

Abstract
Long-term inhibition of extracellular vascular endothelial growth factor (VEGF) in the treatment of age-related macular degeneration (AMD) may induce retinal neuronal toxicity and risk other side effects. We developed a novel strategy which inhibits retinal pigment epithelium (RPE)-derived VEGF, sparing other highly sensitive retinal tissues. Flt23k, an intraceptor inhibitor of VEGF, was able to inhibit VEGF in vitro. Adeno-associated virus type 2 (AAV2)–mediated expression of Flt23k was maintained for up to 6 months postsubretinal injection in mice. Flt23k was able to effectively inhibit laser-induced murine choroidal neovascularization (CNV). VEGF levels in the RPE/choroid complex decreased significantly in AAV2.Flt23k treated eyes. Neither retinal structure detected by Heidelberg Spectralis nor function measured by electroretinography (ERG) was adversely affected by treatment with AAV2.Flt23k. Hence AAV2.Flt23k can effectively maintain long-term expression and inhibit laser-induced CNV in mice through downregulation of VEGF while maintaining a sound retinal safety profile. These findings suggest a promising novel approach for the treatment of CNV.
Introduction
Age-related macular degeneration (AMD) is the leading cause of vision loss in Americans over the age of 65.1,2 Nearly 15 million Americans over the age of 65 suffer from AMD, and 10–15% of them will lose central vision due to choroidal neovascularization (CNV). CNV is principally driven by vascular endothelial growth factor (VEGF). Currently, most drugs on the market and in clinical trials for treating AMD associated CNV function through direct inhibition of extracellular VEGF.3 The first-line therapy for CNV is monthly or bimonthly injections of anti-VEGF reagents, e.g., bevacizumab (Avastin), ranibizumab (Lucentis), or aflibercept (Eylea). While indefinite intravitreal injections of these anti-VEGF agents are commonly used, they pose not only a significant financial burden but also serious injection-associated risks including hemorrhage, infection, traumatic injury, and retinal detachment.4,5 In addition, subretinal fibrosis and photoreceptor/retinal pigment epithelium (RPE) atrophy may be increased by chronic anti-VEGF therapies as evidenced by recent results from the seven-up trial.3,6 Development of novel therapeutics to improve long-term inhibition of angiogenesis and reduce the risk profile while decreasing the financial burden is necessary.
To overcome these challenges, we propose adeno-associated viral (AAV)-mediated intracellular targeting of VEGF, as AAV delivery allows for long-term gene expression without replication or infectivity, and intracellular VEGF targeting allows for disruption of important VEGF autocrine loops.7,8,9 These VEGF autocrine loops remain unaffected by current extracellular anti-VEGF therapies. In addition to intracellular disruption of VEGF signaling through inhibition of VEGF autocrine loops, intracellular targeting also reduces VEGF secretion, further enhancing its efficacy. Finally, by targeting intracellular VEGF, we may be able to mitigate the unwanted side effects, which limit current extracellular anti-VEGF therapies.
Currently, AAV vectors are widely used for gene therapy due to their nonpathogenicity, minimal toxicity and immunogenicity, ability to transduce nondividing cells, and their potential for long-term expression.7,8,9 To date, most studies investigating gene therapy approaches in treating eye disease have used type 2 AAV (AAV2) vectors.10,11,12 In mouse retinal disease models, AAV2 has been shown to successfully transfect the ganglion,13 photoreceptor, and RPE layers of the retina.14,15 Several strategies to inhibit VEGF via AAV vectors exist such as AAV2.PlGF1-DE,16 type 8 AAV-sFlt-1,17 AAV2 combined with anti-VEGF shRNA,18 AAV5.sFLT01,19 and AAV2-sFLT01, which is currently undergoing a phase 1 clinical trial20 (http://www.clinicaltrials.gov/ct2/show/NCT01024998). Furthermore, clinical trials involving AAV2 vectors administered to the eye are in progress for the treatment of Leber's congenital amaurosis.21,22,23 However, with increasing reports of the potential risks of long-term extracellular anti-VEGF therapy including retinal and choroidal atrophy,24,25,26,27,28 retinal ganglion cell loss, retinal inner and outer nuclear layer (ONL) apoptosis, and decline in retinal function,24,28,29,30 many current virus-mediated therapies relying on extracellular VEGF inhibition may also pose significant risks to retinal neurons.
We have developed a novel intracellular approach to inhibiting VEGF, AAV2.Flt23k. Our intraceptor, Flt23k, consists of the VEGF-binding domains 2–3 of Flt-1 coupled to KDEL, a tetrapeptide (Lys-Asp-Glu-Leu) which binds endoplasmic reticulum (ER) retention receptors, thereby sequestering associated proteins. Flt23k binds intracellular VEGF and sequesters it in the ER where it is eventually degraded. Our prior studies demonstrated that nanoparticle delivery of the Flt23k plasmid was able to inhibit corneal angiogenesis,31,32 increase corneal transplant survival,33 and regress CNV in primate and murine laser-induced CNV models.34 However, like many current anti-VEGF therapies, this strategy is limited by the need for repeat injections. Therefore, we utilized the replication deficient AAV2 to mediate the long-term expression of Flt23k (AAV2.Flt23k).
Results
Flt23k inhibits VEGF in vitro
First, we confirmed Flt23k function in HeLa cells. Western blot analysis revealed over-expression of Flt23k and strong suppression of VEGF (Figure 1a). We then analyzed VEGF levels in cell culture media using enzyme-linked immunosorbent assay (ELISA). In normoxic conditions, VEGF was downregulated in pGENIE.Flt23k transfected cell culture media (486.40 ± 28.00 pg/mg) compared to control cell media (1,174.86 ± 75.13 pg/mg, P = 0.000009, n = 4–6). In hypoxic conditions, VEGF levels were dramatically decreased in pGENIE.Flt23k transfected cell culture media (515.76 ± 37.37 pg/mg) compared to control cell media (1,811.08 ± 25.46 pg/mg, P = 0.00000, n = 4–7) (Figure 1b).
Figure 1.

