapy. Ultimately, prior prevention of Aβ deposition could prove to be the most effective treatment to target Aβ but whether such an approach will be tested is unclear. These studies, along with similar trials in sAD such as the A4 trial, may dictate in part, the future of Aβ-targeted therapies. There are also several ongoing trials in very mild and mild dementia due to AD using either BACE inhibition or anti-Aβ antibodies that will also be important to evaluate. Considering the therapeutic regimens employed in diseases such as diabetes, coronary atherosclerosis, and cancer, it seems reasonable that a multi-target therapeutic approach will be needed for AD, with the selection of targets dictated by the stage of the pathology. Thus, regardless of the accuracy of the amyloid hypothesis, target elucidation and therapy development for a variety of pathogenic processes in AD, from tau aggregation to microglial activation to mitochondrial dysfunction, is critical.
Conclusions
In the past 22 years, it has become clear that the idea that Aβ caused AD via a simple, linear model of toxicity is very likely incorrect. Our interpretation of the data places Aβ not as the primary direct neurotoxin that itself alone causes AD, but rather as the initiator of complex network of pathologic changes in the brain, many of them tau-dependent, which culminates years later in neurodegeneration. To successfully forestall the progression of this cascade, multiple therapies targeting several nodes of the neurodegenerative network may be needed, and the timing of these therapies in the disease progression will likely be critical. Many important questions remain to be answered, among them:
-
-
What instigates Aβ aggregation in sAD? Are there earlier precipitating events which precede Aβ aggregation and are related to age or other disease processes? How can we prevent this?
-
-
How does Aβ aggregation trigger spread of tau and synuclein pathology in the human brain? Are there specific therapeutic targets to prevent this process?
-
-
At what point in the cascade is it too late to intervene on Aβ? Once Aβ plaques are apparent by imaging or CSF studies, has Aβ already triggered the critical downstream cascades?
-
-
Which downstream pathologies are legitimate therapeutic targets, and which are epiphenomena? Post-mortem AD brain tissue reveals a dizzying array of cellular and biochemical alterations, but which can be successfully targeted to mitigate disease?
-What is the pathogenesis of AD-like dementias without amyloid plaques (such as “suspected non-amyloid pathology” (SNAP), or “tangle-predominant AD”? Are these new subtypes of dementia, which appear clinically similar to AD, biologically separate entities which will require unique therapeutic approaches? If there are no plaques, should we still call it AD?
The answers to these questions, as well the outcomes of the many anti-Aβ therapeutic trials currently in progress, should bring us closer to a comprehensive understanding of the role and therapeutic potential of Aβ in AD, and guide the development of the next generation of AD therapeutic regimens.
Acknowledgements
E.S.M. is supported by NINDS grant K08NS079405 and Alzheimer’s Association grant NIRG-305476. D.M.H. is supported by NIH grants R01 NS090934, R01 AG047644, P01 NS074969, P01 NS080675, PO1-AG03991, R01 NS034467, P01-AG026276,
Footnotes
Conflicts of Interest: DMH reports being a co-founder of C2N Diagnostics, LLC; being on the scientific advisory board of C2N Diagnostics, Genentech, AstraZeneca, and Neurophage; and being a consultant for Eli Lilly. Washington University receives grants to the lab of DMH from the NIH, C2N Diagnostics, Eli Lilly, and Janssen. ESM reports no conflicts.
