

Abstract
Antiretroviral therapy (ART) is unable to eradicate human immunodeficiency virus type 1 (HIV-1) infection. Therefore, there is an urgent need to develop novel therapies for this disease to augment anti-HIV immunity. T cell therapy is appealing in this regard as T cells have the ability to proliferate, migrate, and their antigen specificity reduces the possibility of off-target effects. However, past human studies in HIV-1 infection that administered T cells with limited specificity failed to provide ART-independent, long-term viral control. In this study, we sought to expand functional, broadly-specific cytotoxic T cells (HXTCs) from HIV-infected patients on suppressive ART as a first step toward developing cellular therapies for implementation in future HIV eradication protocols. Blood samples from seven HIV+ patients on suppressive ART were used to derive HXTCs. Multiantigen specificity was achieved by coculturing T cells with antigen-presenting cells pulsed with peptides representing Gag, Pol, and Nef. All but two lines were multispecific for all three antigens. HXTCs demonstrated efficacy as shown by release of proinflammatory cytokines, specific lysis of antigen-pulsed targets, and the ability to suppress HIV replication in vitro. In conclusion, we are able to generate broadly-specific cytotoxic T cell lines that simultaneously target multiple HIV antigens and show robust antiviral function.
Introduction
Antiretroviral therapy (ART) prolongs the life of human immunodeficiency virus (HIV)–infected individuals by preventing progression to severe immunodeficiency, but ART cannot cure infection, and lifelong therapy is necessary to provide continuous viral suppression. Populations that are at high risk for treatment nonadherence are vulnerable to drug resistance and further transmission of HIV. Furthermore, the long-term use of ART may lead to side effects to the renal, hepatic, and cardiovascular systems.1
Recent efforts to develop therapies to eradicate HIV infection seek to avoid the lifelong dependency on ART by inducing the expression of persistent latent proviral genomes under the cover of continued ART, with the assistance of an augmented antiviral immune response.2 T cell therapy using cytotoxic T lymphocytes (CTLs) have proven successful in the treatment of virus-associated cancers and viral reactivation posttransplant.3,4,5,6,7 This type of therapy consists of expanding preexisting antigen-specific T cells from patients ex vivo until a sufficient number of cells are obtained for reinfusion. While the use of CTLs has proved effective in the cancer and posttransplant settings, CTL therapy for HIV infection appears to be safe but has, thus far, failed to durably control viremia in the absence of ART.8,9,10
One difference that may account for the previously observed lack of clinical efficacy of HIV-specific CTL is that those administered in HIV clinical trials thus far have largely been single-epitope–specific T cell clones expanded in the presence of mitogens and administered without the benefit of ART in actively viremic patients.8,9,10 This contrasts with the polyclonal virus-specific CTLs expanded in the presence of multiple, whole antigens and growth cytokines that have been successful at targeting Epstein-Barr virus (EBV),3,4 cytomegalovirus, and adenovirus5,6,7 in immunocompromised settings and EBV-positive lymphoma outside the hematopoietic stem cell transplantation setting. Hence, we proposed that developing an HIV-specific T cell product with broader antigen recognition would increase the ability of the T cells to target and clear HIV-infected cells, in the setting of an antilatency reagent to induce expression of quiescent viral genomes, and continued ART to prevent spread and viral epitope escape.
In this study, we have developed a novel strategy to expand cytotoxic T cells targeting multiple HIV antigens (HIV-specific T cells (HXTC)). We show that by using both autologous dendritic cells and phytohemagglutinin (PHA)-blasts as antigen-presenting cells (APCs), we can successfully expand HXTC lines from seven ART-established HIV patients who demonstrate robust cytotoxic and antiviral function in vitro.
Results
HXTCs expand to clinically relevant numbers and comprise mixed CD4+ and CD8+ T cells with an effector memory phenotype
To evaluate whether T cells could be expanded from the peripheral blood of HIV-infected patients on ART regardless of their human leukocyte antigen (HLA) type or medical history (Table 1), peripheral blood mononuclear cells (PBMCs) from seven patients were primed with autologous dendritic cell (DC) pulsed with Gag, Pol, and Nef PepMix in the presence of interleukin (IL)-7, IL-15, and IL-12 (Figure 1). PepMix is the commercialized term for peptide libraries spanning regions of the antigens of interest: gag, pol, and nef. (JPT Peptide Technologies). The PepMixes consist of 150 peptides that are 155 amino acids in length. These 15mers were selected based on a proprietary algorithm held by JPT that was used to determine which peptides provide the broadest coverage across all clades of HIV. After two more rounds of stimulation (24–26 days) using autologous PepMix-pulsed PHA-blasts, ex vivo expanded HXTCs derived from seven patients showed a mean expansion of 145.6-fold (range: 37.2–287.0) starting from 1 million T cells on day 0 quantified using cell counting (Figure 2a). While the expansion was a wide range, even the lower end of this range provides robust enough expansion to achieve the numbers required for clinical use. Furthermore, previous clinical trials using adoptively transferred EBV-specific T cells showed efficacy despite a lower fold expansion observed during the manufacturing process.3,11
Table 1. Characteristics of patient samples used to generate HXTCs.

