

Abstract
Three type-1 repeat (3TSR) domain of thrombospondin-1 is known to have anti-angiogenic effects by targeting tumor-associated endothelial cells, but its effect on tumor cells is unknown. This study explored the potential of 3TSR to target glioblastoma (GBM) cells in vitro and in vivo. We show that 3TSR upregulates death receptor (DR) 4/5 expression in a CD36-dependent manner and primes resistant GBMs to tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)-induced caspase-8/3/7 mediated apoptosis. We engineered human mesenchymal stem cells (MSC) for on-site delivery of 3TSR and a potent and secretable variant of TRAIL (S-TRAIL) in an effort to simultaneously target tumor cells and associated endothelial cells and circumvent issues of systemic delivery of drugs across the blood–brain barrier. We show that MSC-3TSR/S-TRAIL inhibits tumor growth in an expanded spectrum of GBMs. In vivo, a single administration of MSC-3TSR/S-TRAIL significantly targets both tumor cells and vascular component of GBMs, inhibits tumor progression, and extends survival of mice bearing highly vascularized GBM. The ability of 3TSR/S-TRAIL to simultaneously act on tumor cells and tumor-associated endothelial cells offers a great potential to target a broad spectrum of cancers and translate 3TSR/TRAIL therapies into clinics.
Introduction
Human glioblastoma (GBMs) are highly vascularized tumors, and their growth relies on the formation of newly generated tumor-associated blood vessels.1 The resulting vessels are structurally immature and make a harsh tumor microenvironment such as hypoxia, acidosis, and high interstitial fluid pressure that selects for tumor malignancy.2,3,4 However, preclinical and clinical studies reveal that antiangiogenic therapy targeting vascular endothelial growth factor (VEGF) can suppress tumor growth but may also promote tumor invasion and recurrent tumor growth in GBMs.5,6,7,8 Thus, there is an urgent need for new treatments that target both tumor cells and tumor-associated vasculature in both primary and secondary invasive tumor sites.
Thrombospondin-1 (TSP-1) is a natural inhibitor of angiogenesis, and the antiangiogenic potential of TSP-1 is mainly driven by its three type-1 repeats (3TSR) domain.9,10 3TSR has been shown to induce apoptosis in human dermal microvascular endothelial cells through the interaction with CD36 receptor that directly induces p59fyn/p38/caspase-3 mediated apoptosis or upregulates TRAIL receptors, death receptor (DR) 4/5.9,11 3TSR also binds β1 integrin and mediates antiangiogenic effect in human umbilical vein endothelial cells that lack CD36 in a phosphoinositide 3-kinase–dependent manner.12 3TSR was shown to inhibit the growth of experimental B16F10 melanoma indirectly through transforming growth factor β–dependent mechanism.13 Elevated levels of transforming growth factor β were detected in the blood serum of patients with malignant glioma and also in culture supernatants of GBM.14,15 Transforming growth factor β signaling in GBM was found not to render antitumorigenic response but to enhance cell growth, migration, and invasion.16 In this study, we reasoned that upregulation of TRAIL receptors through engagement of CD36 by 3TSR might be conserved in GBM lines and thus explored the potential therapeutic role of 3TSR on GBMs. This type of direct effect on cellular apoptosis has not been reported in any other cancer cell type.
TRAIL is a very promising targeted cancer therapeutic since it induces apoptosis in a tumor selective manner.17 However, tumor heterogeneity brings about unavoidable drug resistance to TRAIL18 and about half of the known tumor lines have been reported as resistant to TRAIL.19 A number of chemotherapeutic agents have been shown to be able to sensitize resistant tumor cells to TRAIL-mediated apoptosis.20,21,22,23,24,25,26,27 None of them, however, have been successful in clinical trials in combination with TRAIL and its agonists.28,29,30,31 This study explored the potential of 3TSR to sensitize GBM tumor cells to TRAIL-mediated apoptosis.
The major challenges of utilizing TRAIL, its sensitizers, as well as antiangiogenic agents in cancer therapy are their short half-life, rapid clearance, dose-limiting toxicity, and poor delivery of therapeutics to the tumor sites.29 Additionally, the presence of the blood–brain barrier prevents most of current cancer therapeutic agents from reaching the tumors in the brain.32 A number of research groups including us have previously shown that stem cells can be engineered to express receptor-targeted therapeutics at high levels and have extensive migratory capacity toward GBMs.33,34,35 Stem cell–mediated delivery of therapeutics bypasses the blood–brain barrier and delivers drugs at the site of the tumor.33,35 This strategy has been successful in preclinical studies, by which stem cells secreting TRAIL can enhance treatment efficacy for a longer period with the advantage of a sustained local delivery of TRAIL directly to tumor deposits.36 In this study, we engineered human mesenchymal stem cells (MSCs) to release 3TSR and a potent and secretable variant of TRAIL (S-TRAIL) to target a broad spectrum of GBMs with varied sensitivity to TRAIL in vitro and in mouse models of TRAIL-resistant GBMs.
