

Abstract
Clinical therapy with T cells shows promise for cancer patients, but is currently challenged by incomplete responses and tumor relapse. The exact mechanisms that contribute to tumor relapse remain largely unclear. Here, we treated mouse melanomas with T cell receptor-engineered T cells directed against a human peptide-major histocompatibility complex antigen in immune-competent mice. T cells resulted in significant tumor regression, which was followed by relapse in about 80–90% of mice. Molecular analysis revealed that relapsed tumors harbored nonmutated antigen genes, not silenced by promoter methylation, and functionally expressed surface antigen at levels equal to nontreated tumors. Relapsed tumors resisted a second in vivo T cell treatment, but regained sensitivity to T cell treatment upon retransplantation in mice. Notably, relapsed tumors demonstrated decreased levels of CD8 T cells and monocytes, which were substantiated by downregulated expression of chemoattractants and adhesion molecules. These observations were confirmed when using T cells specific for a less immunogenic, endogenous mouse melanoma antigen. We conclude that tumors, when exposed to T cell treatment, can relapse without loss of antigen and develop a milieu that evades recruitment of effector CD8 T cells. Our findings support the concept to target the tumor milieu to aid T cell therapy in limiting tumor relapse
Introduction
Adoptive therapy with tumor-infiltrating T cells (TIL) shows significant and long lasting clinical responses in melanoma patients.1,2 In an effort to make T cell therapy a more universally applicable and controlled treatment, T cells have been engineered to express tumor-specific T cell receptors (TCR) directed against antigens such as MART-I, gp100, CEA, NY-ESO-1, or MAGE-A3 and clinical responses have been observed in patients with metastatic melanoma, colorectal, and synovial carcinoma.3 Clinical responses with TCR-engineered T cells, although variable and based on relatively small numbers of patients, are promising but challenged by toxicity and, despite effective initial regression, a transient nature of the antitumor response.
Further development of TCR gene therapy depends on choice of target antigen, optimization of the TCR transgene, and procedures to yield fit T cells.3,4 Equally important to the development of TCR gene therapy is to advance our understanding of the underlying cause of incomplete responses and tumor relapse. In the present study, we questioned whether loss of antigen is a requirement for tumors to relapse, and investigated other immune-evasive strategies that relapsed tumors may have developed. Currently, reports on antigen loss in tumor relapse are inconclusive and under debate. Clinical studies have suggested selective loss of MART-I expression in relapsed and residual tumors after infusion of MART-I-specific T cells.5,6 In addition, in nonmanipulated hosts, decreased antigen expression and immune evasion of tumors may be a consequence of molecular alterations in tumor cells, such as genetic and epigenetic alterations in antigen genes, major histocompatibility complex (MHC) genes and genes related to antigen processing and presentation.7,8 Specifically, in melanoma patients, selective loss of antigen or HLA-A2 expression in primary and metastatic lesions has been described in numerous reports.9,10 In contrast, preclinical models have recently suggested that relapsed tumors retained expression of both antigen and MHC.11,12,13
Here, we treated mouse melanomas with TCR-engineered T cells in two immune-competent mouse models. In a first model, T cells targeted a human gp100/HLA-A2 (gp100/A2) antigen that was expressed by melanoma transplanted onto HLA-A2 tg mice, and regressed and relapsed tumor variants are evaluated. Maximal T cell pressure did not prevent tumor relapse in the majority of mice. Extensive molecular analysis of the gp100/A2 target antigen demonstrated that relapsed tumors contained intact and nonmutated antigen DNA and functionally expressed antigen at levels equal to progressed tumors. Relapsed tumors resisted a second in vivo T cell treatment and, interestingly, regained therapy sensitivity upon retransplantation in mice. Further analysis revealed decreased levels of CD8 T cells and monocytes in relapsed tumors, which was substantiated by downregulated expression of chemoattractants and adhesion molecules. In a second model, T cells targeted an endogenous mouse TRP2 antigen and, also in a less immunogenic setting, tumors relapsed despite continued antigen expression and harbored decreased levels of adoptively transferred CD8 T cells.
Results
TCR T cells mediate highly effective, yet mostly transient regression of established tumors, and induce the generation of memory T cells in cured mice
We set out to maximize T cell therapy directed against a human antigen in an immune competent setting according to three lines. First, T cells were derived from HLA-A2 tg mice and transduced with gp100/A2-specific TCR-α and -β chains that were codon optimized, separated by T2A ribosome skipping sequence and cloned into pMP71 vector (TCR T cells, see Supplementary Text and Supplementary Figure S1a,c). Second, we generated a mouse melanoma B16 cell clone, obtained from a single cell, that stably expressed HLA-A2 genetically linked to human gp100 peptide (YLEPGPVTA) (B16:A2-YLEP; Supplementary Figure S1b–d). Third, mice were conditioned prior to T cell treatment with combined injections of busulfan and cyclophosphamide (Bu/Cy), which reduced absolute numbers of lymphocytes (Supplementary Figure S2).