Figure 1.

HXTC manufacturing process. Peripheral blood mononuclear cells (PBMCs) are isolated from 60–100 ml of whole blood samples from aviremic HIV+ patients. Monocytes are separated using plastic adherence and used to generate dendritic cells. Dendritic cells are pulsed with PepMixes spanning Gag, Pol, and Nef before being used to stimulate the nonadherent PBMC fraction which contains T cells. T cells undergo two more weekly stimulations by coculture with autologous, PepMix-pulsed PHA-blasts. During the third stimulation, irradiated K562 cells that have been genetically modified to express costimulatory molecules CD80, CD83, CD86, and 41-BBL are cocultured with the T cells in addition to the PepMix-pulsed PHA-blasts.
Figure 2.

CD8, effector memory HIV-specific T cells expand in response to Gag, Pol, and Nef stimulation. (a) 1 × 106 T cells were stimulated with Gag, Pol, and Nef PepMixes on day 0. Expansion is shown in absolute cell counts and was measured 10 days after the third stimulation. (b) Nonadherent PBMCS containing T cells (n = 3, error bars defined by SEM) and (c) HXTCs were phenotyped using extracellular flow cytometry for T cell markers (CD3, CD4, and CD8), NK cell markers (CD56+), and memory subsets (CD45RA and CD62L) 7–10 days after the third stimulation (n = 5, error bars defined as the SEM). (d) PD-1 expression on HXTCs or EBV-specific CTL generated from healthy, HIV-negative donors measured 7–10 days after third stimulation (representative of n = 3). (e) Comparison of percentage of PD-1+ T cells found in HXTCs versus EBV-specific CTLs generated from healthy, HIV-negative donors (n = 3, error bars defined by SEM).
These lines were predominantly CD3+CD8+ T cells (mean: 84.2%; range: 65.97–97.14%). However, we did retain a proportion of CD4+ T cells (mean: 16.9% (2.9–34.0%)). Despite the presence of CD4+ T cells, viral outgrowth was not observed in the cell cultures, and HXTC culture media was regularly supplemented with amprenavir (data not shown). After three stimulations (days 24–26), T cell lines contained a subpopulation of CD3−CD56+ NK cells (mean: 8%; range: 0–23.9%). Moreover, we observed the majority of the expanded T cells had converted to an effector memory phenotype (CD3+CD45RA−CD62L−; mean: 74.0% (48.8–93.3%); n = 5; Figure 2b,c) which was encouraging due to studies showing the role of functional HIV-specific effector memory CD8 T cells in instances of ART-independent viral control.12,13
The overexpression of PD-1 is well characterized on the surface of HIV-specific T cells from chronically infected patients and is considered a marker of T cell activation and immune dysfunction.14,15 We measured PD-1 expression on HXTCs that were expanded ex vivo for 21 days, 7 days after the third stimulation. As shown in Figure 2d,e, the percentage of PD-1+ cells in HXTCs was not different from that detected on EBV-specific CTL products that were expanded similarly from HIV-negative, healthy donors who have already shown efficacy in clinical trials.3,4
HXTC are specific for multiple HIV antigens
To confirm the multi HIV antigen-specific activity of the HXTCs, the cultures were evaluated for interferon (IFN)-γ secretion in response to individual PepMixes for each HIV antigen in ELIspot. Nonadherent PBMCs containing T cells from ART patients prior to expansion did not secrete IFN-γ in response to HIV antigens (Figure 3a). In contrast, HXTC lines secreted IFN-γ in response to HIV antigens Gag (median: 129.5 spot-forming cell (SFC)/1 × 105 cells; range: 32–327.5), Pol (median: 97.5 SFC; range: 4–402.5, and Nef (median: 310.5 SFC; range: 0–863.5; Figure 3b) but did not respond to negative control actin or Oct4 PepMixes (median: 2.0 SFC; range: 0–20) or media alone (median: 3.5 SFC; range: 0–60). To confirm that the observed responses were T cell mediated, we used MHC class I and class II blocking antibodies in the enzyme-linked immunospot assay (ELIspot) assay. As shown in Figure 3c, the IFN-γ responses to Gag, Pol, and Nef were decreased in the presence of class I or class II antibodies, suggesting that there are HIV-specific CD8 and CD4 T cells. We further confirmed the presence of HIV-specific T cells within HXTC cultures by measuring intracellular IFN-γ. A mean of 6.36% of HXTCs were IFN-γ+ after a 12-hour exposure to HIV PepMixes (normalized to negative control, Figure 3d,e). Importantly, the HXTCs expanded from all seven HIV+ patients showed recognition of at least one HIV antigen. Furthermore, five HXTCs recognized all three HIV antigens, demonstrating the feasibility of generating a multiantigen-specific T cell product from a single culture (Figure 3f and Table 1).