Results
MSC secreting 3TSR and TRAIL have synergistic cytotoxic effect on GBM cells in a CD36-dependent manner
To study the effect of 3TSR and TRAIL in vitro and in vivo on GBM cells, we first generated a secretable variant of 3TSR which encodes the regions of all three type 1 repeats of TSP-1 (AA 361–530). The 3TSR lentiviral plasmid construct (diagrammed in Supplementary Figure S1a) was packaged into lentivirus (LV) virions, and MSC expressing secretable 3TSR (MSC-3TSR) were engineered (Supplementary Figure S1b). Western blotting using the conditioned medium (CM) of MSC-3TSR revealed that 3TSR was secreted into the medium (Supplementary Figure S1c). To test the antiangiogenic activity of MSC-3TSR in vitro, we analyzed branch point formation of human brain microvascular endothelial cells (HBMVEC), the most widely used surrogate to estimate angiogenesis in vitro. MSC-3TSR significantly inhibited the formation of branch points in HBMVEC (Supplementary Figure S1d). Next, we screened a cohort of both established GBM cell lines (U373, LN229, U138, U251, Gli36, and U87) and patient-derived primary GBM stem cell lines (GBM4, GBM6, and BT74) for their response to TRAIL (Supplementary Figure S2). The GBM lines showed varied response to TRAIL (Supplementary Figure S2). To test combination effect of 3TSR and TRAIL on GBM cells, we engineered MSC to express a secretable and highly potent variant of TRAIL, S-TRAIL34 (from here on referred as TRAIL) (MSC-TRAIL), 3TSR (MSC-3TSR), or 1:1 mix of MSC expressing TRAIL or 3TSR (from here on referred as MSC-3TSR/TRAIL). Single treatment of MSC-3TSR CM did not affect viability of the GBM lines tested (Figure 1a). However, pretreatment with 3TSR CM for 24 hours sensitized two TRAIL-resistant cell lines LN229 and GBM6, two semi-resistant lines U138 and U251 and had an additional effect on TRAIL-sensitive line U87 to TRAIL (Figure 1a). Combined treatment with MSC-3TSR CM and CM-containing quantified TRAIL significantly reduced cell viability of LN229, U138, U251, U87, and GBM6 compared to TRAIL alone (P < 0.01; Figure 1a). To further confirm the therapeutic effect of MSC-3TSR and MSC-TRAIL, we cocultured MSCs with GBM lines, LN229 (TRAIL resistant), U251 (TRAIL semi-resistant), and U87 (TRAIL sensitive). The combination treatment with MSC-3TSR/TRAIL significantly reduced cell viability of all three GBM cell lines as compared to MSC-3TSR and MSC-TRAIL treatment (Figure 1b–d), even with TRAIL-resistant line LN229 when cocultured for 120 hours. These data indicate that MSC-3TSR can sensitize TRAIL-resistant GBMs to TRAIL-mediated apoptosis.
Figure 1.
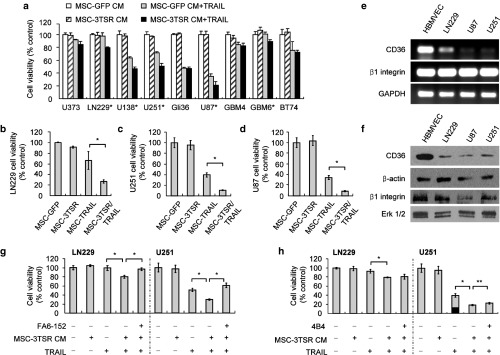
MSC secreting 3TSR and TRAIL have synergistic cytotoxic effect on GBM cells in a CD36-dependent manner. (a) Cell viability was estimated using Celltiter Glow assay in different GBM lines. GBM lines were preincubated with MSC-3TSR-conditioned media for 24 hours and incubated with TRAIL (500 ng/ml for resistant lines, U373, LN229, U138, GBM4, GBM6, and BT74 and 100 ng/ml for sensitive lines, U251, Gli36, and U87) for additional 48 hours. *P < 0.01 between MSC-GFP CM + TRAIL and MSC-3TSR CM + TRAIL (Student's unpaired t-test). (b–d) Cell viability of LN229-mCherry-Fluc, U251-mCherry-Fluc, and U87-mCherry-Fluc was measured by bioluminescence imaging 120 hours (for LN229; TRAIL resistant), 72 hours (for U251; TRAIL semi-resistant), and 48 hours (for U87; TRAIL sensitive) after coculture with indicated MSCs in 1:4 ratio of MSCs to tumor cells. *P < 0.01 (e) Gene expression levels of CD36 and β1 integrin in GBM lines were measured by RT-PCR. (f) Western blotting showing the protein levels of glycosylated CD36 and β1 integrin. Glycosylated CD36 and β1 integrin were indicated by an arrowhead around 88 and 120 kD, respectively. ERK was used for loading control for β1 integrin. (g–h) LN229 and U251 GBM lines were preincubated with anti–CD36-blocking antibody FA6-152 or anti-β1 integrin blocking antibody 4B4 for 2 hours, cultured with MSC-GFP (control) or MSC-3TSR-conditioned media for 24 hours, and further incubated for 48 hours with TRAIL, 500 ng/ml for LN229 and 100 ng/ml for U251. Celltiter glow was used for cell viability assay. *P < 0.01 and **P < 0.05.
Furthermore, we determined the mechanism by which 3TSR sensitizes GBM cells to TRAIL-mediated apoptosis. Although the antiangiogenic effect of 3TSR is mainly driven by the induction of apoptosis through the sequential activation of CD36 receptor,9 3TSR is known to bind β1 integrin and mediate antiangiogenic effects in human umbilical vein endothelial cells that lack CD36.12 Thus, we measured gene expression level of CD36 and β1 integrin in LN229, U251, and U87 lines. RT-PCR result showed that CD36 is transcriptionally much less expressed in GBM lines compared to HBMVEC, whereas there was no difference in the level of β1 integrin transcripts between GBM lines and HBMVEC (Figure 1e). CD36 and β1 integrin are glycosylated while they are transported to cell surface. The protein expression levels of glycosylated CD36 and β1 integrin determined by western blotting analysis also correlated with RT-PCR results while β1 integrin of U87 was expressed less than HBMVEC (Figure 1f and Supplementary Figure S3). Next, we preincubated TRAIL-resistant LN229 and semi-resistant U251 GBM cells with CD36 and β1 integrin–blocking antibodies, FA6-152 and 4B4, respectively, then exposed them to CM from MSC-3TSR for 24 hours and further incubated with CM-containing quantified S-TRAIL (500 ng/ml for LN229 and 100 ng/ml for U251) for 48 hours. CD36-blocking antibody treatment rescued cell viability reduction induced by 3TSR and TRAIL to the level of TRAIL treatment alone both in LN229 and U251 cells (Figure 1g) but β1 integrin blocking antibody had no effect (Figure 1h). These data suggest that 3TSR sensitizes TRAIL-resistant GBMs to TRAIL-mediated apoptosis through the interaction with CD36 receptor.