Flt23k reduces vascular endothelial growth factor (VEGF)-A protein levels in HeLa cells. (a) Western blot analysis reveals Flt23k over expression and VEGF suppression in pGENIE.Flt23k transfected cells compared to control. (b) Enzyme-linked immunosorbent assay (ELISA) analysis demonstrates reduced Hela culture medium VEGF levels in pGENIE.Flt23k transfected cells compared to control in both normoxic and hypoxic conditions.
Reporter gene expression by AAV2 vectors
To evaluate the expression of AAV2.Flt23k in vivo, we designed AAV2.AcGFP as a reporter gene. One microliter subretinal injections per eye were performed containing 5 × 108 vector genomes of AAV2. Spectralis HRA funduscopic screening at four different time points, 2 weeks, 1 month, 3 months, and 6 months post-AAV2.AcGFP subretinal injection revealed pan-retinal GFP expression (Figure 2, top). Mouse eye cryosections showed expression of AAV2.AcGFP to be localized most strongly to the RPE and photoreceptor layers (Figure 2, bottom). AAV2.AcGFP was first detected at 2 weeks postinjection, increased gradually until plateauing at 1 month, and persisted in the RPE and photoreceptor layer for at least 6 months.
Figure 2.

Time course of green fluorescent protein (GFP) expression following subretinal injection of AAV2.AcGFP. (Top) Representative Heidelberg Spectralis images showing AAV2.AcGFP expression at different time points. (Bottom) Representative confocal images revealing GFP expression to be most localized to the RPE and photoreceptor layers. Green: GFP; GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium.
AAV2.Flt23k decreases VEGF expression in the RPE/choroid in vivo
To determine the effect of Flt23k on VEGF in vivo, mouse retina and RPE/choroid complexes were collected and analyzed via western blot and ELISA. Treatment mice received a subretinal injection of either phosphate-buffered saline (PBS), AAV2.AcGFP, or AAV2.Flt23k 4 weeks prior to laser treatment and tissue was harvested at 3 days postlaser. Normal, age-matched mice receiving neither subretinal injection nor laser were analyzed to establish a baseline. Western blot analysis revealed no significant difference in retinal VEGF levels between all groups, however there was a trend toward normal physiologic levels in AAV2.Flt23k treated mice (Figure 3a,b). ELISA analysis also showed a similar trend with average retinal VEGF levels of 59.0 ± 8.8, 68.5 ± 12.7, 69.5 ± 11.4, and 63.5 ± 12.4 pg/mg in the normal, PBS, AAV2.AcGFP, and AAV2.Flt23k groups, respectively (Figure 3c). In the choroid, western blot analysis revealed a marked reduction in VEGF levels in the AAV2.Flt23k-treated group compared to the PBS- and AAV2.AcGFP-treated groups, to the extent that the levels were below the detectable limit (Figure 3d). Due to the low amount of protein loading (2 µg per well), the VEGF levels in the normal mice were also undetectable. ELISA data corroborated these findings with average RPE/choroid complex VEGF levels of 226.3 ± 4.9, 494.1 ± 28.6, 576.7 ± 67.5, and 318.5 ± 52.3 pg/mg in the normal, PBS, AAV2.AcGFP, and AAV2.Flt23k groups, respectively (Figure 3e). In the RPE/choroid complex, the VEGF levels in AAV2.Flt23k-treated mice were significantly lower than those of PBS (P = 0.032) and AAV2.AcGFP (P = 0.038) treated mice, respectively.
Figure 3.
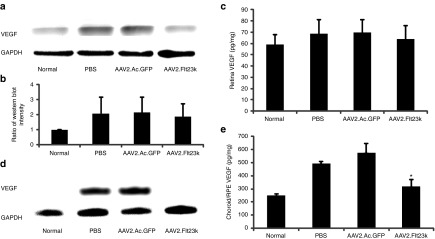
AAV2.Flt23k diminishes vascular endothelial growth factor (VEGF) protein levels in the RPE/choroid but not in the retina. Normal baseline samples are from wild-type C57BL/6 mice without laser injury and serve as baseline. Phosphate-buffered saline (PBS), AAV2.AcGFP, and AAV2.Flt23k mice underwent subretinal injection of reagents 4 weeks prior to laser injury, and tissue was harvested at 3 days postlaser. N = 3 per group. Data are presented as mean ± SD. *P < 0.05. (a) Representative retina western blot results. glyceraldehyde 3-phosphate dehydrogenase was used as a loading control. (b) Relative protein expression of (a) was determined by the semi-quantitative analysis. (c) Enzyme-linked immunosorbent assay (ELISA) analysis for VEGF levels in the retina. Both WB and ELISA analysis of the retina showed no statistical significance in VEGF levels between all groups. (d) Representative western blot analysis of VEGF in the retinal pigment epithelium (RPE)/choroid. Compared to the normal baseline (due to low amount of protein loaded, VEGF levels were below the level of detection) VEGF levels were increased in PBS and AAV2.AcGFP treated control groups. VEGF expression was drastically reduced in the AAV2.Flt23k treatment mice compared to PBS and AAV2.AcGFP treated mice, to the extent that it was undetectable. (e) ELISA analysis of the RPE/choroid shows a significant reduction in VEGF levels in AAV2.Flt23k treated mice compared to both PBS and AAV2.AcGFP treated mice, with levels approaching those seen in normal mice.
AAV2.Flt23k inhibits laser-induced CNV
To investigate the efficacy of AAV2.Flt23k in a murine laser-induced CNV model, mice were given a single, subretinal injection of 1 µl (5 × 108 vector genomes) of AAV2.Flt23k. Control groups received a single injection of an equivalent amount of AAV2.AcGFP or PBS. CNV was induced 4 weeks after subretinal injection to allow sufficient time for transgene expression. Laser-induced CNV was also performed in wild-type mice which did not receive and any subretinal injection; these served as an untreated control group. Two weeks post-CNV induction, mouse eyes were harvested and CNV volumes were analyzed by the choroidal flatmount method.35 We found that CNV was significantly inhibited in mice treated with AAV2.Flt23k (7.60 ± 6.33 × 104 µm3) compared to untreated mice (18.93 ± 10.80 × 104 µm3, P < 0.01), and mice treated with either PBS (16.11 ± 17.37 × 104 µm3, P < 0.05), or AAV2.AcGFP (19.81 ± 17.42 × 104 µm3, P < 0.01) (Figure 4a,b). Otherwise stated, AAV2.Flt23k was able to reduce CNV volumes by 59.86%, 52.9%, and 61.7%, respectively, compared to the lasered mice with no treatment, PBS, and AAV2.AcGFP groups.