Figure 3.

HXTCs have CD8 and CD4-mediated responses to multiple HIV antigens. (a) Nonadherent PBMCs on day 0 (n = 3, error bars defined by SEM) and (b) HXTCs (n = 7, error bars defined by the SEM) 10 days after the third stimulation were tested for IFN-γ release as a marker of T cell activation using IFN-γ ELIspot assay against media as a negative control, irrelevant antigens actin or Oct4 PepMixes as negative controls, HIV PepMixes, or staphylococcus enterotoxin B (SEB) as a positive control. (c) HXTCs were pretreated for 1 hour with no antibodies (black bar), MHC class I (solid gray bar), and class II (striped bar) blocking antibodies before being plated in an IFN-γ ELIspot assay against Gag, Pol, and Nef PepMixes (representative example of n = 3, error bars defined by the SD). (d,e) Intracellular IFN-γ staining in HXTCs after 12-hour exposure to HIV PepMixes, SEB (positive control), or no antigen (media, negative control) (n = 4, error bars defined by SD). (f) Summary of the number of antigens each HXTC line simultaneously recognized based on IFN-γ ELIspot assay. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
HXTCs are polyfunctional and polyclonal
To determine the polyfunctionality and polyclonality of the ex vivo expanded HXTC, we evaluated their Vβ repertoire using flow cytometry and cytokine release in Elispot and ELISA assays. We found that HXTCs not only released IFN-γ but also tumor necrosis factor (TNF)-α and IL-2 upon exposure to Gag, Pol, and Nef (Figure 4a,b). Distinguishing this approach from strategies that utilize single-epitope–specific T cells, HXTCs were shown to be polyclonal by flow cytometry–based Vβ analysis (Figure 4c). HXTCs also recognized multiple epitopes within HIV antigens. Using IFN-γ ELIspot, we tested HXTCs against the Nef Group M consensus region broken up into multiple peptide pools containing 7–8 peptides each and observed that HXTCs recognized multiple epitopes, suggesting that they are broadly epitope specific within each antigen (Figure 4d).
Figure 4.

HXTCs release multiple proinflammatory cytokines and are polyclonal. (a) IL-2 and (b) TNF-a release by HXTCs in response to no antigen (neg), Gag, Pol, and Nef PepMix was measured by ELISA as described in the Materials and Methods section (representative of n = 3, error bars represent SD). (c) Vβ usage of HXTCs was analyzed using a flow cytometry–based assay, IOTest Beta Mark TCR V Kit (BD Biosciences). (d) HXTCs were tested against the Nef group M consensus region broken up into peptide pools containing seven to eight peptides each in an IFN-γ ELIspot assay. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
HXTCs can lyse antigen-pulsed and HIV-infected targets
To evaluate the cytolytic specificity of the HXTCs, we incubated the HXTCs with a panel of 51Chromium (51Cr)-labeled autologous target cells. HXTCs robustly lysed target cells pulsed with Gag (mean: 90.4 ± 6.68% lysis; n = 2), Pol (mean: 69.6 ± 1.22% lysis; n = 2), and Nef (mean: 44.4 ± 29.65% lysis; n = 3) at an effector to target ratio of 5:1. Importantly, HXTCs were specific since lysis of activated autologous target cells alone was not observed (1.9 ± 2.38% lysis; n = 4; Figure 5a,b, representative of n = 4). To further evaluate the antiviral activity of the HXTCs in vitro, we determined if they could inhibit HIV replication in autologous T cells. HXTCs or unexpanded autologous CD8+ T cells were cocultured with autologous CD8-depleted PBMCs infected with a laboratory strain of HIV JR-CSF, and p24 release was measured after 7 days. At a HXTC to target ratio of 1:10, cocultures with HXTCs produced significantly less p24 than cocultures with unexpanded CD8 T cells (mean: 50.9; range: 9.5–86.4% p24 recovery versus unexpanded CD8: mean: 92.8; range: 42.0–135.0% p24 recovery; P = 0.035), indicative of inhibition of HIV replication in vitro (Figure 5c).