MSC-3TSR upregulates the expression of TRAIL receptors and primes GBMs to caspase-8/3/7–mediated apoptosis
We next examined the mechanism of combination effect of 3TSR and TRAIL in resistant GBM. Western blot analysis of lysates from TRAIL-resistant LN229 and semi-resistant U251 cells treated with MSC-3TSR CM greatly increased expression level of DR5, whereas DR4 expression was slightly increased (Figure 2a). We also investigated whether 3TSR affects downstream molecules of DR4/5 signaling pathway. Western blot analysis of lysates from TRAIL-resistant LN229 and semi-resistant U251 cells treated with MSC-3TSR CM did not change the protein levels of procaspase-8 and FLIP long form (FLIP L) both in U251 and LN229 (Supplementary Figure S4). Detectable amounts of FLIP short form (FLIP S) were not observed in either U251 or LN229 (Supplementary Figure S4). To further confirm the link between 3TSR and TRAIL receptors, we used a lentiviral-based DR4 or DR5 promoter-Fluc (Firefly luciferase) and CMV-Rluc (Renila luciferase)-DsRed reporter system that simultaneously allows real-time monitoring of DR4 and DR5 promoter activity (schemed in Figure 2b). We generated LN229 and U251 lines in which DR4 and DR5 promoter activity can be measured by Fluc signal intensity normalized by the corresponding changes in cell viability measured by Rluc activity. Consistent with the data shown in Figure 2a, treatment with MSC-3TSR CM for 48 hours increased DR4 and DR5 promoter activity similar to their protein expression level both in LN229 and U251, suggesting that 3TSR can upregulate transcriptional level of DR5 more than twofold in TRAIL-resistant LN229 and semi-resistant U251 GBM cells (Figure 2b). DR4 transcriptional level was slightly increased in both lines, but this increase was still significant (P < 0.01; Figure 2b). Furthermore, a combination of MSC-3TSR CM and CM-containing quantified TRAIL resulted in a decrease in the procaspase-8 indicating the cleavage of caspase-8 and enhanced activity of caspase-3/7 apoptosis executioners (Figure 2c,d), which led to significantly increased cleavage of poly-ADP ribose polymerase in both U251 and LN229 cells (Figure 2d). These results suggest that 3TSR sensitizes TRAIL-resistant GBM cells to TRAIL-mediated apoptosis through upregulation of TRAIL receptor DR4/DR5 expression.
Figure 2.
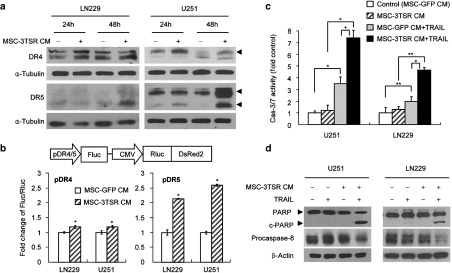
MSC-3TSR upregulates the expression of TRAIL receptors and primes TRAIL-resistant GBMs to caspase-8/3/7–mediated apoptosis. (a) LN229 and U251 were incubated with MSC-3TSR-conditioned media for indicated time, and DR4 and DR5 protein level was detected by western blotting. Arrowheads indicate DR4 (55 kD) and two different forms of DR5 (43 and 48 kD). (b) Schematic representation of polycistronic lentiviral vectors for dual bioluminescence reporter system to measure promoter activity of DR4 and DR5. Firefly luciferase (Fluc) and Renilla luciferase (Rluc) are expressed under the control of DR4 or DR5 promoter (pDR4/5) and CMV promoter, respectively. U251 and LN229 cells expressing the reporter system were incubated with MSC-3TSR-conditioned media for 48 hours. Transcriptional regulation of DR4 (bottom left) and DR5 (bottom right) by 3TSR were estimated by measuring bioluminescence of Fluc normalized with that of Rluc by Dual-Glo luciferase assay. *P < 0.01. (c) Caspase3/7 activation was assessed by caspase3/7 Glo assay in LN229 and U251 cells which were cultured with conditioned media from MSCs (GFP or 3TSR) for 48 hours and followed by treatment with TRAIL (50 ng/ml for U251 for 7 hours and 100 ng/ml for LN229 for 14 hours). *P < 0.01 and **P < 0.05 (Student's unpaired t-test). (d) Cleaved poly-ADP ribose polymerase and procaspase-8 protein level were western blotted to monitor apoptosis induction. Cell lysates were prepared from U251 and LN229 cells cultured in same condition as described in panel c.