Figure 4.

AAV2.Flt23k inhibits laser-induced choroidal neovascularization (CNV) in a mouse model. (a) Representative images of laser-induced CNV lesions on RPE/choroidal flatmounts in mice show a reduced volume of CNV lesions in AAV2.Flt23k treated mice compared to untreated mice and mice treated with either PBS or AAV2.AcGFP. (b) CNV volume quantification demonstrates a significant reduction in CNV volumes in mice treated with AAV2.Flt3k compared to untreated, and both PBS and AAV2.AcGFP treated mice. Scale bar: 30 µm. Error bar represents the SD. P values were calculated using the two-tailed student's T-test.
AAV2.Flt23k does not change the morphology and function of mouse retina
To evaluate the retinal safety of AAV2.Flt23k, retinal morphology and function were analyzed 6 months after subretinal injection of AAV2.Flt23k and compared to the retinal morphology and function in mice having received an equivalent subretinal injection of either AAV2.AcGFP or PBS. Whole retinas were screened by Heidelberg Spectralis and retinal thickness was measured by spectral domain optical coherence tomography (SD-OCT) imaging along the vertical and horizontal axis respectively. There was no retinal degeneration observed in vivo except at the injection site. Measurement of the ONL and whole retinal thickness in vivo along the vertical meridian did not reveal any changes in the AAV2.Flt23k group compared to the AAV2.AcGFP and PBS groups (Figure 5). In the horizontal axis, retinal thickness at the nasal injecton site (3.5 mm from the optic nerve) was significantly thinner in all injected subgroups with no significant difference between subgroups. Additionally, electroretinography (ERG) demonstrated that both the a-waves and b-waves of AAV2.Flt23k injected eyes were similar to those of wild-type, AAV2.AcGFP-treated and PBS-treated eyes (Figure 6). Thus, we found no structural or functional evidence of retinal toxicity in mice treated with subretinal injections of AAV2.Flt23k.
Figure 5.

AAV.Flt23k does not affect retinal thickness. (a) Retinal sections of AAV2.Flt23k treated mice and control mice. Top: High magnification (×40) of normal, control and AAV2.Flt23k treated eye retinal sections shows no toxicity of retinal and outer nuclear layer. Middle: High magnification (×40) of subretinal injection site. In the injection site, the retinal structure was damaged and the outer nuclear cells degenerated. Bottom: The whole retinal section (×10) shows only injection site retinal degeneration, other parts of retina remain normal. (b) Representative optical coherence tomography (OCT) subretinal injection section. The OCT image shows the whole retinal structure is normal except the injection site. (c) Top left: The schematic of method of OCT assesses the retinal thickness along the vertical axle. Top right: The thickness of the whole retina and outer nuclear layer (ONL) in normal C57 mice and mice who received subretinal injection of phosphate-buffered saline (PBS), AAV2.AcGFP, or AAV2.Flt23k. In each group, n = 5. The error bar represents the SD. Bottom left: The schematic of method of OCT assesses the retinal thickness along the horizontal axis. Bottom right: The thickness of the whole retina and ONL in normal C57 mice and mice who received subretinal injection of PBS, AAV2.AcGFP, or AAV2.Flt23k. In each group, n = 5. The error bar represents the SD.
Figure 6.
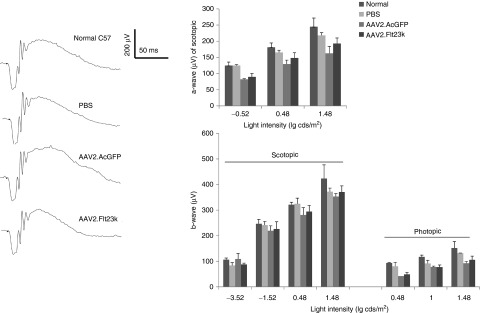
AAV2.Flt23k does not affect electroretinography (ERG) responses. (Left) Representative scotopic ERG tracings at the light intensity of 0.48 lg cds/m2. (Right) Bar charts showing the a-wave and b-wave amplitudes of scopotic ERGs and b-wave amplitudes of photopic ERGs at different light intensities. The number of eyes measured at each light intensity in each group was between 3 and 5. The error bar represents the SD.
Fl23k did not induce apoptosis in the RPE or choroid
To evaluate whether the blockade of VEGFA in RPE cells affected the survival of choroidal endothelial cells, we performed an apoptosis assay using retina sections from the same mice tested with ERG. Supplementary Figure S1 shows TUNEL staining in the retina, choroidal, and scleral layers of the control retina with no evidence of apoptotic figures in the RPE or choroid of untreated mice or mice treated with subretinal injection of either PBS, AAV2.AcGFP, or AAV2.Flt23k.
Discussion
Our data demonstrate that subretinal injection of AAV2.Flt23k can efficiently and locally express Flt23k for at least 6 months, reduce VEGF levels in the RPE/choroid complex, and inhibit CNV development while avoiding retinal toxicity. We recently employed an alternative intravenous approach using nanoparticle targeted delivery of our novel anti-VEGF therapy, which has shown promising results35; however, frequent doses with this strategy are still required and systemic toxicity profiles need to be further investigated. Taken together, our findings suggest a potential therapeutic role for AAV2.Flt23k gene therapy in the treatment of AMD.