Figure 5.
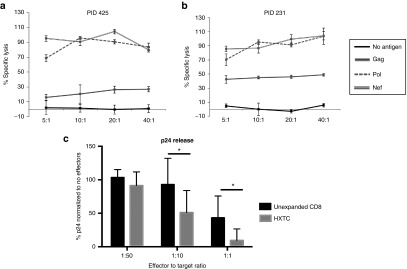
HXTCs are cytotoxic and inhibit viral outgrowth in infected cultures. (a,b) Ability of HXTCs to lyse antigen-expressing targets was measured using an 8-hour 51Chromium-release assay. Target cells consisted of autologous PHA-blasts that were pulsed with either media or Gag, Pol, or Nef PepMixes and were loaded with chromium. HXTCs were cultured with target cells pulsed with only the antigens they were specific for, which was predetermined by IFN-γ ELIspot assay. (c) Autologous CD8 depleted PBMCs were infected with HIV strain, JR-CSF by spinoculation. HXTCs or unexpanded CD8 T cells were cocultured with the infected target cells for 7 days before p24 release was measured using ELISA. *P ≤ 0.05. All error bars represent the SD.
Discussion
Although ART has increased the lifespan of HIV-infected patients, this treatment cannot fully eradicate the virus. The strict adherence necessary for continuous suppression and associated costs, side effects resulting from long-term use, and the populations at high risk for drug resistance make finding alternative therapies that could provide a cure imperative. Due to our previous successes treating immune-compromised patients with polyclonal T cells for other latent viruses (EBV and CMV),3,4,5,6,7 we sought to determine the feasibility of expanding polyclonal T cells to target HIV infection in acutely and chronically diagnosed HIV+ patients on suppressive ART and to characterize their function and determine their potential as an immunotherapy strategy.
In this study, we demonstrated that by the use of a novel expansion protocol, we produced HXTCs that comprised CD8+ and CD4+ T cells had an effector memory phenotype, and the majority of the T cell lines generated demonstrated reactivity to all three HIV antigens. This is in contrast to ex vivo unexpanded peripheral blood T cells, which failed to produce IFN-γ in response to stimulation with Gag, Pol, and Nef.
One limitation of previous studies utilizing T cells targeting HIV is their specificity for a restricted number of epitopes within a single antigen.8,9,10 The majority of our HXTC lines recognized multiple epitopes within three HIV antigens, We achieved this using a single-culture methodology which is simple and cost effective for clinical scale production. To achieve sufficient expansion of HXTCs for clinical use, we used genetically modified K562 cells (GM-K562), which have been engineered to express costimulatory molecules to drive T cell proliferation. Although the incorporation of these cells may pose a risk, tumor-specific T cells expanded using these cells are already being used clinically.16 Prior to culturing GM-K562 cells with HXTCs, these cells are irradiated at 100 Gy which is enough to lose replicative ability. Also, our FDA-approved release criteria requires that the final product be less than 2% CD3negCD83pos cells and <0.1% CD3−, CD16/56−, CD32+, and CD83+ (GM-K562 cells) cells.
We intentionally selected Gag, Pol, and Nef antigens to target both early and late antigens17,18,19 so that the HXTCs would have the potential to target HIV+ cells at different stages of infection. Moreover, all three of these antigens have been correlated with viral control.20,21,22 Our data also suggests that HXTCs are CD8 and CD4 restricted and recognize a broad number of epitopes. Since CD4 T cells are known to provide CD8 T cell help, aiding in memory, persistence, and effector functions.13,23 It is critical that any T cell immunotherapy approach for HIV utilizes CD4 T cells. While the infusion of a small percentage of CD4 T cells may seem counterintuitive, non-CD4–depleted T cells have been infused in the past, and no significant increase in viral load was observed.8 We envision administering HXTCs while maintaining the patient on ART therapy. Furthermore, it may be interesting to evaluate whether recent technologies that render CD4 T cells HIV resistant could be used in combination with our approach.24 Additionally, we eliminated the need to deplete CD4+ T cells prior to the expansion process by incorporating the use of antiretroviral drugs during the manufacturing process. Amprenavir, a protease inhibitor, was used to grow HXTCs, as well as the PHA-blasts used as APCs. This did not impact the ability to yield functional T cells against HIV antigens, and no viral outgrowth was observed.