MSC-3TSR inhibits angiogenesis and sensitizes brain endothelial cells to TRAIL in a CD36-dependent manner
Glioma cells including established GBM lines U87 and LN229 have been reported to secrete factors that promote angiogenesis8,37 and U87 and LN229 have been used frequently in a variety of antiangiogenic studies since they form highly vascularized tumors in vivo. To determine the combined potency of 3TSR and TRAIL on HBMVEC in the presence of glioma cells, we cocultured HMVEC-mCherry-Fluc and U87-GFP-Rluc cells with MSC-GFP (control), MSC-3TSR, MSC-TRAIL, and MSC-3TSR/TRAIL. Coculture analysis revealed that MSC-3TSR inhibited branch point formation of HBMVEC and significantly reduced cell viability of HBMVEC (P < 0.01; Figure 3a,b). Combination of MSC-3TSR and MSC-TRAIL showed even greater antiangiogenic activity and suppressed cell viability more than the single treatment with MSC-3TSR or MSC-TRAIL (Figure 3a,b). MSC-3TSR–treated HBMVEC upregulated DR5 in a time-dependent manner (Figure 3c), suggesting that 3TSR also sensitizes HBMVEC to TRAIL-mediated apoptosis. We next determined whether 3TSR could sensitize HBMVEC to TRAIL through a CD36-dependent manner. HBMVEC was preincubated with CD36-blocking antibody (FA6-152) followed by treatment with MSC-conditioned media for 48 hours. As shown in Figure 3d, CD36-blocking antibody FA6-152 rescued cell viability of HBMVEC treated with a combination of 3TSR and TRAIL to the level of TRAIL alone suggesting that 3TSR sensitizes HBMVEC to TRAIL-mediated apoptosis in a CD36-dependent manner. This data demonstrates that 3TSR effectively sensitizes both brain endothelial cells and GBM lines to TRAIL-mediated apoptosis in vitro. We next examined the antiangiogenic ability of MSC secreting 3TSR and TRAIL in vivo. MSC-GFP, MSC-3TSR, MSC-TRAIL, or MSC-3TSR/TRAIL were implanted intratumorally into mice bearing intracranial LN229-mCherry-Fluc tumors, and mice were sacrificed 5 days post stem cell implantation. Immunofluorescent staining analysis of brain tumor sections revealed that MSC-3TSR and MSC-3TSR/TRAIL improved tumor-associated vessels significantly in contrast to MSC-GFP and MSC-TRAIL which still displayed thick and irregular shaped vessels in the treated mice (Figure 3e,f). We also assessed the expression levels of CD36 in MSC-GFP and MSC-3TSR/TRAIL treated tumor bearing mice brain. In both treated groups, tumor area is less stained for CD36, which is consistent with our in vitro data (Figure 1e,f). However, CD36 is much strongly stained in normal brain area and CD36-positive spots do not colocalize with CD31-positive areas (Supplementary Figure S5). This result is consistent with a previous finding,38 which demonstrated that microglia expresses CD36 most abundantly in brain. Anti-CD36 mostly stains microglia that consists of up to 15–20% of brain cells and less frequently stained endothelial cells.38 The control group often showed a lot of blood vessels within and near tumors, while fewer vessels and/or CD31-positive spots were observed in the MSC-3TSR/TRAIL-treated group (Supplementary Figure S5). Since we observed that MSC-3TSR/TRAIL reduced the cell viability of HBMVEC in vitro (Figure 3b), we investigated whether the MSC-3TSR/TRAIL may harm normal brain microvasculature and normal brain tissues. MSC-GFP and MSC-3TSR/TRAIL were implanted intracranially into non–tumor-bearing mice, and mice were sacrificed 5 day post stem cell implantation. MSC-3TSR/TRAIL did not induce apoptosis in the normal brain tissue (Supplementary Figure S6a,c). However, the number of blood vessels near MSC-3TSR/TRAIL implantation site were reduced (Supplementary Figure S6a,b). These data support the concept that MSC secreting 3TSR or 3TSR/TRAIL possesses strong antiangiogenic activity against tumor vasculature in vivo without damaging normal brain tissues.
Figure 3.

MSC-3TSR inhibits angiogenesis and sensitizes brain endothelial cells to TRAIL in a CD36-dependent manner. (a) Photomicrographs and average branch points of HBMVEC after 18 hours coculture with U87 cells and MSCs expressing GFP (control), 3TSR, TRAIL, or 3TSR/TRAIL. *P < 0.01 between MSC-3TSR and MSC-3TSR/TRAIL. (b) Cell viability of HBMVEC was measured by Fluc bioluminescence after 48 hours post coculture with HBMVEC-Fluc-mCherry cells, U87 cells, and indicated MSCs. *P < 0.05 between MSC-TRAIL and MSC-3TSR/TRAIL. (c) Death receptor 5 expression was detected by western blot analysis using whole cell lysates of HBMVEC which was incubated with conditioned medium from MSC-3TSR or MSC-GFP (control) for indicated times. (d) HBMVEC was preincubated with anti-CD36 antibody FA6-152 for 1 hour before adding MSC-3TSR-conditioned media. Cell viability was measured by Celltiter Glo assay after 48 hours. *P < 0.05. (e) Immunofluorescence staining images of endothelial cell marker CD31 on brain sections from LN229-Fluc-mCherry GBM-bearing mice treated with indicated MSCs. MSCs were intratumorally injected 10 days after tumor implantation. Brain sections were prepared from mice sacrificed at day 5 after MSC injection. Bar = 100 µm. (f) Comparison of total area of tumor associated vessels obtained from anti-CD31 stained brain sections described in panel e (n = 3). *P < 0.01 (Student's unpaired t-test). The area of tumor associated vessels was measured using ImageJ.
MSC-3TSR/TRAIL inhibits growth of TRAIL-resistant GBM and prolongs survival of mice bearing TRAIL-resistant GBM
We next assessed the therapeutic efficacy of a combination of MSC-3TSR and MSC-TRAIL in TRAIL-resistant GBM in vivo. Mice bearing established intracranial TRAIL-resistant LN229-Fluc-mCherry GBM were implanted intratumorally with MSC-GFP (control), MSC-3TSR, MSC-TRAIL, or MSC-3TSR/TRAIL and followed up for changes in tumor volumes by Fluc imaging and survival (schematic diagram in Figure 4a). A combination treatment of GBMs with MSC-3TSR/TRAIL resulted in significant suppression of tumor growth compared to MSC-TRAIL and MSC-3TSR treatment alone (Figure 4b) and prolonged survival of mice bearing intracranial TRAIL-resistant GBM (Figure 4a). Immunofluorescent staining analysis of brain tumor sections showed that significantly higher number of cleaved caspase-3–positive tumor cells were observed in brain sections of mice treated with a combination of MSC-3TSR and MSC-TRAIL compared to control (MSC-GFP), single MSC-3TSR or MSC-TRAIL treated mice (Figure 4c,d). Taken together, our results demonstrate that MSC-3TSR and MSC-TRAIL combination treatment reveals significant therapeutic efficacy in TRAIL-resistant GBMs in vivo.