The RPE maintains the choriocapillaris (CC) in the normal eye and is involved in the pathogenesis of CNV in age-related macular degeneration. RPE cells communicate not only through paracrine signaling to maintain the CC or induce CNV, but also through autocrine loops.36,37 In pathologic conditions such as AMD, RPE cells produce high levels of VEGF, inducing CNV.38 Our results demonstrated AAV2.Flt23k expression to be highest in the RPE layer, giving it a crucial therapeutic advantage for effective targeting of VEGF inhibition as evidenced by a greater than 50% reduction in CNV volumes compared to controls.
While not significant, there was a trend toward reduced VEGF in the retina of mice treated with AAV2.Fl23k compared to PBS and AAV2.AcGFP treated mice. The more modest decrease in VEGF levels in the retina compared to the choroid is consistent with the limited retinal expression unique to AAV2.Fl23k therapy. Alternatively, this could be explained by the difference in protein levels in the retinal sample, which are diluted by the high number of cells that are not transduced (e.g., inner retinal bipolar cells and RGCs). Additionally, the total yield of protein obtained from the retina is higher than from the RPE/choroid, further confounding the analysis of the cell specific effects of AAV2.Flt23k in the retina and RPE/choroidal samples. Nevertheless, our data are consistent with the expected effects, including the upregulation of VEGFA in response to injury (PBS and GFP), and a decrease in VEGFA levels by treatment with Flt23k.
A key advantage of our AAV2.Flt23k gene therapy over current extracellular approaches to inhibit VEGFA is that AAV2.Flt23k inhibits only RPE and photoreceptor-derived intracellular VEGFA. By utilizing a subretinal approach, we are able to maximize the dose reaching the targeted RPE/choroid complex and avoid the global suppression of retinal VEGF induced by current intravitreal injections of anti-VEGF biologics and AAV.sFlt01. Recent evidence implies that long term anti-extracellular VEGFA treatment may induce the development of geographic atrophy.6,39 Current approaches rely on monthly or bimonthly injections, which entail delivery of a bolus of anti-VEGF drug, followed by a delayed concentration until the next injection. This “sawtooth” pattern of drug delivery may result in extra toxicity at the peak anti-VEGF levels right after the bolus and insufficient angiogenic control in the troughs; this peak-trough problem would be avoided by a constant rate of delivery which could be achieved by a long-acting AAV. A concern about choroidal endothelial cell atrophy is warranted; we did not observe this issue. Regarding the delivery of AAV, subretinal administration restricts the tissues which are exposed. With respect to clinical translation, subretinal AAV delivery can be localized surgically.40 Another future approach that merits further investigation would be the utilization of a novel micro-stent developed for the treatment of glaucoma, the CyPass Micro-Stent (Transcend Medical, Menlo Park, CA), for the delivery of more targeted anti-VEGF therapy. The stent is placed in the supracilliary region and could provide permanent, direct access to the choroid for extremely targeted and consistent treatment. Further focal delivery modalities may be developed in the future as well. With respect to our treatment strategy, we have identified two primary limitations. First, subretinal injections may induce retinal damage. However, this was not a significant issue in our study and has been performed safely for gene therapy in human clinical trials. Second, our strategy may still permit excessive VEGF inhibition in photoreceptor cells which could ultimately lead to retinal atrophy. While our ERG and retinal thickness data show no evidence of such atrophy in our current study, future studies might be improved by the use of a VMD2 promoter to constrict Flt23k expression to the RPE and avoid photoreceptor expression.
We used AAV2.AcGFP as the control for comparison of AAV2.Flt23k in our study. It is possible that AcGFP expression could affect the experimental outcomes although we did not observe this in our current study. AAV2.Flt23k.AcGFP could theoretically be an additional subgroup for a more-controlled experiment, however, bicistronic cassettes are exceptionally difficult to create within AAV2, and it is unknown how a GFP coding sequence might affect Flt23k expression or activity by AAV2. Further, from a regulatory perspective, having the minimum number of constituents in an active gene therapy drug would facilitate future drug development.
Increasing evidence has demonstrated the safety and efficacy of AAV viral vectors in retinal gene therapy targeting RPE and photoreceptor cells, which is critical for an AMD mouse model.20,41,42 Currently, several strategies to inhibit VEGF via AAV vectors exist including AAV2.PlGF1-DE,16 type 8 AAV-sFlt-1,17 AAV2 combined with anti-VEGF shRNA,18 AAV5.sFLT01,19 and AAV2-sFLT01, which is currently undergoing a phase 1 clinical trial20 (http://www.clinicaltrials.gov/ct2/show/NCT01024998). Although each strategy is unique, all function via neutralization of extracellular VEGF except for anti-VEGF shRNA.18 Furthermore, AAV2.Flt23k functions through a novel mechanism of intracellular VEGF inhibition, uniquely suppressing both autocrine loops and VEGF secretion.
VEGF is a key regulator of angiogenesis and tumor growth. In addition to benefiting patients with ocular neovascularization, anti-VEGF targeted therapy has also benefited many cancer patients.43 However, current anti-VEGF therapies are challenged by insufficient efficacy, intrinsic refractoriness, and resistance. One mechanism through which cancer cells have been known to develop resistance to anti-VEGF therapies is through VEGF autocrine loops.44,45 Future studies should explore whether AAV2.Flt23k's ability to inhibit these autocrine loops may benefit management of cancer models.
Previous studies have shown that in the nonpathologic state, endogenous VEGF plays an important role as a survival factor for both the retina and choroid.29,30,38,46,47,48 Thus, despite the deleterious increase in vascular permeability and angiogenesis associated with VEGF upregulation in certain pathologic states, some baseline level of VEGF is still required for proper functioning of both the retina and choroid. Currently most available anti-VEGF therapies, which are administered with intravitral injection, indiscriminately inhibit released VEGF, suppressing the pathologic as well as the physiologic functions of VEGF, giving rise to the potential risk of retinal and choroidal toxicity.24,25,26,27,28 With this concern, recent studies focusing on the physiologic roles of VEGF in the eye have questioned the safety of long-term extracellular anti-VEGF therapy, revealing significant ocular toxicities including: retinal ganglion cell loss, chorioretinal atrophy, increased apoptosis in cells of the inner and ONLs, and a decline in retinal function,24,28,29,30 none of which we observed with AAV.Flt23k. By selectively inhibiting intracellular VEGF and localizing most strongly to the RPE, AAV2.Flt23k subretinal therapy permits more physiologic retinal and choroidal VEGF signaling and thus greatly minimizes the risk for retinal and choroidal toxicity. Through ERG analysis, we found that the retinal function was preserved in mice subjected to long-term AAV2.Flt23k therapy. Additionally, through further analysis using Spectralis, we found the retinal thickness to be preserved with no evidence of dystrophy or morphological change. Future studies investigating the correlation between cell-type specific extracellular VEGF levels, CNV inhibition, and retinal toxicity are needed to guide future neovascular treatment. However, as more research continues to support the goal of normalization of VEGF levels, as opposed to total VEGF inhibition, and as targeted therapies are refined, new strategies such as AAV2.Flt23k will likely come into prominence, elevating AMD treatment for future generations.