HXTCs demonstrated strong antiviral effector functions in vitro. When exposed to Gag, Pol, and Nef antigens, HXTCs produced large amounts of IFN-γ, TNF-α, and IL-2 cytokines important for the antiviral response. Previous studies also showed the importance of polyfunctional cytokine production in T cell-mediated control of HIV.25 HXTCs were also able to specifically lyse target cells presenting Gag, Pol, and Nef epitopes, even at low effector to target ratios, and they showed greater suppression of HIV replication in infected T cell cocultures compared to unexpanded CD8 T cells.
Although we generated seven HXTC lines, we did not preselect which patient to derive these lines from based on HLA type or disease prognosis. These patients varied in their time to ART initiation, and some had reached severe T cell deficiency and AIDS in the past. Despite this, we were able to expand HXTCs from all seven HIV+ patients on suppressive ART irrespective of the clinical history, thus highlighting the feasibility and broad applicability of our approach in the clinical setting.
In summary, functionally active multi-HIV antigen-specific T cells can easily be manufactured using the same protocol for any patient, and we envision that any ART-established HIV patient would be eligible for future clinical studies. Furthermore, this T cell therapy platform is also complementary to other eradication strategies such as the use of histone deacetylase inhibitors that can induce viral reactivation and unmask the latent reservoir so that persistent infection may be cleared by immune effectors, such as HXTCs.
Materials and Methods
Patient samples. Peripheral blood was obtained from seven aviremic HIV patients on ART at the University of North Carolina–Chapel Hill (Chapel Hill, NC). All patients had provided written informed consent, and samples were obtained through a blood collection protocol approved by the University of North Carolina–Chapel Hill Institutional Review Board in accordance with the Declaration of Helsinki. Use of coded peripheral blood samples was approved by the Baylor College of Medicine and Children's National Medical Center Institutional Review Boards. Approximately, 60–100 ml of blood was drawn, and PBMCs were isolated by density gradient centrifugation and cryopreserved.
Generation of APCs. For DC generation, fresh or cryopreserved PBMCs were plated on 6-well plates and incubated for 2 hours in DC media (CellGenix DC media; CellGenix, Freiberg, Germany) supplemented with 2 mmol/l GlutaMax (Invitrogen, Carlsbad, CA). Nonadherent cells were harvested and cryopreserved. Adherent cells were cultured in dendritic cell media in the presence of IL-4 (1,000 U/ml) and granulocyte macrophage colony-stimulating factor (800 U/ml; both from R&D Systems, Minneapolis, MN). On day 5, immature DCs were matured using a cytokine cocktail consisting of IL-4 (1,000 U/ml), granulocyte macrophage colony-stimulating factor (800 U/ml), IL-6 (10 ng/ml), TNF-α (10 ng/ml), IL-1β (10 ng/ml; all from R&D Systems), and PGE2 (1 μg/ml; Sigma-Aldrich, St Louis, MO)26 and were harvested after 24–48 hours of maturation for use as APCs.
To generate PHA-blasts, PBMCs were stimulated with the mitogen PHA-P (5 μg/ml; Sigma-Aldrich) in the presence of IL-2 to promote blast formation. PHA-blasts were cultured in RPMI-1640 supplemented with 10% human serum (Valley Medical, Winchester, VA), 2 mmol/l GlutaMax, and IL-2 (100 U/ml; R&D Systems). To prevent possible viral outgrowth, PHA-blast cultures were treated with 0.5 ng/ml of amprenavir every other day (GlaxoSmithKline, Philadelphia, PA).