We observed that using these conditions in a preventive setting, adoptive T cell therapy did not allow tumors to grow in up to 70% of mice and promoted survival in a T cell dose-dependent manner (Supplementary Figures S3 and S4a). Numbers of gp100/A2 pMHC-binding CD8 T cells in peripheral blood were detected with flow cytometry up to 3 weeks after T cell transfer (Supplementary Figure S3c), and gp100/A2-specific effector memory T cells were detected in spleens of tumor-free mice following tumor cell rechallenge (Supplementary Figure S3d). When testing adoptive T cell therapy in a curative setting (average size of established tumors: 300 mm3) (Figure 1a), we observed that mice receiving TCR but not mock T cells tumors rapidly regressed to volumes that were either not detectable or < 15 mm3 (Figure 1b). In 10–20% of mice, tumors remained absent, whereas in the majority of mice tumors relapsed by day 30–35 post-T cell transfer. TCR T cell treatment did not result in a significant increase in survival (Supplementary Figure S4b), which was related to treatment-related weight loss in 40–50% of mice and which can be alleviated by decreasing the Bu/Cy dosing (Straetemans et al., unpublished data). Numbers of peripheral gp100/A2 pMHC-binding CD8 T cells were most pronounced between days 4 and 11 after T cell transfer (Figure 1c). In analogy to a preventive setting, frequencies of TCR T cells decreased 2–3 weeks after T cell transfer below the detection level of flow cytometry (Figure 1c). Again, memory T cells were detectable in mice that remained tumor-free > 75 days following T cell transfer, as evidenced by detectable gp100/A2 pMHC-binding and interferon (IFN)-γ production by splenocytes upon stimulation with B16:A2-YLEP cells (Figure 1d).
Figure 1.

T cell receptor (TCR) T cells result predominantly in transient tumor regression, and in generation of antigen-specific memory T cells in case of durable regression. (a) Tumor curative model: HLA-A2 tg mice were subcutaneously (s.c.) transplanted with 0.5 × 106 B16:A2-YLEP cells at day 0. Ten days later, mice were conditioned with a total of four intraperitoneal (i.p.) Bu injections, twice daily on 2 consecutive days, followed by a single i.p. Cy injection. At day 13, 20 × 106 TCR T cells that bind gp100/A2 pMHC or mock T cells were injected (number of mock T cells equal to number of TCR T cells). (b) Tumor sizes were measured three times a week with a caliper. Data are expressed as mean mm3 ± SEM; % of mice with tumor relapse is indicated in parenthesis. (c) Peripheral blood was collected from mice at the indicated time points and absolute numbers of gp100/A2 pMHC-binding CD8 T cells were determined by flow cytometry. Data are presented as mean numbers per µl blood ± SEM. (d) Splenocytes were isolated from tumor-bearing and tumor-free mice following transfer of TCR T cells and from mice without tumor and not receiving T cell treatment, and cultured in vitro in the presence or absence of gp100 peptide and analyzed for gp100/A2 pMHC binding and IFN-γ production. Representative dot plots showing data from 1 of 4 mice are shown and the % of cells in each quadrant is indicated. Statistical significances were calculated with Student's t-tests: *P < 0.05, **P < 0.005.
Relapsed tumors functionally express antigen but do not respond to a second T cell treatment
To investigate antigen expression in relapsed tumors, we studied the following parameters. First, we completely sequenced A2-YLEP DNAs from these tumors and demonstrated no gene mutations in HLA-A2 or gp100 peptide sequences. Second, we assessed the methylation status of the retroviral promoter and observed that this was not different between relapsed and progressed tumors (Figure 2a). Third, we measured antigen expression and found that relapsed tumors express A2-YLEP mRNA at levels comparable to B16:A2-YLEP tumor cells in vitro (Figure 2b) and, when cultured ex vivo, show surface expression of antigen protein (Figure 2c). Treatment of these tumor cells with azacitidine (AZA), a demethylating agent, did not further upregulate expression of antigen protein, whereas IFN-γ treatment and extended culture periods did upregulate expression of antigen protein. Furthermore, relapsed tumors show equal levels of surface expressed antigen when compared to progressed tumors (Figure 3a,b). In contrast to these two types of tumors (20–30% HLA-A2), regressed tumors revealed significantly upregulated expression of antigen (95% HLA-A2, Figure 3b). In addition to percentage of cells, surface-expression of antigen per cell was also significantly increased in regressed tumors (mean fluorescent intensity). Finally, we studied if antigen expression could trigger a T cell response and showed that both relapsed and progressed tumors were equally able to induce the production of high levels of IFN-γ by TCR T cells ex vivo (20–30 ng/ml IFN-γ) (Figure 3c), whereas regressed tumors induced the production of significantly lower levels of IFN-γ (5 ng/ml, Figure 3c). In addition, cells derived from relapsed tumors are sensitive to TCR T cell-mediated cytotoxicity (Supplementary Figure S5). Given these observations, we subjected mice with a relapsed tumor to a second T cell treatment. A second cycle of conditioning and T cell transfer delayed tumor growth, which was, however, independent of the TCR transgene (Figure 3d). After a second treatment with TCR T cells, relapsed and progressed tumors again expressed equal levels of antigen (Supplementary Figure S6a) and were equally potent in inducing T cell IFN-γ production (Supplementary Figure S6b). More so, in tumor-bearing mice that experienced effective and curative T cell responses, we were unable to detect T cell reactivity to B16 antigens other than gp100/A2 (Supplementary Figure S7). To further confirm the nonedited antigen status of relapsed tumor cells, and rule out a potential “resistant tumor cell phenotype”, we injected short-term cultures of relapsed tumor cells, almost exclusively consisting of B16 cells, into naive mice and subjected re-established tumors to T cell treatment. Following retransplantation, we observed that therapy-resistant tumors regained sensitivity to treatment with TCR T cells (Figure 3e).
Figure 2.

Relapsed tumors do neither demonstrate methylation of antigen gene nor Azacitidine-mediated upregulated expression of antigen protein. HLA-A2 tg mice bearing B16:A2-YLEP tumors were conditioned and treated with T cell receptors (TCR) or mock T cells as described in legend to Figure 1a. (a) Relapsed and progressed tumors derived from B16:A2-YLEP clones were analyzed for methylation of the antigen gene promoter. Following methylation-specific PCR, products were analyzed by gel electrophoresis. U and M denote PCR products specific for unmethylated and methylated promoter sequences, respectively. (b) Relapsed tumors derived from either B16:A2-YLEP clone or cell line (the latter corresponding to group 3, Figure 4) were analyzed for levels of A2-YLEP DNA and mRNA. In vitro cultured B16:A2-YLEP clone and cell line were included as controls. DNA and mRNA levels are presented relative to the endogenous reference gene TRP2, with n = 3–4 per group. (c) HLA-A2 surface expression was measured by flow cytometry following ex vivo treatment of tumor cells with the demethylating agent azacitidine (AZA) (n = 9), IFN-γ (n = 11), or prolonged culture times (n = 4). Data are presented as mean % positive cells in viable gate + SEM. Statistical significances were calculated with Student's t-tests: *P < 0.05.