Figure 4.
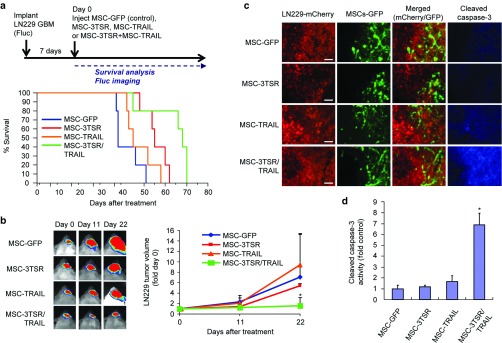
MSC-3TSR/TRAIL inhibits growth of TRAIL-resistant GBM and prolongs survival of mice bearing TRAIL-resistant GBM. (a) Timeline and survival curves of mice bearing intracranial LN229-Fluc-mCherry GBM. MSC-GFP (control), MSC-3TSR, MSC-TRAIL, or MSC-3TSR/TRAIL were intratumorally injected 1 week after tumor implantation (n = 5 mice each group). Median survival is 38, 45, 55, and 68 days for MSC-GFP, MSC-TRAIL, MSC-3TSR, and MSC-3TSR/TRAIL, respectively. P < 0.05 between 3TSR/TRAIL group and other groups in log-rank test. (b) Representative bioluminescence images of tumor-bearing mice for each group used in panel a were shown (left). Change in tumor volumes was estimated by comparing Fluc bioluminescence intensity of mice at days 11 and 22 with that of day 0 (right). *P < 0.05 between MSC-3TSR/TRAIL and each individual group at day 22. (c) Immunofluorescence stained images of cleaved caspase-3 on brain sections from LN229-Fluc-mCherry GBM-bearing mice treated with indicated MSCs. MSCs were intratumorally injected 1 week after tumor implantation. Brain sections were prepared from mice sacrificed at 72 hours after MSC injection. Bar = 100 µm. (d) Comparison of immunofluorescence intensity of cleaved caspase-3 staining on brain sections imaged in panel c (n = 3). *P < 0.01 between MSC-3TSR/TRAIL and each individual group. Immunofluorescence intensity of cleaved caspase-3 from tumors was measured using ImageJ.
Discussion
In this study, we explored the potential of 3TSR to target both GBM tumor cells and GBM-associated endothelial cells. We show that 3TSR upregulates TRAIL receptor DR4/5 expression in a CD36-dependent manner and sensitizes GBMs to caspase-3/7–mediated apoptosis. MSC expressing 3TSR/TRAIL targets the vascular component of GBMs and induces apoptosis in an expanded spectrum of GBMs with varied sensitivity to TRAIL in vitro and in vivo.
Human GBMs are highly vascularized tumors that actively release a substantial amount of stimulating factors and promote either by direct interaction with endothelial cells or by secretion of a large number of factors.8 In our coculture and in vivo experiments, the combination of secreted 3TSR and TRAIL was able to overcome the support of GBM cells on endothelial cells, which use complex mechanisms to promote angiogenesis in their vicinity.1 A number of clinical trials are ongoing to target tumor angiogenesis and sustained levels of angiogenic inhibitors are a potential key to improve the efficiency of antiangiogenic cancer therapy due to their short blood circulating half-life.8 Using the tumor-homing properties of stem cells,33,35 our study shows that engineered stem cells can provide a sustained and continuous delivery of 3TSR to tumor deposit and improve tumor vasculature significantly. Since stem cells chase tumor cells and migrate through tissues, stem cell–mediated delivery of 3TSR may overcome possible drawbacks of current antiangiogenic therapies: (i) tumor invasiveness and metastasis caused by sustained antiangiogenic inhibitor treatment5,39 and (ii) decreased delivery of antiangiogenic therapeutics after loss of tumor vasculature.40
Antiangiogenesis is an effective strategy to suppress tumor growth; however, treatment with antiangiogenic inhibitors alone cannot be expected to eradicate tumors8 (also see Figure 4b). Thus, activating apoptosis on tumor cells is a favorable combinational approach to cancer treatment as it has the potential to induce tumor regression. The ability of TRAIL to selectively target tumor cells while remaining harmless to most normal cells19 best fits to an apoptotic therapy for highly malignant GBMs. Although TRAIL is a selective and potent antitumor agent, many tumor lines, including some established and patient-derived tumor lines, have varying response to TRAIL-induced apoptosis.19 To address GBMs that do not respond to TRAIL monotherapy, a number of in vitro studies have shown the potential of chemotherapeutic agents that can sensitize TRAIL-resistant GBM cell lines to TRAIL-mediated apoptosis.29 Although these studies hold promise, it would be ideal to use stem cells that simultaneously secrete different therapeutic proteins that target multiple pathways in GBMs as the single source of therapy. Our results demonstrate that antiangiogenic 3TSR is a new class of sensitizer to prime GBM lines to TRAIL-mediated apoptosis by upregulating TRAIL receptors in a CD36-dependent manner, which can expand the spectrum of tumors that can be treated by TRAIL.
Systemically delivered TRAIL is rapidly cleared by the kidneys due to its small size.41 As such, a number of recombinant versions of human TRAIL have been created to increase the size of the molecule to avoid a rapid renal clearance and enhance its tumor-killing activity.42 S-TRAIL bears an isoleucine zipper in the N terminus of TRAIL to facilitate trimerization to mimic phamacodynamics of natural TRAIL on TRAIL receptors.43 Similar to many other stem cells, MSC exhibit inherent tumor tropism44 and anti-tumor effects because of factors released from stem cells and physical interaction between the stem cell and the tumor cell.45 In this study, we engineered MSC to secrete TRAIL and 3TSR to further enhance the tumor-killing potential of MSC, which can accomplish continuous delivery of drugs in close proximity to tumor in vivo.