Another limitation of current anti-VEGF therapy is the method of drug delivery. While CNV is a process originating from aberrant VEGF signaling in the RPE and subretinal space, intravitreal treatments deliver the highest drug concentration to the supraretinal space, relying on diffusion through the highly sensitive retinal layers to reach the RPE and choroid. To circumvent this limitation, subretinal injections were used to administer AAV2.Flt23k in our study.
In summary, subretinal injection of AAV2.Flt23k can provide significant long-term suppression of VEGF in RPE cells and inhibit CNV development without evidence of retinal toxicity. These findings could translate to improve efficacy and decreased toxicity in the future treatment of AMD. Finally, this strategy may prove to be beneficial for the future treatment of select cancers with anti-VEGF resistance profiles.
Materials and Methods
Animals. Male wild-type C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) between 6 and 8 weeks of age were used to minimize variability. All animal experiments were performed in accordance with the guidelines of the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah.
Generation of AAV2 vectors. Previously, we reported the intraceptor plasmid Flt23k construction.49 By integrating cDNA of Flt23k to pAAV2-MCS (Agilent Technologies, Santa Clara, CA) using BamHI/HindIII restriction enzyme sites, pAAV2-Flt23k was generated. AAV2.Flt23k was generated in the Vector Core Gene Therapy Center, University of Massachusetts Medical School. As a control, we used AAV2.AcGFP which was used previously.50
Flt23k function in vitro. First we checked the expression of Flt 2-3k in HeLa cells. The Flt23k gene was transfected into HeLa cells using transposon based helper-independent piggybac plasmid (pmGENIE). Flt23k and DSRed Express 2 cDNA were cloned in pmGENIE. The plasmid lacking transposase (pBt gene) gene was used as a control. To examine genomic integration and stable expression of transgenes in vitro, we transfected the plasmids into HeLa cells and cultured up to 10 passages. Western blot was done to detect Flt23k. We then checked Flt23k plasmid function in normoxia and hypoxia treated HeLa cells. Flt23k transfected and control cells were cultured with Dulbecco's modified Eagle's medium with 10% fetal bovine serum. For hypoxia treatment, media was preconditioned with a hypoxic gas mixture (1%O2, 5%CO2, and 94%N2). Monolayer cultures of both cultures were replenished with the hypoxic medium, placed in the hypoxia incubator and maintained for 48 hours at 37 °C. Control monolayer cultures of both HeLa cell lines were maintained in a normoxic environment, which refers to standard cell culture in a humidified incubator (20% O2, 5% CO2, and 75% N2). Culture media was then collected, centrifuged to remove cellular debris, and preserved for VEGF ELISA.
Western blot analysis. To determine the plasmid Flt23k function, the retina and RPE/choroid complex was isolated and immediately put into 200 µl RIPA buffer (R0278, Sigma, St Louis, MO) contained with 1% protease inhibitor cocktail (P8340, Sigma) on ice respectively. After sonication, lysates were placed on ice for 15 minutes and centrifuged at 14,000 rpm for 15 minutes at 4 °C. The supernatants were collected and preserved at −80 °C for future use. The protein concentrations were measured using a BCA protein assay (Thermo Scientific, Waltham, MA). Five micrograms of retinal pretein and 2 µg of RPE/choroid protein denatured in NuPAGE sample buffer (Invitrogen, Grand Island, NY) were loaded onto 4–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and gels were transferred onto polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ). The membranes were blocked for 1 hour in Tris-buffered saline containing 5% (W/V) non-fat milk powder. Blots were treated overnight at 4 °C with rabbit anti-VEGF antibody 1:200 (Cat. sc-507, Santa Cruz, Dallas, TX). After a series of washings, the blots were incubated with 1:2,000 dilution of goat anti-rabbit antibody conjugated to horseradish peroxidase (Cat. 65612, Invitrogen) and developed using the chemiluminescnce western blot detection kit (Thermo Scientific). After stripping, the same blot was treated with an anti-glyceraldehyde 3-phosphate dehydrogenase antibody 1:3,000 (Cat. ab9845, Abcam, Cambridge, MA) as a loading control.
ELISA of VEGF. To quantify VEGF protein levels, retinal and RPE/choroid tissues were harvested. VEGF was detected by a commercially available Mouse VEGF ELISA kit (Cat. No: SMMV00, R&D Systems, Minneapolis, MN). The assay was performed according to the manufacturer's instructions. All measurements were performed in duplicate. Tissue sample concentrations were calculated from the standard curve and corrected by total protein. One RPE/choroid complex was isolated for each sample, and four samples in each group were examined. Both retinas from each mouse were pooled in one tube.
Subretinal injection of either AAV2. Flt23k or AAV2.AcGFP. Normal male C57BL/6 mice were placed under general anesthesia with an intraperitoneal injection of ketamine/xylazine (90 mg/10 mg per kg body weight). Pupils were dilated with topical application of 1% tropicamide (Bausch & Lomb, Tampa, FL). For local anesthesia, a 0.5% proparacaine solution was applied to the cornea. Under a stereo microscope, a small incision was made behind the limbus with a 30.5-gauge needle. A blunt 33-gauge needle (Hamilton Company, Reno, NV) was then inserted through the incision, vitreous, and retina into the subretinal space at the posterior pole of the retina. Extra care was taken to avoid damaging the lens. Subretinal administration of 1 µl of AAV2.Flt23k or control AAV2.AcGFP virus at the titer of 5 × 1011 GC/ml was performed into both eyes of each mouse. Subretinal injection of PBS buffer was performed in additional mice as another control. Visualization of partial retinal detachment around the injection site by fundus examination at the conclusion of injection confirmed successful subretinal delivery.