Generation of HXTCs. We define the use of “T cell line” in this manuscript as a single-culture of ex vivo expanded T cells derived from an individual donor. These cells are not immortalized. To generate HXTC lines, matured DCs were harvested and pulsed with Gag, Pol, and Nef peptide libraries (0.2 μg/ml; Ultra PepMix; JPT Peptide Technologies, Berlin, Germany) and used as APCs at a stimulator-to-responder ratio of 1:10 with nonadherent PBMCs, which contain T cells. T cells were cultured in RPMI-1640 supplemented with 40% Clicks media (Irvine Scientific, Santa Ana, CA), 10% human AB serum, and 2 mmol/l GlutaMax. For the first HIV stimulation, a cytokine mix containing IL-7 (10 ng/ml), IL-12 (10 ng/ml), IL-15 (5 ng/ml; all from R&D Systems) was added. Nine days later, T cells were restimulated with peptide-pulsed autologous irradiated (30 Gy) PHA-blasts at a stimulator-to-responder ratio of 1:4 on days 10 to 12 and maintained in IL-15 (5 ng/ml)–supplemented media on the second stimulation or IL-2 (50 U/ml)–supplemented media for the third stimulation cycle. On the third stimulation, irradiated (100 Gy) K562 cells that have been genetically modified to express costimulatory molecules, CD80, CD83, CD86, and 41-BBL (K562-cs) were added to the T cell:PHA-blast coculture to provide further costimulation (K562-cs cells were a gift from Dr Cliona Rooney at Baylor College of Medicine and described in ref. 16). HXTCs were expanded in presence of amprenavir (0.5 ng/ml) which was added to the cultures on the day of each stimulation and on days 3 and 5 poststimulation.
Phenotyping of HXTCs. HXTCs or EBV-specific CTLs from healthy donors (generated as described in refs. 3,4) were phenotyped with extracellular antibody staining with anti-CD3, CD4, CD8, CD45RA and CD45RO, CCR7, CD62L, CD56, CD16, and PD-1 or IOTest Beta Mark TCR V Kit (all from BD Biosciences, Franklin Lakes, NJ) and analyzed on a BD FACSCalibur, FACSCanto, or Accuri Flow Cytometer. Control samples labeled with appropriate isotype-matched antibodies were included in each experiment. Data were analyzed using FCS Express software (De Novo Software, Los Angeles, CA).
IFN-γ ELIspot assay. The antigen specificity of the HXTCs was tested in an IFN-γ ELIspot assay using peptide pools as antigen. Recognition of the pooled HIV antigens was compared with no-peptide (media) control and irrelevant peptide pools not used for T cell generation (Oct4 or actin; JPT Peptide Technologies). Staphylococcus enterotoxin B (SEB; Sigma) was used as a positive control. Nonadherent PBMCS were tested on day 0 prior to expansion, and HXTCs were tested 10 days after the third stimulation.
Millipore Multi Screen HTS filter plates (Millipore, Billerica, MA) were coated with IFN-γ capture antibody (Mabtech, Cincinnati, OH) at a concentration of 10 μg/ml overnight at 4 °C. Plates were washed with phosphate-buffered saline and blocked for 1 hour at 37 °C to rule out nonspecific protein binding. Nonadherent PBMCs were plated at 2 × 105/well, and HXTCs were plated at 1 × 105/well. Cells were washed and resuspended and stimulated with PepMix or single peptides at a concentration of 0.1 mg/ml. The plates were incubated for 16–20 hours at 37 °C. For development, plates were washed six times with phosphate-buffered saline /0.05% Tween 20 and incubated with biotinylated IFN-γ detection antibody (0.5 μg/ml; Mabtech) for 2 hours at 37 °C, followed by incubation with streptavidin-coupled alkaline phosphatase complex (Vectastain; Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. Spots were developed by incubation with 3-amino-9-ethylcarbazole substrate solution. SFC were counted and evaluated by Zellnet Consulting (Fort Lee, NJ) using an automated plate reader system (Carl Zeiss, Thornwood, NY). A T cell line positive for a given antigen is defined as having greater than double the SFC obtained in the negative control but at least 50 SFC/1e5 cells (or 2e5 cells in the case of nonadherent PBMCS).
HLA-blocking experiments. The HLA-restriction of antigen recognition was tested in IFN-γ–ELIspot using blocking antibodies against HLA class I and II (both from BD Biosciences, San Jose, CA). HXTCs were incubated with HLA class I or II blocking antibodies for 1 hour prior to being plated with media (negative control), Gag, Pol, or Nef PepMix, and SEB (positive control). ELIspot plates were incubated and developed as described earlier.
Intracellular cytokine staining for IFN-γ. 1 × 106 HXTCs were stimulated with media alone (negative control), Gag or Nef PepMixes, or SEB (positive control). After 3–4 hours, brefeldin A (BD Biosciences, San Jose, CA) was added to block Golgi transport. After 8 hours, cells were washed, permeabilized, and stained with extracellular (CD3-FITC; BD Biosciences) and intracellular antibodies (IFNy-APC; BD Biosciences). Samples were analyzed on a BD Accuri Flow Cytometer. Control samples labeled with appropriate isotype-matched antibodies were included in each experiment. Data were analyzed using FCS Express software.