References
- 1.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer's disease. Lancet Neurol. 2013;12:92–104. doi: 10.1016/S1474-4422(12)70259-4. [DOI] [PubMed] [Google Scholar]
- 3.Levy E, et al. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 4.Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 5.Tsubuki S, Takaki Y, Saido TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet. 2003;361:1957–1958. doi: 10.1016/s0140-6736(03)13555-6. [DOI] [PubMed] [Google Scholar]
- 6.Tomiyama T, et al. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 7.Rovelet-Lecrux A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 8.Sleegers K, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- 9.Cabrejo L, et al. Phenotype associated with APP duplication in five families. Brain. 2006;129:2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- 10.Prasher VP, et al. Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann Neurol. 1998;43:380–383. doi: 10.1002/ana.410430316. [DOI] [PubMed] [Google Scholar]
- 11.Citron M, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 12.Eckman CB, et al. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43) Hum Mol Genet. 1997;6:2087–2089. doi: 10.1093/hmg/6.12.2087. [DOI] [PubMed] [Google Scholar]
- 13.Chavez-Gutierrez L, et al. The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shepherd C, McCann H, Halliday GM. Variations in the neuropathology of familial Alzheimer's disease. Acta Neuropath. 2009;118:37–52. doi: 10.1007/s00401-009-0521-4. [DOI] [PubMed] [Google Scholar]
- 15.Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryman DC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 18.Chiang GC, et al. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 2010;75:1976–1981. doi: 10.1212/WNL.0b013e3181ffe4d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 2011;10:241–252. doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castellano JM, et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fagan AM, et al. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- 22.Hudry E, et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5:212ra161. doi: 10.1126/scitranslmed.3007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris JC, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vemuri P, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67:308–316. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verghese PB, et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci USA. 2013;110:E1807–E1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bales KR, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 27.Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med. 2012;209:2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunderland T, et al. Cerebrospinal fluid beta-amyloid1–42 and tau in control subjects at risk for Alzheimer's disease: the effect of APOE epsilon4 allele. Biol Psych. 2004;56:670–676. doi: 10.1016/j.biopsych.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 31.Jonsson T, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 32.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropath. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 33.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomez-Isla T, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 35.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer's disease. Neurology. 1992;42:1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 36.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 37.Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging. 1991;12:295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 38.Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neurolpath. 2011;121:171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- 39.Price JL, Morris JC. Tangles and plaques in nondemented aging and"preclinical" Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 40.Elobeid A, Soininen H, Alafuzoff I. Hyperphosphorylated tau in young and middle-aged subjects. Acta Neuropath. 2012;123:97–104. doi: 10.1007/s00401-011-0906-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knopman DS, et al. Neuropathology of cognitively normal elderly. J Neuropath Exp Neurol. 2003;62:1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 42.Petersen RC, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 43.Gomez-Isla T, et al. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 45.Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 46.Reed LA, et al. Autosomal dominant dementia with widespread neurofibrillary tangles. Annals of neurology. 1997;42:564–572. doi: 10.1002/ana.410420406. [DOI] [PubMed] [Google Scholar]
- 47.Lindquist SG, et al. Alzheimer disease-like clinical phenotype in a family with FTDP-17 caused by a MAPT R406W mutation. Eur J Neurol. 2008;15:377–385. doi: 10.1111/j.1468-1331.2008.02069.x. [DOI] [PubMed] [Google Scholar]
- 48.Kauwe JS, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8050–8054. doi: 10.1073/pnas.0801227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira A, Lu Q, Orecchio L, Kosik KS. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol Cell Neurosci. 1997;9:220–234. doi: 10.1006/mcne.1997.0615. [DOI] [PubMed] [Google Scholar]
- 50.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta - amyloid-induced neurotoxicity. Proc Natl Acad Sci USA. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin M, et al. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi SH, et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014 doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hurtado DE, et al. A{beta} accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol. 2010;177:1977–1988. doi: 10.2353/ajpath.2010.100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewis J, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 56.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 57.Roberson ED, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 58.Roberson ED, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2010;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lippa CF, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–1370. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hashimoto M, Masliah E. Alpha-synuclein in Lewy body disease and Alzheimer's disease. Brain Pathol. 1999;9:707–720. doi: 10.1111/j.1750-3639.1999.tb00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masliah E, et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larson ME, et al. Soluble alpha-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci. 2012;32:10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Josephs KA, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropath. 2014;127:811–824. doi: 10.1007/s00401-014-1269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jack CR, Jr, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jack CR, Jr, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol. 2011;68:1526–1535. doi: 10.1001/archneurol.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roe CM, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology. 2013;80:1784–1791. doi: 10.1212/WNL.0b013e3182918ca6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vos SJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Villemagne VL, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 69.Chen X, et al. Pittsburgh compound B retention and progression of cognitive status--a meta-analysis. Eur J Neurol. 2014;21:1060–1067. doi: 10.1111/ene.12398. [DOI] [PubMed] [Google Scholar]