Generation of murine laser photocoagulation-induced CNV model. To generate the laser-induced murine CNV model, mice were deeply anesthetized in the same fashion as described above. The beam from a diode laser (532 nm; Oculight Glx, Iridex, Mountain View, CA) was shone onto to the retina through a slit lamp, using a 22-mm coverslip as a contact lens. The treatment parameters were chosen to produce a cavitation bubble in the choroid without hemorrhage (spot size, 100 µm2; intensity, 120 mW; duration, 100 msec). Four laser burns were made for CNV volume calculation at the 3, 6, 9, and 12 o'clock positions of the posterior pole, around the optic nerve in both eyes. Eight laser burns were created around the optic nerve for protein analyses.
CNV volume calculation. CNV laser induction was performed in untreated, control mice and treated mice 1 month after subretinal injection of either PBS, AAV2.AcGFP, AAV2.Flt23k. Two weeks after laser injury, the animals were euthanatized with CO2, and the eyes were enucleated. After removing the cornea and lens, the eye cup was fixed in 4% paraformaldehyde for 2 hours at 4 °C. The retina was then removed and the Sclera/choroid/RPE complex washed three times in phosphate-buffered saline (PBS), permeated for 30 minutes in 1% triton X-100, blocked in 5% FBS/PBS with 0.02% tritonX-100 and 2 mmol/l MgCl2, and stained with 5 mg/ml Alexa594 conjugated isolectin GS-IB4 (Invitrogen) 1:200 in blocking buffer overnight. After three additional washings, samples were flat mounted on glass slides and cut into four to six pieces, taking care to avoid damaging the laser treated areas. CNV volumes were measured using scanning laser confocal microscopy (Olympus America, Center Valley, USA).
Histology. For histology, mouse eyes were immersion-fixed overnight in a fixative containing 2.5% glutaraldehyde/1% formaldehyde and resin embedded as described http://prometheus.med.utah.edu/~marclab/marclab_09_tools-protocols.html. Samples were sectioned at 500 nm along the horizontal axis including the injection site. Sections were stained with Richardson stain according to http://depts.washington.edu/rubelab/protocols/Richardson-Stain.html.
Retinal thickness calculation. Spectral domain optical coherence tomography (SD-OCT) imaging was performed using a commercially available Spectralis HRA+OCT device from Heidelberg. SD-OCT provides high resolution analysis of retinal architecture previously achieved only through histology. Using SD-OCT, we measured the thickness of the whole retina and ONL along the vertical axle and horizontal axis including the injection site. Mice were anesthetized as described above. 1000 linear A-scans were used to produce each horizontal B-scan, and 20 B-scans were averaged to minimize the background and achieve higher resolution. To produce surface images of the fundus, a rectangular volume of 100 B-scans was obtained and analyzed. Whole retina and ONL thickness were measured manually using the distance measuring tool of the system software InVivoVue. We recorded three measurements at the same distance from the optical nerve head in each eye and used this data to determine the average final whole retina and ONL thickness.
ERG analysis. ERG was performed on normal 6-month-old C57BL/6 mice after subretinal injected PBS, AAV2.AcGFP or AAV2.Flt23k. All mice were kept in darkness overnight prior to analysis. Mice were anesthetized as described above. They were then kept in dim, red light and placed on a chemical hand warmer that maintained body temperature between 37 and 38 °C for the duration of the experiment. Pupils were dilated with 1% tropicamide, and ERGs were measured (UTAS E-3000; LKC Technologies, Gaithersburg, MD) between a gold corneal and a stainless-steel scalp electrode with a 0.3-500 Hz band-pass filter. Scotopic ERGs were recorded using white flashes in darkness and photopic ERGs were elicited by full-field bright white flashes with 35 cd/m2 background illumination. For both scotopic and photopic ERG recordings, increasing flash intensities were used and a total of five to eight mice of each treatment group were analyzed. Five to eight a-wave and b-wave traces were averaged for every stimulus intensity. The mean value at each stimulus intensity was compared using an unpaired two-tailed t-test with Bonferroni correction for multiple comparisons.
TUNEL. TUNEL assay was performed on retina cryosections. Slides were dried at room temperature for 30 minutes. Sections were processed for apoptosis with the Click-iT TUNEL Alexa Fjuor image assay kit (Cat. No. C10246, Invitrogen). A positive control was set up by treatment with DNase I. Retina sections were furthered counterstained with Hoechst. An Inverted Fluorescence microscope (Carl Zeiss MicroImaging, Thornwood, NY) was used to take images.
Statistical analysis. Data are presented as mean ± SEM or mean ± SD. Statistical analysis was performed using the student two-tail T-test. A P value < 0.05 was considered significant. All data collection was performed by technicians blinded to treatment groups.
SUPPLEMENTARY MATERIAL Figure S1. Apoptosis was assessed with TUNEL staining, performed on C57Bl/6 wild-type mice and mice injected subretinally with PBS, AAV2.AcGFP, or AAV.Flt23k.
Acknowledgments
The authors thank Ryan Watkins for his assistance with this study. This work was supported by the RPB Unrestricted Award and NEI 5R01EY017182 and in part by an Unrestricted Grant from Research to Prevent Blindness, New York, NY, to the Department of Ophthalmonlogy & Visual Sciences, University of Utah. B.K.A. has patent rights related to this technology, but there are no other financial conflicts of interest among any of the authors.