Figure 3.

Tumors that relapse following T cell treatment functionally express antigen, resist a second T cell treatment, but are responsive to T cell treatment after serial transplantation. HLA-A2 tg mice bearing established tumors from B16:A2-YLEP clone were conditioned and treated with 7.5 × 106 T cell receptors (TCR) or mock T cells according to legend to Figure 1a. (a) Following T cell treatment, mice with regressing (treated with TCR T cells, group 1), progressing (treated with mock T cells, group 2), and relapsing (treated with TCR T cells, group 3) tumors were sacrificed at the indicated time points. Tumors were isolated, single cell suspensions were prepared and short-term cultures (4–7 days) were set up (n = 3–5 mice per group). (b) Tumor cell HLA-A2 surface expression was measured by flow cytometry and data are presented as mean % positive cells in viable gate + SEM. (c) IFN-γ production by TCR T cells upon a 20 hours exposure to tumor cells ex vivo was analyzed by enzyme-linked immunosorbent assay. Data are presented as mean ng/ml + SEM. Statistical significances were calculated with Student's t-tests: *P < 0.05, ***P < 0.0005. (d) HLA-A2 tg mice bearing relapsed B16:A2-YLEP tumors were treated with TCR T cells for a second time (conditioned at days 30–32 and treated with T cells at day 34). A second treatment with mock T cells and no second treatment were included as controls. (e) Relapsed tumors from mice were isolated, cultured for 4 days, stored frozen (to allow synchronization of multiple samples), and retransplanted upon thawing (0.5 × 106 viable cells) in naïve HLA-A2 tg mice (retransplanted tumors, n = 5). When tumors re-established, recipient mice were subjected to treatment with TCR T cells as described in legend to Figure 3a. HLA-A2 tg mice bearing established tumors from B16:A2-YLEP clone were used as controls (transplanted tumors, n = 4). Tumor sizes in Figure 3d,e were measured and expressed as described in legend to Figure 1b.
Antigen-negative tumor cells in relapsed tumors originate from pre-treatment variants and have neither lost nor altered antigen DNA
To introduce heterogeneity with respect to antigen expression, and allow the tracing of antigen-negative tumor cells, we treated tumors derived from B16:A2-YLEP cell lines rather than clones. Tumors that were derived from a cell line relapsed to the same extent and with the same kinetics when compared with tumors that were derived from a clone (Supplementary Figure S8). The former tumors were analyzed for the presence and quantities of antigen protein, mRNA and DNA. Quantitative analyses of the A2-YLEP antigen revealed a significantly lowered level of protein expression in relapsed tumors (Supplementary Figure S9d) and reduced levels or in some cases even absence of A2-YLEP-specific mRNA and DNA (Supplementary Figure S9e,f). These data extended our findings in a preventive setting showing absent A2-YLEP DNA, mRNA and protein with high T cell doses (Supplementary Figure S9a–c). To understand the decrease of A2-YLEP DNA levels, we polymerase chain reaction (PCR)-amplified and sequenced retrovirally integrated A2:YLEP genes. DNA sequences in six out of nine relapsed tumors, but none of the tumors following treatment with mock T cells, revealed the presence of HLA-A2 without the gp100 peptide sequence (A2 DNA, Supplementary Figure S10, upper part, (b) band)). The A2 DNA sequences in all six samples were identical and showed no mutations other than the omission of peptide and linker sequences. Quantitative PCR reagents that specifically distinguished A2 with and without gp100 peptide (Supplementary Figure S10, lower part) revealed that levels of A2 and A2-YLEP DNA are inversely related (Figure 4). Also, antigen-negative DNA was not observed in relapsed tumors that were originally derived from a B16:A2-YLEP clone (Figures 4 and Supplementary Figure S10). The above data show that A2 tumor cells, which were present in pretreated tumors and had already lost or changed their antigen DNA, became exposed as a consequence of T cell selection against antigen-positive tumor cells.
Figure 4.

Relapsed tumor cells that lack antigen DNA are selected from pretreatment variants. HLA-A2 tg mice bearing B16:A2-YLEP tumors derived either from B16 cell line or clone were conditioned and treated with T cell receptors (TCR) or mock T cells as described in legend to Figure 1a. Relapsed tumors were collected and quantitative PCR was performed to distinguish between A2-YLEP and A2 DNAs (see Supplementary Figure S9 for the primer/probe combinations used). Results identified three groups based on antigen expression in relapsed tumors derived from B16 cell line. Group 1 tumors have no or negligible A2-YLEP DNA; group 2 tumors have intermediate levels of A2-YLEP DNA; and group 3 tumors have normal levels of A2-YLEP DNA (comparable to tumors treated with mock T cells). Group 3 tumors show the same antigen profile as tumors from clonal origin, demonstrating that tumors do relapse despite the presence of antigen DNA.