It will be of high interest to extend our observation to panels of primary GBM lines and utilize similar strategies in other tumor types with varying TRAIL response. In conclusion, our study reveals that antiangiogenic 3TSR can be a modulator of TRAIL response in GBMs and the potential application of 3TSR and TRAIL combination can expand TRAIL therapy to a broad spectrum of tumors.
Materials and Methods
Generation of lentiviral vectors. Lentiviral vector, pLV CSC-IG bearing an internal ribosomal entry site-GFP element, was used as a backbone to generate 3TSR lentiviral vector (LV-3TSR). The cDNA sequence encoding amino acid 361–530 of all three TSRs of TSP-1 (3TSR) were amplified by PCR using primers in which an EcoRI site were introduced in the forward primer (5′ cggaattcgactctgcggacgatggc 3′) and an XhoI site were introduced in the reverse primer (5′ ccgctcgagtcaaattggacagtcctgcttg 3′). The resulting 0.51 kb EcoRI/XhoI fragment was ligated in frame with a 63 bp NheI/EcoRI cDNA fragment encoding the human Flt3L secretion signaling sequence (a.a. 1–21) into the NheI/XhoI digested pLV CSC-IG vector thus resulting in the LV-3TSR construct. LV-Pico2-Fluc-mcherry (a kind gift from Dr Andrew Kung, Dana Farber Cancer Institute, Boston, MA) was used to generate LV-Pico2-Rluc-GFP.
Cell lines and cell culture. Human bone marrow–derived MSCs were obtained from Dr Darwin J. Prockop (Texas A&M Health Science Center, Temple, TX) and were grown in Alpha-MEM (Invitrogen/GIBCO, Carlsbad, CA) with 10% fetal bovine serum, 2–4 mmol/l l-glutamine and penicillin/streptomycin. HBMVECs (Cell Systems, Kirkland, WA) were grown in EGM-2-MV medium (LONZA, Walkersville, MD) supplemented with human epidermal growth factor, Hydrocortisone, GA-1000 (Gentamicin, amphotericin-B), 5% fetal bovine serum, vascular endothelial growth factor, human fibroblast growth factor-B, R3, IGF-1, and ascorbic acid. HBMVEC was passaged before they reached confluence (70–80%) and not grown beyond 10th passage. Established human GBM cell lines were grown in Dulbecco's Modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin/streptomycin, and primary human glioma lines were obtained from Dr Wakimoto (Mass General Hospital, Boston, MA) and were grown in a neurosphere culture medium as described.46
Lentiviral transductions and stable cell lines. The following lentiviral vectors were used in this study: LV-3TSR, LV-GFP, LV-S-TRAIL,47 LV-Pico2-Fluc-mCherry, and LV-Pico2-Rluc-GFP. Both LV-3TSR and LV-S-TRAIL have an IRES-GFP element in the backbone. Lentiviral packaging was performed by transfection of 293T cells as previously described.48 MSC-GFP, MSC-3TSR, and MSC-TRAIL lines were generated by transducing MSC with LV-GFP, LV-3TSR, or LV-S-TRAIL at multiplicity of infection of two in a growth medium containing protamine sulfate (2 µg/ml, Sigma-Aldrich, St Louis, MO), and cells were visualized for GFP expression by fluorescence microscopy. Similarly, GBM cells and HBMVEC were transduced with LV-Pico2-Fluc-mCherry or LV-Pico2-Rluc-GFP at a multiplicity of infection of two in medium containing protamine sulfate (2 µg/ml) and LN229-Fluc-mCherry; U87-Rluc-GFP; HBMVEC-Fluc-mCherry lines were obtained after puromycin (1 µg/ml) selection in culture.
Western blot analysis. Cells were lysed with NP40 buffer supplemented with protease inhibitors (Roche, San Francisco, CA) and phosphatase inhibitors (Sigma-Aldrich). Twenty micrograms of harvested proteins from each lysate were resolved on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotted with antibodies against 3TSR raised in the Lawler lab, cleaved poly-ADP ribose polymerase, FLIP, caspase-8, and β-actin (Cell Signaling, Beverly, MA), DR4 (Abcam, Cambridge, MA), DR5 (Santa Cruz Biotechnology, Santa Cruz, CA), CD36 (Cayman Chemical, Ann Arbor, MI), β1 integrin (a kind gift from Dr Richard Hynes), or α-tubulin (Sigma-Aldrich), and blots were developed by chemiluminescence after incubation with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology).
Matrigel assay. HBMVEC-mCherry-Fluc (Firefly luciferase), U87-GFP-Rluc (Renilla luciferase), and MSCs-GFP were plated on Matrigel (BD Biosciences, San Jose, CA). Eighteen hours later, photomicrographs were taken using the Nikon E400 light microscope (Nikon Instruments, Melville, NY). Endothelial cell branch points were quantified and averaged on each of the photomicrograph (n = 6).
Viability assay. Cell viability was measured by determining the aggregate cell metabolic activity using an ATP-dependent luminescent reagent (Cell Titer Glo; Promega, Madison, WI).
Intracranial GBM cell implantation and in vivo bioluminescence imaging. To establish intracranial GBM, LN229-Fluc-mCherry GBM (5 × 105 cells per mouse) cells were stereotactically implanted into the brains (right striatum, 2.5 mm lateral from bregma and 2.5 mm deep) of severe combined immune deficiency mice (6 weeks of age; Charles River Laboratories, Wilmington, MA). To assess the therapeutic efficacy of MSC-3TSR/TRAIL, mice bearing established intracranial LN229-Fluc-mCherry GBM were implanted intratumorally with MSC-GFP (control), MSC-3TSR, MSC-TRAIL, or MSC-3TSR/TRAIL (2 × 105 cells per mouse; n = 8 per group) and followed up for changes in tumor volumes by Fluc bioluminescence imaging 0, 4, 7, 11, and 22 days post MSC implantation. Specifically, mice were injected with 1 mg d-luciferin per mouse intraperitoneally and imaged for Fluc activity 5 minutes later by recording photon counts over 5 minutes using a cryogenically cooled high-efficiency CCD camera system (Roper Scientific, Trenton, NJ). Images were processed and visualized as described previously,49 and time sequential bioluminescence imaging intensities were compared to that of day 0 to estimate the change of tumor volume. Mice (n = 3 per group) were sacrificed 3 days post MSC implantation for immunostaining of cleaved caspase-3 on brain sections as described below. Remaining mice (n = 5 per group) were followed up for their survival posttreatment.