Supplementary Material
References
- la Cour M, Kiilgaard JF, Nissen MH. Age-related macular degeneration: epidemiology and optimal treatment. Drugs Aging. 2002;19:101–133. doi: 10.2165/00002512-200219020-00003. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O'Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PT, et al. Eye Diseases Prevalence Research Group Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Friedlander M. Fibrosis and diseases of the eye. J Clin Invest. 2007;117:576–586. doi: 10.1172/JCI31030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson TL, Kirkpatrick L. Cost comparison of ranibizumab and bevacizumab. BMJ. 2011;343:d5058. doi: 10.1136/bmj.d5058. [DOI] [PubMed] [Google Scholar]
- Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP) Ophthalmol. 2013;120:2292–2299. doi: 10.1016/j.ophtha.2013.03.046. [DOI] [PubMed] [Google Scholar]
- Salmon F, Grosios K, Petry H. Safety profile of recombinant adeno-associated viral vectors: focus on alipogene tiparvovec (Glybera) Expert Rev Clin Pharmacol. 2013;7:53–65. doi: 10.1586/17512433.2014.852065. [DOI] [PubMed] [Google Scholar]
- Dismuke DJ, Tenenbaum L, Samulski RJ. Biosafety of recombinant adeno-associated virus vectors. Curr Gene Ther. 2013. [DOI] [PubMed]
- Guziewicz KE, Zangerl B, Komáromy AM, Iwabe S, Chiodo VA, Boye SL, et al. Recombinant AAV-mediated BEST1 transfer to the retinal pigment epithelium: analysis of serotype-dependent retinal effects. PLoS One. 2013;8:e75666. doi: 10.1371/journal.pone.0075666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon TJ, Deng WT, Erger K, Cossette T, Pang JJ, Ryals R, et al. Preclinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retinitis pigmentosa. Hum Gene Ther Clin Dev. 2013;24:23–28. doi: 10.1089/humc.2013.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay CN, Ryals RC, Aslanidi GV, Min SH, Ruan Q, Sun J, et al. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS One. 2013;8:e62097. doi: 10.1371/journal.pone.0062097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Huang L, Li X, Qi H, Zhou P, Yu W, et al. Prevalence of neutralizing factors against adeno-associated virus types 2 in age-related macular degeneration and polypoidal choroidal vasculopathy. Curr Gene Ther. 2013;13:182–188. doi: 10.2174/1566523211313030003. [DOI] [PubMed] [Google Scholar]
- Ju WK, Kim KY, Duong-Polk KX, Lindsey JD, Ellisman MH, Weinreb RN. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Mol Vis. 2010;16:1331–1342. [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wu J, Wu X, Xu P, Tian Y, Yi M, et al. Enhancement of rAAV2-mediated transgene expression in retina cells in vitro and in vivo by coadministration of low-dose chemotherapeutic drugs. Invest Ophthalmol Vis Sci. 2012;53:2675–2684. doi: 10.1167/iovs.11-8856. [DOI] [PubMed] [Google Scholar]
- Cronin T, Chung DC, Yang Y, Nandrot EF, Bennett J. The Signalling Role of the avβ5-Integrin Can Impact the Efficacy of AAV in Retinal Gene Therapy. Pharmaceuticals (Basel) 2012;5:447–459. doi: 10.3390/ph5050447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarallo V, Bogdanovich S, Hirano Y, Tudisco L, Zentilin L, Giacca M, et al. Inhibition of choroidal and corneal pathologic neovascularization by Plgf1-de gene transfer. Invest Ophthalmol Vis Sci. 2012;53:7989–7996. doi: 10.1167/iovs.12-10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi T, Miyake K, Masuda I, Takahashi H, Shimada T. Adeno-associated vector (type 8)-mediated expression of soluble Flt-1 efficiently inhibits neovascularization in a murine choroidal neovascularization model. Hum Gene Ther. 2010;21:631–637. doi: 10.1089/hum.2009.153. [DOI] [PubMed] [Google Scholar]
- Askou AL, Pournaras JA, Pihlmann M, Svalgaard JD, Arsenijevic Y, Kostic C, et al. Reduction of choroidal neovascularization in mice by adeno-associated virus-delivered anti-vascular endothelial growth factor short hairpin RNA. J Gene Med. 2012;14:632–641. doi: 10.1002/jgm.2678. [DOI] [PubMed] [Google Scholar]
- Tuo J, Pang JJ, Cao X, Shen D, Zhang J, Scaria A, et al. AAV5-mediated sFLT01 gene therapy arrests retinal lesions in Ccl2(-/-)/Cx3cr1(-/-) mice. Neurobiol Aging. 2012;33:433 e1–10. doi: 10.1016/j.neurobiolaging.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclachlan TK, Lukason M, Collins M, Munger R, Isenberger E, Rogers C, et al. Preclinical safety evaluation of AAV2-sFLT01- a gene therapy for age-related macular degeneration. Mol Ther. 2011;19:326–334. doi: 10.1038/mt.2010.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. 2013;120:1283–1291. doi: 10.1016/j.ophtha.2012.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufnagel RB, Ahmed ZM, Corrêa ZM, Sisk RA. Gene therapy for Leber congenital amaurosis: advances and future directions. Graefes Arch Clin Exp Ophthalmol. 2012;250:1117–1128. doi: 10.1007/s00417-012-2028-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Ashtari M, Wellman J, Marshall KA, Cyckowski LL, Chung DC, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4:120ra15. doi: 10.1126/scitranslmed.3002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano MR, Biagioni F, Besozzi G, Carrizzo A, Vecchione C, Fornai F, et al. Effects of bevacizumab on neuronal viability of retinal ganglion cells in rats. Brain Res. 2012;1478:55–63. doi: 10.1016/j.brainres.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Takeda A, Baffi JZ, Kleinman ME, Cho WG, Nozaki M, Yamada K, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460:225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayanagi K, Sharma S, Kaiser PK. Photoreceptor status after antivascular endothelial growth factor therapy in exudative age-related macular degeneration. Br J Ophthalmol. 2009;93:622–626. doi: 10.1136/bjo.2008.151977. [DOI] [PubMed] [Google Scholar]