Relapse of antigen-positive tumors is related to decreased levels of tumor-specific T cells and monocytes
To study the involvement of the tumor micro-milieu in the occurrence of tumor relapse, we performed genome-wide expression arrays on tumor-derived RNA. Interestingly, the top-20 genes that were most differentially up-regulated in relapsed versus regressed tumors (out of a total of 24,000 genes) were related to gene expression, whereas those that were most differentially downregulated were mostly related to immunity (Supplementary Figure S11). When analyzing genes that mark immune cells, we observed that markers related to CD8 T cells and monocytes showed a decreased expression (Figure 5a), whereas those related to B cells (Ig molecules, CD19), natural killer cells (CD160, Ly108/CD352), dendritic cells (CD11c, Ly75/CD205), granulocytes (CD66a, d), T regulatory cells (CD25, CD127), and myeloid-derived suppressor Cells (CD11b, Ly6G5 and 6) showed a nonchanged expression in relapsed tumors. Moreover, the frequency of pMHC-binding TILs and their ability to recognize antigen-positive tumor cells is decreased in relapsed tumors (Figure 5b). The mRNA expression of molecules that contribute to the recruitment of blood T cells and monocytes, such as CCL and CXCL chemokines, and ligands for T cell integrins,14 was also decreased in relapsed tumors (Figure 6). The expression of vascular ligands for selectins appeared not differentially regulated at the mRNA level, whereas the mRNA expression of adhesion molecules reported to contribute to immune cell-adhesion and trafficking, such as certain galectins, annexins, and tetraspanins,15,16,17 was dramatically decreased in relapsed tumors (Figure 6).
Figure 5.
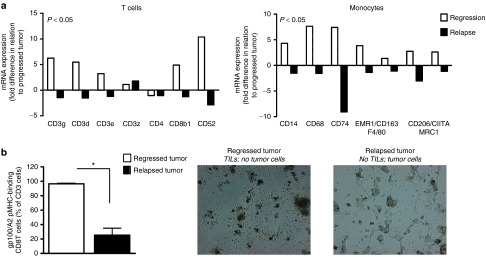
Relapsed tumors show decreased expression of CD8 T cell and monocyte markers, and decreased frequencies and activity of tumor-specific CD8 T cells. Regressed, relapsed and progressed tumors as defined in Figure 3a were collected and analyzed for immune cell infiltration. (a) RNA was isolated, biotin-labeled and used to hybridize onto Illumina MouseWG-6 v2 beads. See Supplementary Text for details on sample processing and data analysis. Analyses presented in this figure were restricted to genes that correspond to T cells and monocytes. Expression of genes (annotation according to www.genecards.org) in either regressed or relapsed tumors (white and black bars, respectively) is presented as fold change in relation to progressed tumors. Calculations were based on mean values, n = 3, per tumor type. Statistical significances for both cell types were calculated with Sign tests. (b) Tumors were prepared into single cell suspensions, cultured for 4 days under T cell conditions and assessed for the presence as well as antitumor activity of TILs (n = 4). TILs were analyzed by flow cytometry for the fraction of pMHC-binding CD8 T cells within CD3 cells and by microscopic evaluation for their ability to eliminate B16 tumor cells (magnification 200×). Statistical significances were calculated with Student's t-test: *P < 0.01.
Figure 6.

Relapsed tumors show decreased expression of markers that define immune cell recruitment. Regressed, relapsed, and progressed tumors as defined in Figure 3a were collected and analyzed for the expression of molecules involved in immune cell recruitment. RNA isolation, processing and data analysis are described in Supplementary Text. Analyses were restricted to chemoattractants, ligands for leukocyte rolling and ligands for leukocyte adhesion. Expression of genes in either regressed or relapsed tumors (white and black bars, respectively) is presented as fold change in relation to progressed tumors. Calculations were based on mean values, n = 3, per tumor type. Statistical significances for all three types of molecules were calculated with Sign tests.
T cell treatment directed against a native antigen also results in relapsed tumors with continued expression of antigen and decreased frequencies of CD8 T cells
To address whether our observations are dependent on the immunogenicity of the target antigen, we have targeted the mouse TRP2 antigen that is naturally expressed by B16 melanoma cells (see Supplementary Text and Supplementary Figure S12a–c for in vitro validation of TCR T cells and B16WT clone). When testing the anti-B16 melanoma activity of TRP2/H2-Kb TCR T cells in C57Bl/6 mice, we observed that 70% of mice showed moderate tumor regression (responder mice, Supplementary Figure S12d), and that numbers of peripheral TCR T cells were low but detectable for up to 2 weeks after T cell transfer (Supplementary Figure S12e). In this less immunogenic model, tumor regression was always followed by tumor relapse, and flow cytometry measurements showed that the majority if not all relapsed tumor cells are positive for TRP2 and H2-Kb proteins (Figure 7a,b). In addition, relapsed tumor cells were clearly able to induce production of IFN-γ by TCR T cells (Figure 7c). When investigating the number of TILs, we observed that relapsed tumors harbor a significantly decreased number of total CD8 T cells as well as TRP2/H2-Kb TCR, CD8 T cells (Figure 7d,e).
Figure 7.
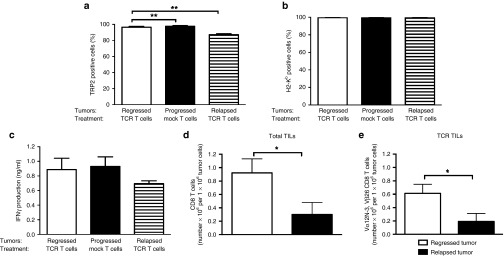
Tumors that relapse following T cell treatment directed against a native antigen show continued expression of antigen and decreased frequencies of tumor-specific CD8 T cells. In a second tumor model, B6 mice bearing established B16WT tumors were conditioned and treated with 7.5 × 106 TRP2/H2-Kb TCR or mock T cells according to legend in Figure 1a. Following T cell treatment, mice with regressing, progressing and relapsing tumors were sacrificed in accordance to the time points indicated in Figure 3a. Tumors were isolated, and short-term tumor cell cultures were assessed for (a) TRP2 expression and (b) H2-Kb surface expression by flow cytometry. Data are presented as mean % positive cells in viable gate + SEM (n = 3–6 mice per group). (c) IFN-γ production by TCR T cells upon a 20 hours exposure to tumor cells ex vivo was analyzed by enzyme-linked immunosorbent assay. Data are presented as mean ng/ml + SEM. In addition, short-term T cell cultures were assessed for the presence of (d) total number of CD8 TILs and (e) TCR-Vα12-N3, TCR-Vβ26-positive CD8 T cells by flow cytometry. Statistical significances were calculated with Student's t-tests: *P < 0.05, **P < 0.005.