To test the antiangiogenic effect of MSC-3TSR or MSC-3TSR/TRAIL, LN229-Fluc-mCherry were intracranially implanted into mice (2 × 105 cells per mouse; n = 12). Tumor-bearing mice were intratumorally implanted with MSC-GFP (control), MSC-3TSR, MSC-TRAIL, or MSC-3TSR/TRAIL (2 × 105 cells per mouse; n = 3 per group). Mice were sacrificed 5 days post MSC implantation for immunostaining of CD31 and CD36 on brain sections as described below. To assess the possible toxicity of 3TSR/TRAIL to normal brain vasculature and tissue, MSC-GFP and MSC-3TSR/TRAIL were intracranially implanted into non–tumor-bearing mice (2 × 105 cells per mouse; n = 3 per group). Mice were sacrificed 5 days post MSC implantation for immunostaining of CD31 and cleaved caspase-3 on brain sections. All in vivo procedures were approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital.
Histology. Brain tumors were dissected and processed for immunohistochemistry as described.48 Twenty micron sections were assessed for GFP and mCherry expression representing MSCs and LN229, respectively. Immunostaining was performed as described previously34 using anti-CD31 antibody (Abcam; 1:100), anti-CD36 (Cayman Chemical; 1:100), and cleaved caspase-3 antibody (Cell Signaling; 1:100) and quantified using ImageJ (US National Institutes of Health, Bethesda, MD).
Statistical analysis. Data were analyzed by Student's t-test when comparing two groups. Data were expressed as mean ± SD, and differences were considered significant at P < 0.05. Kaplan–Meier analysis was used for mouse survival studies, and the groups were compared using the log-rank test.
SUPPLEMENTARY MATERIAL Figure S1. Characterization of SS-3TSR lentiviral vector. Figure S2. S-TRAIL sensitivity was estimated using Celltiter-Glow cell viability assay in established and patient-derived primary GBM lines incubated with different dose of S-TRAIL for 48h. Figure S3. Relative protein levels of CD36 and β1 integrin. Figure S4. LN229 and U251 were incubated with MSC-3TSR conditioned media for indicated time and protein levels of procaspase-8 and FLIP L were detected by western blotting. Figure S5. Immunofluorescence staining images of CD36 and endothelial cell marker CD31 on brain sections from LN229-Fluc-mCherry GBM-bearing mice treated with MSC-GFP or MSC-3TSR/TRAIL. Figure S6. MSC-GFP and MSC-3TSR/TRAIL were implanted intracranially into normal mice brain (n = 3) and brain sections were prepared from mice sacrificed at day 5 after MSC injection.
Acknowledgments
The authors would like to acknowledge the technical assistance of Mark Duquette. No potential conflicts of interest were disclosed. This work was supported by RO1 CA138922 (KS), RO1 NS071197 (KS), RO1 CA130895 (JL) and James McDonnell Foundation (KS).
Supplementary Material
References
- Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- Plate KH, Mennel HD. Vascular morphology and angiogenesis in glial tumors. Exp Toxicol Pathol. 1995;47:89–94. doi: 10.1016/S0940-2993(11)80292-7. [DOI] [PubMed] [Google Scholar]
- Rampling R, Cruickshank G, Lewis AD, Fitzsimmons SA, Workman P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int J Radiat Oncol Biol Phys. 1994;29:427–431. doi: 10.1016/0360-3016(94)90432-4. [DOI] [PubMed] [Google Scholar]
- Valk PE, Mathis CA, Prados MD, Gilbert JC, Budinger TF. Hypoxia in human gliomas: demonstration by PET with fluorine-18-fluoromisonidazole. J Nucl Med. 1992;33:2133–2137. [PubMed] [Google Scholar]
- de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y.et al. (2010Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice Neuro Oncol 12233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F.et al. (2011Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma Proc Natl Acad Sci USA 1083749–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F.et al. (2009Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis Cancer Cell 15220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate KH, Scholz A, Dumont DJ. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012;124:763–775. doi: 10.1007/s00401-012-1066-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res. 2007;74:90–99. doi: 10.1016/j.mvr.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Song K, Parangi S, Jin T, Ye M, Humphreys R.et al. (2009A double hit to kill tumor and endothelial cells by TRAIL and antiangiogenic 3TSR Cancer Res 693856–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SM, Derrien A, Narsimhan RP, Lawler J, Ingber DE, Zetter BR. Inhibition of endothelial cell migration by thrombospondin-1 type-1 repeats is mediated by beta1 integrins. J Cell Biol. 2005;168:643–653. doi: 10.1083/jcb.200407060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao WM, Seng WL, Duquette M, Lawler P, Laus C, Lawler J. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-beta-dependent and -independent mechanisms. Cancer Res. 2001;61:7830–7839. [PubMed] [Google Scholar]
- Rich JN. The role of transforming growth factor-beta in primary brain tumors. Front Biosci. 2003;8:e245–e260. doi: 10.2741/992. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Naganuma H, Satoh E, Nagasaka M, Isoe S, Nakano S.et al. (1995Secretion of transforming growth factor-beta 1 and -beta 2 by malignant glioma cells Neurol Med Chir (Tokyo) 35423–430. [DOI] [PubMed] [Google Scholar]
- Platten M, Wick W, Weller M. Malignant glioma biology: role for TGF-beta in growth, motility, angiogenesis, and immune escape. Microsc Res Tech. 2001;52:401–410. doi: 10.1002/1097-0029(20010215)52:4<401::AID-JEMT1025>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Yerbes R, Palacios C, López-Rivas A. The therapeutic potential of TRAIL receptor signalling in cancer cells. Clin Transl Oncol. 2011;13:839–847. doi: 10.1007/s12094-011-0744-4. [DOI] [PubMed] [Google Scholar]
- Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijlen JM, Bremer E, Mooij JJ, den Dunnen WF, Helfrich W. Review: on TRAIL for malignant glioma therapy. Neuropathol Appl Neurobiol. 2010;36:168–182. doi: 10.1111/j.1365-2990.2010.01069.x. [DOI] [PubMed] [Google Scholar]
- Evdokiou A, Bouralexis S, Atkins GJ, Chai F, Hay S, Clayer M.et al. (2002Chemotherapeutic agents sensitize osteogenic sarcoma cells, but not normal human bone cells, to Apo2L/TRAIL-induced apoptosis Int J Cancer 99491–504. [DOI] [PubMed] [Google Scholar]
- Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Büchler P, Haas TL.et al. (2006Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs Clin Cancer Res 122640–2646. [DOI] [PubMed] [Google Scholar]
- Gliniak B, Le T. Tumor necrosis factor-related apoptosis-inducing ligand's antitumor activity in vivo is enhanced by the chemotherapeutic agent CPT-11. Cancer Res. 1999;59:6153–6158. [PubMed] [Google Scholar]
- Jacquemin G, Granci V, Gallouet AS, Lalaoui N, Morlé A, Iessi E.et al. (2012Quercetin-mediated Mcl-1 and survivin downregulation restores TRAIL-induced apoptosis in non-Hodgkin's lymphoma B cells Haematologica 9738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S. Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res. 1999;59:734–741. [PubMed] [Google Scholar]
- Lacour S, Micheau O, Hammann A, Drouineaud V, Tschopp J, Solary E.et al. (2003Chemotherapy enhances TNF-related apoptosis-inducing ligand DISC assembly in HT29 human colon cancer cells Oncogene 221807–1816. [DOI] [PubMed] [Google Scholar]
- Ravi R, Jain AJ, Schulick RD, Pham V, Prouser TS, Allen H.et al. (2004Elimination of hepatic metastases of colon cancer cells via p53-independent cross-talk between irinotecan and Apo2 ligand/TRAIL Cancer Res 649105–9114. [DOI] [PubMed] [Google Scholar]
- Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–5400. [PubMed] [Google Scholar]
- Chen W,, Hou J, Zhao Y, Qiu L, Ke X, Wang Z.et al. (2012. Circularly permuted TRAIL (CPT) combined with thalidomide for the treatment of relapsed or refractory multiple myeloma: an open-label, multicenter phase II clinical trial. 54 ASH Annual Meeting and Exposition, abstr 2958.
- Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br J Pharmacol. 2013;169:1723–1744. doi: 10.1111/bph.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria JC, Márk Z, Zatloukal P, Szima B, Albert I, Juhász E.et al. (2011Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer J Clin Oncol 294442–4451. [DOI] [PubMed] [Google Scholar]
- Yee L,, Burris HA, Kozloff M, Wainberg Z, Pao M, Skettino S, et al. Phase Ib study of recombinant human Apo2L/TRAIL plus irinotecan and cetuximab or FOLFIRI in metastatic colorectal cancer (mCRC) patients (pts): preliminary results. J Clin Oncol. 2009;27:abstra 4129. [Google Scholar]
- Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W.et al. (2000Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas Proc Natl Acad Sci USA 9712846–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA, Mohapatra G.et al. (2009Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy Proc Natl Acad Sci USA 1064822–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Bureau E, Kim DE, Yang K, Tang Y, Weissleder R.et al. (2005Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression Ann Neurol 5734–41. [DOI] [PubMed] [Google Scholar]
- Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat Rev Cancer. 2014;14:683–691. doi: 10.1038/nrc3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godard S, Getz G, Delorenzi M, Farmer P, Kobayashi H, Desbaillets I.et al. (2003Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes Cancer Res 636613–6625. [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK.et al. (2002CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils Am J Pathol 160101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J.et al. (2001Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety J Pharmacol Exp Ther 29931–38. [PubMed] [Google Scholar]
- Stuckey DW, Shah K. TRAIL on trial: preclinical advances in cancer therapy. Trends Mol Med. 2013;19:685–694. doi: 10.1016/j.molmed.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Tung CH, Yang K, Weissleder R, Breakefield XO. Inducible release of TRAIL fusion proteins from a proapoptotic form for tumor therapy. Cancer Res. 2004;64:3236–3242. doi: 10.1158/0008-5472.can-03-3516. [DOI] [PubMed] [Google Scholar]
- Corsten MF, Shah K. Therapeutic stem-cells for cancer treatment: hopes and hurdles in tactical warfare. Lancet Oncol. 2008;9:376–384. doi: 10.1016/S1470-2045(08)70099-8. [DOI] [PubMed] [Google Scholar]
- Schichor C, Albrecht V, Korte B, Buchner A, Riesenberg R, Mysliwietz J.et al. (2012Mesenchymal stem cells and glioma cells form a structural as well as a functional syncytium in vitro Exp Neurol 234208–219. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Kesari S, Farrell CJ, Curry WT, Jr, Zaupa C, Aghi M.et al. (2009Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors Cancer Res 693472–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- Shah K, Hingtgen S, Kasmieh R, Figueiredo JL, Garcia-Garcia E, Martinez-Serrano A.et al. (2008Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model J Neurosci 284406–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Tang Y, Breakefield X, Weissleder R. Real-time imaging of TRAIL-induced apoptosis of glioma tumors in vivo. Oncogene. 2003;22:6865–6872. doi: 10.1038/sj.onc.1206748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.