ELISA for cytokine release. Detection of cytokines release in the supernatants of HXTC cocultures were analyzed by an ELISA specific for TNF-α (Invitrogen) or IL-2 (BD Biosciences), following the manufacturers' protocols. Seven to 10 days after the third stimulation, 1 × 105 HXTCs were stimulated in duplicates with Gag, Pol, and Nef PepMix, media, or SEB as a positive control. Twelve hours later, supernatant was collected for ELISA analysis.
51Cr-release cytotoxicity assay. The cytolytic activity of HXTCs was assessed in 51Cr-release assays 10 days after the third stimulation. Unpulsed autologous PHA-blasts (negative control) or HIV PepMix-pulsed PHA-blasts were labeled with 51Cr sodium chromate for 1 hour at 37 °C, washed, and resuspended with HXTCs at effector-to-target ratios from 40:1 to 5:1 and incubated for 6–8 hours. Spontaneous lysis was determined by measuring 51Cr-release into the supernatant on a gamma-counter from each target cell in the absence of effectors. Maximum lysis was measured by addition of 1% Triton X-100 (Sigma-Aldrich) to target cells. Specific lysis was calculated as follows: specific lysis (%) = (experimental release − spontaneous release)/(maximum release − spontaneous release) × 100.
Viral inhibition assay. CD8 depleted, PBMCs were cultured in complete media containing IL-2 (50 U/ml) and PHA (2 µg/ml) for 2 days before being superinfected with HIV laboratory strain JR-CSF via spinoculation. The infected target cells were cocultured for 7 days with autologous HXTCs or unexpanded autologous CD8 T cells (that were isolated using magnetic beads (Stem Sep; Stem Cell Technologies, Vancouver, British Columbia, Canada) added at varying effector-to-target ratios. Seven days later, supernatant was assayed for HIV-1 gag p24 concentration by ELISA (ABL, Rockville, MD); results are shown as %p24 production normalized to a positive infection control with no effector cells added.
Statistical analysis. Data are summarized as mean ± SD or mean (range), unless noted otherwise in the text or figure legends. Student's t-test was used to determine whether there was a statistically significant difference between samples, with two-tailed P values less than 0.05 indicating a significant difference. Two-way ANOVA with Holm-Sidak correction was used to determine statistical significance of IFN-γ release in response to HIV antigens compared to the negative control.
Acknowledgments
This study was funded by: The Center for Cell and Gene Therapy Trainee Award T32HL092332-10 to SL; Baylor-UT Houston Center for AIDS Research Core Support Grant A136211 from the National Institute of Allergy and Infectious Disease to C.B.; DC-Center for AIDS Research Grant M-0018R to C.B., U19-AI-096113 to D.M.; and T32-AI007001 to J.S.
The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 Consensus Group M Nef Peptides—Complete Set.
S.L., J.S., D.M., and C.B. designed experiments with input from N.A. and C.R. S.L. optimized protocol for expansion for HXTC lines and expanded HXTC lines for experiments. S.L., J.S., C.C., P.C.-C., and C.G. performed experiments. M.N. performed studies on the use of genetically modified, costimulatory K562 cells used in the expansion. J.K. coordinated study patients and obtainment of patient samples.