Discussion
In this study, we tested TCR gene therapy in immune-competent mice directed against human gp100/A2 and mouse TRP2/H2-Kb and questioned whether loss of target antigen expression contributes to the occurrence of tumor relapse. TCR-engineered T cells resulted in regression of established tumors, but not in prevention of tumor relapse. Even in case of highly avid gp100/A2 T cells, the majority of established tumors reappeared after initial regression. These observations extend data from animal models reporting on tumor recurrence despite the presence of high T cell pressure18 and clinical trials with metastatic melanoma patients, where tumors relapsed after initial responses to vaccination10 or adoptive T cell therapy.2,5,6
Our findings provide evidence that tumors relapse following T cell therapy despite continued antigen expression. We could not detect gene mutations, and our results argue against involvement of epigenetic mechanisms that could have regulated antigen expression in relapsed tumors. On the one hand, levels of antigen surface expression and abilities to induce IFN-γ or target cell killing by TCR T cells ex vivo did not differ between relapsed and progressed tumors. On the other hand, promoter methylation was similar for both types of tumors and AZA, a demethylating drug, was not able to further upregulate antigen expression ex vivo. Although other studies have demonstrated that methylation of the antigen gene promoter results in progression of antigen-negative tumor variants,18 our data are in line with studies in which either skin, lung or ovarium tumors were induced and progressed despite continued antigen expression.11,12,13 In addition, relapsed tumors resisted a second treatment with TCR T cells, which again did not result in loss of antigen. To complement the above studies, and allow tracing of antigen-negative tumor cells, we treated tumors derived from B16:A2-YLEP cell lines. These studies demonstrated that antigen-negative tumors escaped from treatment with TCR T cells and were derived from rare, yet pre-existing antigen-negative tumor cells. In fact, extended quantitative PCRs showed that frequencies of antigen-negative tumor cell variants present in B16:A2-YLEP tumor cell lines are between 1 in 50,000 and 500,000 cells. T cell selection as a driving force to determine the antigen profile of tumors, although not a novel concept in itself,19 may have been underappreciated when assessing the genetic status of antigens from heterogeneous tumors or tumor cell lines. In fact, “antigen-loss” or “tumor escape” variants may even be misleading terms, and we would prefer to use the term “antigen-negative” variants unless evidence is provided for active tumor-editing.
There is compelling evidence that tumor escape is initiated by the interaction between CD8 T cells and highly immunogenic antigens, potentially resulting in growth of pre-existing tumor variants that are antigen negative or the occurrence of tumor variants that epigenetically silence an MHC class I allele.20,21 Our observations extend these findings and postulate that in a T cell therapy setting, loss of immunogenicity does not need to be a driving mechanism in tumor escape and may represent a rare event.22 It is noteworthy that in tumor-bearing HLA-A2 tg mice that experienced an effective in vivo treatment with TCR T cells, we were unable to detect T cell responses to B16 antigens other than gp100/A2 nor demonstrate involvement of antigen cross-presentation (Supplementary Figure S7). Although upon an initial immune response, antigen spreading and cross-presentation have both been implicated to broaden the response to tumor antigens,23,24,25,26 these results imply that, at least in this model, T cell specificities toward new and additional antigens do not explain tumor cure. Observations from our tumor models are compatible with tumor-induced suppression, an aspect that has recently been added to the cancer immunoediting hypothesis.27 First, progression of antigen-positive tumors was found in a curative rather than preventive setting. Second, regressed tumors showed upregulated expression of MHC class I antigen, but induced downregulated production of T cell IFN-γ when compared with progressed tumors. An ongoing T cell response is likely to be responsible for increased MHC class I expression by tumor cells. In regressed tumors, we detected enhanced numbers of TCR T cells, and enhanced expression of genes that correspond to of CD8, IFN-γ-positive T cells, and IFN-γ-downstream molecules, such as MHC class I (data not shown). However, at the same time, CD8 T cell-derived IFN-γ may ensue negative feedback mechanisms, such as upregulated expression of PD-L1 and IDO and enhanced presence of regulatory T cells in the tumor environment,28 and be responsible for impairment of T cell function. Finally, when retransplanting short-term cultures of relapsed B16 cells, therapy-resistant tumors changed into therapy-sensitive tumors. This latter observation suggests that cues from the micro-environment of established tumors contribute to the occurrence of therapy resistance. Landsberg and colleagues demonstrated in a gene-engineered model of melanoma that a tumor cell resistant phenotype may be directed by tumor necrosis factor-α, derived from the inflammatory milieu, which may lead to decreased expression of melanoma antigens.29 We were not able to confirm changed levels of tumor necrosis factor-α in relapsed B16 tumors.