- Yodoi Y, Tsujikawa A, Nakanishi H, Otani A, Tamura H, Ojima Y, et al. Central retinal sensitivity after intravitreal injection of bevacizumab for myopic choroidal neovascularization. Am J Ophthalmol. 2009;147:816–24, 824 e1. doi: 10.1016/j.ajo.2008.11.020. [DOI] [PubMed] [Google Scholar]
- Leon CS, Leon JA. Microendoscopic ocular surgery: a new intraoperative, diagnostic and therapeutic strategy. Part II: Preliminary results from the study of glaucomatous eyes. J Cataract Refract Surg. 1991;17:573–576. doi: 10.1016/s0886-3350(13)81043-7. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, Maharaj AS, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, et al. Endogenous VEGF is required for visual function: evidence for a survival role on müller cells and photoreceptors. PLoS One. 2008;3:e3554. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Amin S, Richter E, Rashid S, Scoglietti V, Jani PD, et al. Flt-1 intraceptors inhibit hypoxia-induced VEGF expression in vitro and corneal neovascularization in vivo. Invest Ophthalmol Vis Sci. 2005;46:1647–1652. doi: 10.1167/iovs.04-1172. [DOI] [PubMed] [Google Scholar]
- Jani PD, Singh N, Jenkins C, Raghava S, Mo Y, Amin S, et al. Nanoparticles sustain expression of Flt intraceptors in the cornea and inhibit injury-induced corneal angiogenesis. Invest Ophthalmol Vis Sci. 2007;48:2030–2036. doi: 10.1167/iovs.06-0853. [DOI] [PubMed] [Google Scholar]
- Cho YK, Uehara H, Young JR, Tyagi P, Kompella UB, Zhang X, et al. Flt23k nanoparticles offer additive benefit in graft survival and anti-angiogenic effects when combined with triamcinolone. Invest Ophthalmol Vis Sci. 2012;53:2328–2336. doi: 10.1167/iovs.11-8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Zhang X, Hirano Y, Tyagi P, Barabás P, Uehara H, et al. Targeted intraceptor nanoparticle therapy reduces angiogenesis and fibrosis in primate and murine macular degeneration. ACS Nano. 2013;7:3264–3275. doi: 10.1021/nn305958y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen LA, Uehara H, Cahoon J, Huang W, Simonis J, Ambati BK. Morpholino-mediated increase in soluble Flt-1 expression results in decreased ocular and tumor neovascularization. PLoS One. 2012;7:e33576. doi: 10.1371/journal.pone.0033576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klettner A, Roider J. Comparison of bevacizumab, ranibizumab, and pegaptanib in vitro: efficiency and possible additional pathways. Invest Ophthalmol Vis Sci. 2008;49:4523–4527. doi: 10.1167/iovs.08-2055. [DOI] [PubMed] [Google Scholar]
- Deudero JJ, Caramelo C, Castellanos MC, Neria F, Fernández-Sánchez R, Calabia O, et al. Induction of hypoxia-inducible factor 1alpha gene expression by vascular endothelial growth factor. J Biol Chem. 2008;283:11435–11444. doi: 10.1074/jbc.M703875200. [DOI] [PubMed] [Google Scholar]
- Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH, Chu YK, et al. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol Vis Sci. 2010;51:1190–1197. doi: 10.1167/iovs.09-4144. [DOI] [PubMed] [Google Scholar]
- Grunwald JE, Daniel E, Huang J, Ying GS, Maguire MG, Toth CA, et al. CATT Research Group Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2014;121:150–161. doi: 10.1016/j.ophtha.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarra GM, Stephens C, Schlichtenbrede FC, Bainbridge JW, Thrasher AJ, Luthert PJ, et al. Kinetics of transgene expression in mouse retina following sub-retinal injection of recombinant adeno-associated virus. Vision Res. 2002;42:541–549. doi: 10.1016/s0042-6989(01)00230-9. [DOI] [PubMed] [Google Scholar]
- Guy J, Qi X, Koilkonda RD, Arguello T, Chou TH, Ruggeri M, et al. Efficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual system. Invest Ophthalmol Vis Sci. 2009;50:4205–4214. doi: 10.1167/iovs.08-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Boye SL, Aleman TS, Conlon TJ, Zeiss CJ, Roman AJ, et al. Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in Leber congenital amaurosis. Hum Gene Ther. 2006;17:845–858. doi: 10.1089/hum.2006.17.845. [DOI] [PubMed] [Google Scholar]
- Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Yeger H, Tsuchida R, Torkin R, Gee MF, Thorner PS, et al. A hypoxia-driven vascular endothelial growth factor/Flt1 autocrine loop interacts with hypoxia-inducible factor-1alpha through mitogen-activated protein kinase/extracellular signal-regulated kinase ½ pathway in neuroblastoma. Cancer Res. 2005;65:7267–7275. doi: 10.1158/0008-5472.CAN-04-4575. [DOI] [PubMed] [Google Scholar]
- Aesoy R, Sanchez BC, Norum JH, Lewensohn R, Viktorsson K, Linderholm B. An autocrine VEGF/VEGFR2 and p38 signaling loop confers resistance to 4-hydroxytamoxifen in MCF-7 breast cancer cells. Mol Cancer Res. 2008;6:1630–1638. doi: 10.1158/1541-7786.MCR-07-2172. [DOI] [PubMed] [Google Scholar]
- Ford KM, Saint-Geniez M, Walshe T, Zahr A, D'Amore PA. Expression and role of VEGF in the adult retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2011;52:9478–9487. doi: 10.1167/iovs.11-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3'-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D'Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci USA. 2009;106:18751–18756. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Jani PD, Suthar T, Amin S, Ambati BK. Flt-1 intraceptor induces the unfolded protein response, apoptotic factors, and regression of murine injury-induced corneal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:4787–4793. doi: 10.1167/iovs.06-0419. [DOI] [PubMed] [Google Scholar]
- Luo L, Uehara H, Zhang X, Das SK, Olsen T, Holt D, et al. Photoreceptor avascular privilege is shielded by soluble VEGF receptor-1. Elife. 2013;2:e00324. doi: 10.7554/eLife.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.