References
- Carr A. Toxicity of antiretroviral therapy and implications for drug development. Nat Rev Drug Discov. 2003;2:624–634. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- Margolis DM, Hazuda DJ. Combined approaches for HIV cure. Curr Opin HIV AIDS. 2013;8:230–235. doi: 10.1097/COH.0b013e32835ef089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. 2014;32:798–808. doi: 10.1200/JCO.2013.51.5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard CM, Gottschalk S, Huls MH, Molldrem J, Przepiorka D, Rooney CM, et al. In vivo expansion of LMP 1- and 2-specific T-cells in a patient who received donor-derived EBV-specific T-cells after allogeneic stem cell transplantation. Leuk Lymphoma. 2006;47:837–842. doi: 10.1080/10428190600604724. [DOI] [PubMed] [Google Scholar]
- Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–5123. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Christin A, Myers GD, Liu H, Cruz CR, Hanley PJ, et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood. 2009;114:4283–4292. doi: 10.1182/blood-2009-07-232454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- Lieberman J, Skolnik PR, Parkerson GR, 3rd, Fabry JA, Landry B, Bethel J, et al. Safety of autologous, ex vivo-expanded human immunodeficiency virus (HIV)-specific cytotoxic T-lymphocyte infusion in HIV-infected patients. Blood. 1997;90:2196–2206. [PubMed] [Google Scholar]
- Chapuis AG, Casper C, Kuntz S, Zhu J, Tjernlund A, Diem K, et al. HIV-specific CD8+ T cells from HIV+ individuals receiving HAART can be expanded ex vivo to augment systemic and mucosal immunity in vivo. Blood. 2011;117:5391–5402. doi: 10.1182/blood-2010-11-320226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie SJ, Lewinsohn DA, Patterson BK, Jiyamapa D, Krieger J, Corey L, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med. 1999;5:34–41. doi: 10.1038/4716. [DOI] [PubMed] [Google Scholar]
- Bollard CM, Straathof KC, Huls MH, Leen A, Lacuesta K, Davis A, et al. The generation and characterization of LMP2-specific CTLs for use as adoptive transfer from patients with relapsed EBV-positive Hodgkin disease. J Immunother. 2004;27:317–327. doi: 10.1097/00002371-200407000-00008. [DOI] [PubMed] [Google Scholar]
- Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15:293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SJ, Lacabaratz C, Lambotte O, Perez-Patrigeon S, Vingert B, Sinet M, et al. Preserved central memory and activated effector memory CD4+ T-cell subsets in human immunodeficiency virus controllers: an ANRS EP36 study. J Virol. 2007;81:13904–13915. doi: 10.1128/JVI.01401-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokey DA, Johnson FB, Smith J, Weber JL, Yan J, Hirao L, et al. Activation drives PD-1 expression during vaccine-specific proliferation and following lentiviral infection in macaques. Eur J Immunol. 2008;38:1435–1445. doi: 10.1002/eji.200737857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham LR, Jain V, Sinclair E, Glidden DV, Hartogenesis W, Hatano H, et al. Programmed death-1 expression on CD4+ and CD8+ T cells in treated and untreated HIV disease. AIDS. 2014;28:1749–1758. doi: 10.1097/QAD.0000000000000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo MC, Ando J, Leen AM, Ennamuri S, Lapteva N, Vera JF, et al. Complementation of antigen-presenting cells to generate T lymphocytes with broad target specificity. J Immunother. 2014;37:193–203. doi: 10.1097/CJI.0000000000000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- Wu Y. HIV-1 gene expression: lessons from provirus and non-integrated DNA. Retrovirology. 2004;1:13. doi: 10.1186/1742-4690-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland M, Nickle DC, Mullins JI. HIV-1 group M conserved elements vaccine. PLoS Pathog. 2007;3:e157. doi: 10.1371/journal.ppat.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk G, Ghiglione Y, Falivene J, Socias ME, Laufer N, Coloccini RS, et al. Early Gag immunodominance of the HIV-specific T-cell response during acute/early infection is associated with higher CD8+ T-cell antiviral activity and correlates with preservation of the CD4+ T-cell compartment. J Virol. 2013;87:7445–7462. doi: 10.1128/JVI.00865-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther. 2014;22:464–475. doi: 10.1038/mt.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adland E, Carlson JM, Paioni P, Kløverpris H, Shapiro R, Ogwu A, et al. Nef-specific CD8+ T cell responses contribute to HIV-1 immune control. PLoS One. 2013;8:e73117. doi: 10.1371/journal.pone.0073117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almedia JR, Price DA, Papagno L, Arkoub ZA, Sauce D, Bornstein E, et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med. 2007;204:2473–2485. doi: 10.1084/jem.20070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Cruz CR, Savoldo B, Leen AM, Stanojevic M, Khalil M, et al. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood. 2009;114:1958–1967. doi: 10.1182/blood-2009-03-213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Rahman T, Chakravorty R.2014Characterization of the protective HIV-1 CTL epitopes and the corresponding HLA class I alleles: a step towards designing CTL based HIV-1 vaccine Adv Virolepub ahead of print). [DOI] [PMC free article] [PubMed]
- McKellar MS, Cope AB, Gay CL, McGee KS, Kuruc JD, Kerkau MG, et al. Duke-Unc Acute HIV Infection Consortium Acute HIV-1 infection in the Southeastern United States: a cohort study. AIDS Res Hum Retroviruses. 2013;29:121–128. doi: 10.1089/aid.2012.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]