To explore, in a nonbiased manner, by which mechanism the tumor milieu contributes to tumor progression, we performed genome-wide expression arrays. Remarkably, the expression of markers of CD8 T cells and monocytes, but not those of other immune cells, was markedly decreased in relapsed versus regressed tumors. These findings were substantiated by a lowered frequency of pMHC-binding TILs and a lowered ability of TILs to recognize antigen-positive tumor cells in relapsed tumors. Clinical studies have already demonstrated the unfavorable prognostic value of a limited CD8 T cell infiltration in melanoma, colorectal and ovarium carcinomas.30,31,32 Poor patient prognosis has also been reported to correlate with high numbers of macrophages with a tumor-promoting, but not those with a tumor-inhibiting phenotype.33 Findings here suggest that a deceased infiltration of CD8 T cells and monocytes is related to tumor relapse following T cell therapy. Notably, relapsed tumors demonstrated a decreased mRNA expression of chemoattractants that contribute to recruitment of CD8 T cells, such as CCL5, CXCL9 and CXCL10,34 and monocytes, such as CCL2.33 Interestingly, Hong et al.35 have demonstrated that the chemotherapeutic drugs dacarbazine, temozolomide, and cisplatin enhanced the expression of T-cell-attracting chemoattractants in patient melanoma, which in turn correlated with improved tumor control. The expression of vascular ligands for selectins, expected to contribute to the tethering of T cells and monocytes to endothelium, was not differentially regulated at the mRNA level in relapsed tumors. Since the activity of these ligands is primarily regulated via glycosylation,36 further research is needed to clarify the involvement of these molecules in relapsed tumors. The expression of integrin ligands, such as ICAM1, expected to contribute to the adhesion of T cells to endothelium, was downregulated at the mRNA level in relapsed tumors. Furthermore, the expression of certain galectins, annexins, and tetraspanins, with reported contributions to leukocyte trafficking and tumor development,37,38 was dramatically down-regulated in relapsed tumors. Currently, the exact role of the above-mentioned molecular interactions in the decreased infiltration of immune effector cells in relapsed tumors is part of our ongoing research.
Since the level of immunogenicity of the target antigen may determine the outcome of our results, we have repeated part of our experiments using an endogenously expressed target antigen. Targeting mouse TRP2 resulted in a less stringent T cell response, yet did show that tumor relapse again occurred with continued expression of antigen and decrease of CD8 T cell numbers. Collectively, our results propose a model for target antigen expression and immune evasion in tumor relapse after TCR T cell treatment, illustrated in Figure 8. According to this model, tumor relapse does not depend on loss of immunogenicity, but rather on the strength of initial T cell selection. Relapsed tumors, despite continued expression of antigen, are not cured by a second T cell treatment in vivo. Instead, these tumors develop a milieu that evades recruitment of effector CD8 T cells and monocytes. TCR gene therapy qualifies as a valid primary therapy to target relapsed tumors, but may benefit from supportive treatments. Three lines of examples are given below. First, supportive therapy may include the targeting of multiple tumor antigens.39,40 For future trials, preferred target antigens are those that contribute to the process of oncogenesis, such as driver neoantigens or selected cancer testis antigens.41,42 Second, supportive therapy may include the targeting of molecules or cells that are involved in T cell extravasation and T cell migration into tumor tissues. In addition to chemoattractants and adhesion molecules, targeting may include endothelial cells, fibroblasts or regulatory T cells that can impose barriers and refrain T cells to enter and deeply penetrate into tumor tissues.43,44,45 Third, supportive therapy may include strategies to improve T cell fitness, such as those that enhance T cell costimulation and/or the use of less-differentiated T cells (reviewed in ref. 3). In example, antibodies that block T cell coinhibitory molecules, such as CTLA4 or PD1 showed clear clinical successes in the treatment of patients with advanced melanoma and other tumors.46,47 Also, T cells that have been stimulated via CD28 and/or gene-engineered with chimeric antigen receptors that contain costimulatory molecules prior to T cell administration also resulted in significant responses in patients with B cell leukemia as well as advanced melanoma.48,49 Furthermore, it has become evident that true stem cells are contained within the CD62L central memory T cell pool and these cells may provide powerful therapeutic potential.50,51 The current manuscript further builds on the concept of targeting the tumor milieu and provides a rationale to develop of strategies to re-establish ligand-receptor interactions and enhance recruitment of immune effector cells into tumors.
Figure 8.

Relapsed tumors after T cell receptor (TCR) T cell treatment: a model for antigen expression and immune evasion. Treatment of established tumors with TCR T cells can result in rapid tumor regression. Tumor regression may be durable in a small proportion of mice and is associated with the formation of memory T cells, but in most mice tumors will relapse. Antigen-positive tumor cells (black cells) show continued antigen expression, with no changes in gene sequence, promoter methylation or functional expression. Antigen-negative tumor cells (white cells) are present in tumors prior to treatment, and do not arise as a consequence of T cell treatment. Instead, the relative quantity of antigen-negative tumor cells is determined by the strength of the T cell response against antigen-positive tumor cells. Relapsed tumors, despite expression of antigen, are not cured by a second T cell treatment in vivo. These tumors develop a milieu that is different from regressed tumors and which evades recruitment of effector CD8 T cells and monocytes, which is potentially due to downregulated expression of chemoattractants and adhesion molecules.
Materials and Methods
See Supplementary Text for cell culture; mice; construction, retroviral transduction, and validation of TCR and tumor antigen; measurements of tumor growth, number of T cells, and T cell memory formation; tumor prevention model; assessment of promoter methylation; cytotoxicity assays; and measurement of T cell responses to antigens other than gp100/A2.
Adoptive T cell therapy. At day 0, HLA-A2 tg mice were injected subcutaneously (s.c.) with 0.5 × 106 B16:A2-YLEP cells (from a clone or in some cases from a cell line) and at day 10 and 11 mice received a total of two or four Bu (Duchefa Farma, Haarlem, The Netherlands) injections intraperitoneally (i.p.) (16.5 µg/kg ea.), followed a day later by a single Cy (Sigma-Aldrich, St Louis, MO) injection i.p. (200 mg/kg). Mice received 7.5 or 20 × 106 TCR or mock T cells at day 13 and IL-2 at days 14 to 17 (1 × 105 IU per dose per day). In some experiments, mice with relapsed tumors received a second treatment with chemotherapeutics and TCR or mock T cells (see legend to Figure 3d). In other experiments, mice were retransplanted with relapsed tumor cells prior to treatment (see legend to Figure 3e).