- 70.Villemagne VL, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Knopman DS, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012;78:1576–1582. doi: 10.1212/WNL.0b013e3182563bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tarawneh R, et al. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011;70:274–285. doi: 10.1002/ana.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Donohue MC, et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 2014;71:961–970. doi: 10.1001/jamaneurol.2014.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chetelat G, et al. Accelerated cortical atrophy in cognitively normal elderly with high beta-amyloid deposition. Neurology. 2012;78:477–484. doi: 10.1212/WNL.0b013e318246d67a. [DOI] [PubMed] [Google Scholar]
- 75.Shankar GM, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 77.Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Esparza TJ, et al. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73:104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–358. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McLean CA, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 81.Lesne SE, et al. Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Handoko M, et al. Correlation of specific amyloid-beta oligomers with tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol. 2013;70:594–599. doi: 10.1001/jamaneurol.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Y, et al. A lifespan observation of a novel mouse model: in vivo evidence supports abeta oligomer hypothesis. PloS One. 2014;9:e85885. doi: 10.1371/journal.pone.0085885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomiyama T, et al. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lesne S, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 86.Ma QL, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29:9078–9089. doi: 10.1523/JNEUROSCI.1071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ittner LM, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 88.Haass C, Mandelkow E. Fyn-tau-amyloid: a toxic triad. Cell. 2010;142:356–358. doi: 10.1016/j.cell.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Z, et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat Med. 2014;20:1254–1262. doi: 10.1038/nm.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin L, et al. Tau protein kinases: involvement in Alzheimer's disease. Ageing Res Rev. 2013;12:289–309. doi: 10.1016/j.arr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 91.Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 92.Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PloS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guo JL, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Giasson BI, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 97.Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med. 2013;62:170–185. doi: 10.1016/j.freeradbiomed.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 98.De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev. 2010;90:465–494. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- 99.Nixon RA, Yang DS. Autophagy failure in Alzheimer's disease--locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu T, et al. REST and stress resistance in ageing and Alzheimer's disease. Nature. 2014;507:448–454. doi: 10.1038/nature13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benzing WC, et al. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 104.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guix FX, et al. Modification of gamma-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer's disease. EMBO Mol Med. 2012;4:660–673. doi: 10.1002/emmm.201200243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wahlster L, et al. Presenilin-1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer's disease. Acta Neuropath. 2013;125:187–199. doi: 10.1007/s00401-012-1065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kukreja L, Kujoth GC, Prolla TA, Van Leuven F, Vassar R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer's disease. Mol Neurodegener. 2014;9:16. doi: 10.1186/1750-1326-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu Y, et al. IKKbeta Deficiency in Myeloid Cells Ameliorates Alzheimer's Disease-Related Symptoms and Pathology. J Neurosci. 2014;34:12982–12999. doi: 10.1523/JNEUROSCI.1348-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Durazzo TC, Mattsson N, Weiner MW. Smoking and increased Alzheimer's disease risk: a review of potential mechanisms. Alzheimers Dement. 2014;10:S122–S145. doi: 10.1016/j.jalz.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer's disease. Nat Commun. 2013;4:1495. doi: 10.1038/ncomms2494. [DOI] [PubMed] [Google Scholar]
- 111.Ulrich JD, et al. Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cady J, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:449–453. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rayaprolu S, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Melnikova T, et al. Reversible pathologic and cognitive phenotypes in an inducible model of Alzheimer-amyloidosis. J Neurosci. 2013;33:3765–3779. doi: 10.1523/JNEUROSCI.4251-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim J, et al. Normal cognition in transgenic BRI2-Abeta mice. Mol Neurodegener. 2013;8:15. doi: 10.1186/1750-1326-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Berger-Sweeney J, et al. Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Brain Res Mol Brain Res. 1999;66:150–162. doi: 10.1016/s0169-328x(99)00014-5. [DOI] [PubMed] [Google Scholar]
- 117.Gao Y, Pimplikar SW. The gamma -secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci USA. 2001;98:14979–14984. doi: 10.1073/pnas.261463298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dodart JC, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nature Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 119.Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bero AW, et al. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nature Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kang JE, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xie L, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Siegel SJ, Bieschke J, Powers ET, Kelly JW. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry. 2007;46:1503–1510. doi: 10.1021/bi061853s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Head E, et al. Oxidation of Abeta and plaque biogenesis in Alzheimer's disease and Down syndrome. Neurobiol Dis. 2001;8:792–806. doi: 10.1006/nbdi.2001.0431. [DOI] [PubMed] [Google Scholar]
- 125.Kress BT, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76:845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao W, Zhang J, Davis EG, Rebeck GW. Aging reduces glial uptake and promotes extracellular accumulation of Abeta from a lentiviral vector. Front Aging Neurosci. 2014;6:210. doi: 10.3389/fnagi.2014.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mawuenyega KG, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 129.Holmes C, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 130.Boche D, et al. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer's disease. Acta Neuropath. 2010;120:13–20. doi: 10.1007/s00401-010-0705-y. [DOI] [PubMed] [Google Scholar]
- 131.Serrano-Pozo A, et al. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–1327. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vellas B, et al. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009;6:144–151. doi: 10.2174/156720509787602852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mills SM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN-TU trial. Rev Neurol (Paris) 2013;169:737–743. doi: 10.1016/j.neurol.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Reiman EM, et al. Alzheimer's Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26(Suppl 3):321–329. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]