Molecular characterization and functional expression of target antigen. Tumors were isolated, cut into small pieces, and digested in phosphate-buffered saline containing collagenase (1 mg/ml) (Sigma-Aldrich) at 37 °C for 45 minutes. Digestion was stopped by ethylenediaminetetraacetic acid and the tumor was disrupted over a cell strainer. Tumor cells were frozen down and stored for analyses of A2-YLEP transgene DNA and mRNA. Part of the freshly isolated tumor cells were cultured for 4–7 days (using B16 culture medium, see Supplementary Text) and tested for A2-YLEP surface expression and ability to induce T cell responses. DNA and mRNA isolation, cDNA synthesis and quantitative PCRs were performed as described previously. Sequences of primers and probes used to quantify levels of antigen and control DNAs are provided in Supplementary Text and Supplementary Figure S9. Retrovirally introduced antigens were amplified from tumor DNA using pLXSN-specific primers, and analyzed by gel electrophoresis and sequencing. This allowed design of peptide-specific primers and probes and performance of selective quantitative PCRs (see legend to Supplementary Figure S9 for details). Cultures of isolated tumor cells were analyzed for antigen expression using either a FITC-labeled anti-HLA-A2 mAb (clone BB7.2, Abcam, Cambridge, UK) (in case of gp100/A2 tumors), an anti-mouse TRP2 mAb (Novus Biologicals, Cambridge, UK) followed by second step PE-labeled anti-rabbit IgG (eBioscience, San Diego, CA), or a PE-labeled anti-H2-Kb (clone AF6-88.5, eBioscience) (in case of TRP2/H2-Kb tumors) by flow cytometry. Induction of T cell IFN-γ production was measured in supernatants following coincubation of tumor cells with TCR T cells for 20 hours, and analyzed by enzyme-linked immunosorbent assay (U-CyTech Biosciences, Utrecht, The Netherlands). In some studies, tumor cell cultures were exposed to AZA, IFN-γ, or extended periods of culture times (see Supplementary Text for details). Measurements of target cell lysis were performed with similar cultures of tumor cells and using WST-1 or chromium release assays (Supplementary Text).
Detection of presence and activity of TILs. TILs were obtained from single tumor cell suspensions and cultured for 4 days using complete mouse T cell medium (see Supplementary Text). The presence of TILs was assessed as the total number of CD8 T cells, the number of TCR-positive CD8 T cells (both determined with Flow-Count Fluorospheres), or the fraction of pMHC-binding CD8 T cells by flow cytometry. The functional ability of TILs to eliminate B16 tumor cells was evaluated by microscopy (Leica Microsystems, Rijswijk, The Netherlands: DM-IL, digital camera 300).
mRNA expression of markers for immune cells, chemoattractants, and adhesion molecules. RNA was extracted from tumors and subjected to genome-wide expression analysis using Illumina expression bead chips (ServiceXS, Leiden, The Netherlands). RNA processing, hybridization onto beads and gene expression analyses are described in Supplementary Text.
Statistical analyses. Statistical significances were calculated with the Student's t-test, the Sign test for microarray data, and the Mantel-Cox test for survival data using GraphPad Prism5 (GraphPad Software, La Jolla, CA) software. P values < 0.05 were considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Validation of TCR and antigen gene constructs. Figure S2. Conditioning with Busulphan and Cyclophosphamide results in effective lymphodepletion in vivo. Figure S3. Treatment with gp100/A2 TCR T cells prevents or delays tumor growth and results in the generation of antigen-specific memory T cells. Figure S4. TCR T cells provide a survival advantage in preventive but not curative model. Figure S5. Cells isolated from relapsed tumors are killed by TCR T cells ex vivo. Figure S6. Relapsed tumors that progress following a second T cell treatment have not lost expression of antigen. Figure S7. T cell responses in cured mice are not directed against tumor antigens other than the target antigen. Figure S8. Frequency and kinetics of tumor relapse are neither affected by presence of antigen-negative tumor cells nor cross-presentation of gp100 antigen. Figure S9. TCR T cells select for tumors that lack antigen DNA. Figure S10. Antigen-negative cells in relapsed tumors originate from antigen-negative cells already present in pre-treatment tumors. Figure S11. Ranking of genes that are most differentially expressed in relapsed tumors. Figure S12. Validation of TRP2 TCR in vitro and in vivo.
Acknowledgments
Zsolt Sebestyen is acknowledged for help and expertise in setting up flow cytometric stainings and analyses of mouse T cells. Marcel Smid is acknowledged for expertise and help in analyzing micro-array data. The work was financed in part by the Erasmus MC Cancer Institute Translational Research grant (TR-2004) and the European Union Framework Program 6 grant “Adoptive engineered T cell Targeting to Activate Cancer Killing” (EUFP6 ATTACK, number FP6-018914). The authors declare that they have no conflict of interest.
Supplementary Material
References
- Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L.et al. (2010Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients Clin Cancer Res 162646–2655. [DOI] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP.et al. (2005Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma J Clin Oncol 232346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S.et al. (2013TCR-Engineered T Cells Meet New Challenges to Treat Solid Tumors: Choice of Antigen, T Cell Fitness, and Sensitization of Tumor Milieu Front Immunol 4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govers C, Sebestyén Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med. 2010;16:77–87. doi: 10.1016/j.molmed.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E.et al. (2002Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells Proc Natl Acad Sci USA 9916168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol. 2006;24:5060–5069. doi: 10.1200/JCO.2006.07.1100. [DOI] [PubMed] [Google Scholar]
- Algarra I, Cabrera T, Garrido F. The HLA crossroad in tumor immunology. Hum Immunol. 2000;61:65–73. doi: 10.1016/s0198-8859(99)00156-1. [DOI] [PubMed] [Google Scholar]
- Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21:455–464. doi: 10.1016/s0167-5699(00)01692-3. [DOI] [PubMed] [Google Scholar]
- Maeurer MJ, Gollin SM, Martin D, Swaney W, Bryant J, Castelli C.et al. (1996Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen J Clin Invest 981633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004;27:184–190. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM.et al. (2011Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression Cancer Cell 1972–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PL, Koeppen HK, Hurteau T, Rowley DA, Schreiber H. Major histocompatibility complex class I and unique antigen expression by murine tumors that escaped from CD8+ T-cell-dependent surveillance. Cancer Res. 1990;50:3851–3858. [PubMed] [Google Scholar]
- Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J.et al. (2012Ovarian cancer progression is controlled by phenotypic changes in dendritic cells J Exp Med 209495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Cooper D, Iqbal AJ, Gittens BR, Cervone C, Perretti M. The effect of galectins on leukocyte trafficking in inflammation: sweet or sour. Ann N Y Acad Sci. 2012;1253:181–192. doi: 10.1111/j.1749-6632.2011.06291.x. [DOI] [PubMed] [Google Scholar]
- Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- Yáñez-Mó M, Barreiro O, Gordon-Alonso M, Sala-Valdés M, Sánchez-Madrid F. Tetraspanin-enriched microdomains: a functional unit in cell plasma membranes. Trends Cell Biol. 2009;19:434–446. doi: 10.1016/j.tcb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Goldberger O, Volovitz I, Machlenkin A, Vadai E, Tzehoval E, Eisenbach L. Exuberated numbers of tumor-specific T cells result in tumor escape. Cancer Res. 2008;68:3450–3457. doi: 10.1158/0008-5472.CAN-07-5006. [DOI] [PubMed] [Google Scholar]
- Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482:405–409. doi: 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ.et al. (2012Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting Nature 482400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanski JL, Marchand M, Uyttenhove C, Boon T. Immunogenic variants obtained by mutagenesis of mouse mastocytoma P815. VI. Occasional escape from host rejection due to antigen-loss secondary variants. Int J Cancer. 1983;31:119–123. doi: 10.1002/ijc.2910310119. [DOI] [PubMed] [Google Scholar]
- Corbière V, Chapiro J, Stroobant V, Ma W, Lurquin C, Lethé B.et al. (2011Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases Cancer Res 711253–1262. [DOI] [PubMed] [Google Scholar]
- Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R.et al. (2008Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1 N Engl J Med 3582698–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10:294–298. doi: 10.1038/nm999. [DOI] [PubMed] [Google Scholar]
- Thomas DL, Kim M, Bowerman NA, Narayanan S, Kranz DM, Schreiber H.et al. (2009Recurrence of intracranial tumors following adoptive T cell therapy can be prevented by direct and indirect killing aided by high levels of tumor antigen cross-presented on stromal cells J Immunol 1831828–1837. [DOI] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT.et al. (2013Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells Sci Transl Med 5200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M.et al. (2012Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation Nature 490412–416. [DOI] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C.et al. (2006Type, density, and location of immune cells within human colorectal tumors predict clinical outcome Science 3131960–1964. [DOI] [PubMed] [Google Scholar]
- Kalialis LV, Drzewiecki KT, Klyver H. Spontaneous regression of metastases from melanoma: review of the literature. Melanoma Res. 2009;19:275–282. doi: 10.1097/CMR.0b013e32832eabd5. [DOI] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G.et al. (2003Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer N Engl J Med 348203–213. [DOI] [PubMed] [Google Scholar]
- Lee HW, Choi HJ, Ha SJ, Lee KT, Kwon YG. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim Biophys Acta. 2013;1835:170–179. doi: 10.1016/j.bbcan.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol Immunother. 2012;61:751–759. doi: 10.1007/s00262-012-1253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C.et al. (2011Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control Cancer Res 716997–7009. [DOI] [PubMed] [Google Scholar]
- Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunol Rev. 2009;230:97–113. doi: 10.1111/j.1600-065X.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara S, Raz A. Biological modulation by lectins and their ligands in tumor progression and metastasis. Anticancer Agents Med Chem. 2008;8:22–36. doi: 10.2174/187152008783330833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanska HM, Berditchevski F. Tetraspanins in human epithelial malignancies. J Pathol. 2011;223:4–14. doi: 10.1002/path.2779. [DOI] [PubMed] [Google Scholar]
- Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM.et al. (2012Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma Nat Biotechnol 30337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza KM, Kottke T, Diaz RM, Rommelfanger D, Thompson J, Vile R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum Gene Ther. 2012;23:1054–1064. doi: 10.1089/hum.2012.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccoris M, Straetemans T, Govers C, Lamers C, Sleijfer S, Debets R. T cell receptor (TCR) gene therapy to treat melanoma: lessons from clinical and preclinical studies. Expert Opin Biol Ther. 2010;10:547–562. doi: 10.1517/14712591003614756. [DOI] [PubMed] [Google Scholar]
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B.et al. (2013Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma J Clin Oncol 31e439–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Kishore M, Gittens B, Wang G, Coe D, Komarowska I.et al. (2014Self-recognition of the endothelium enables regulatory T-cell trafficking and defines the kinetics of immune regulation Nat Commun 53436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS.et al. (2014Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors Nat Med 20607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A.et al. (2012Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors J Clin Invest 122899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB.et al. (2010Improved survival with ipilimumab in patients with metastatic melanoma N Engl J Med 363711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R.et al. (2013Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma N Engl J Med 369134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MO, Friedlander P, Milstein MI, Mooney MM, Metzler G, Murray AP.et al. (2011Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells Sci Transl Med 380ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M.et al. (2014Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells Immunity 41116–126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.