

Abstract
Notch receptors guide mammalian cell fate decisions by engaging the proteins Jagged and Delta-like (DLL). The 2.3 angstrom resolution crystal structure of the interacting regions of the Notch1-DLL4 complex reveals a two-site, antiparallel binding orientation assisted by Notch1 O-linked glycosylation. Notch1 epidermal growth factor–like repeats 11 and 12 interact with the DLL4 Delta/Serrate/Lag-2 (DSL) domain and module at the N-terminus of Notch ligands (MNNL) domains, respectively. Threonine and serine residues on Notch1 are functionalized with O-fucose and O-glucose, which act as surrogate amino acids by making specific, and essential, contacts to residues on DLL4. The elucidation of a direct chemical role for O-glycans in Notch1 ligand engagement demonstrates how, by relying on posttranslational modifications of their ligand binding sites, Notch proteins have linked their functional capacity to developmentally regulated biosynthetic pathways.
The Notch signaling pathway is essential for cell-fate determination in all metazoan species (1). In adult mammals, Notch signaling directs neural and hematopoietic stem cell differentiation and development of many immune cell subsets (2–4). Mammalian Notch receptor homologs (Notch1 to 4) encode a Notch extracellular domain (NECD) that engages ligands, a transmembrane domain, and a Notch intracellular domain (NICD) that translocates to the nucleus to serve as a transcriptional cofactor (5). Signaling is initiated when the NECD binds Jagged1, Jagged2, Delta-like 1 (DLL1), or Deltalike 4 (DLL4) ligands on the surface of an apposing cell, triggering proteolysis of the Notch receptor in a process that is dependent on ligand endocytosis (6–9). Mutations in Notch and its ligands are associated with a number of inherited and acquired diseases, including Alagille syndrome (AGS) and T cell acute lymphoblastic leukemia (T-ALL) (10).
Notch-ligand interactions are apparently dependent on posttranslational modification of Notch receptors with O-glucose (O-Glc) and O-fucose (O-Fuc). The epidermal growth factor (EGF) modules of the NECD are specifically targeted by protein O-glucosyltransferase 1 (Poglut) and protein O-fucosyltransferase 1 (Pofut1) enzymes (11, 12). O-glucosylation and O-fucosylation are both required for Notch activation yet play distinct roles in the signaling pathway. O-glucose modifications indirectly affect signaling by increasing susceptibility to protease processing after ligand engagement, whereas O-fucose modifications directly enhance Notch affinity for Jagged/ DLL (11–13). Elongation of O-fucose to a disaccharide by Fringe N-acetyl-glucosaminyltransferases further influences specificity by facilitating preferential interactions between certain receptor-ligand pairs (14–16). A long-standing question has been whether O-glycans directly contact ligands or contribute through allosteric effects. There are few examples of posttranslational modifications engaging in specific interactions between extracellular signaling proteins, although this has been observed in other ancient developmental pathways, such as the lipid-mediated interaction critical to Wnt-Frizzled recognition (17).
There are presently no high-resolution structures describing Notch-ligand complexes, although there are structures of unliganded Notch1 EGF-like repeats 11 to 13 (Notch1(11–13), amino acids 411 to 530) and an unliganded Jagged1 fragment spanning from the N terminus to EGF3 (amino acids 32 to 337) (18, 19). Mammalian NECDs consist of 29 to 36 EGF repeats followed by 3 cysteine-rich LIN repeats. It has been established from many studies that EGF11 and 12 alone are sufficient for binding to Jagged/DLL, and these domains are assumed to represent the core recognition element (18, 20). Additional regions have been implicated in Notch ligand recognition, including EGF domains 6 to 15 (21, 22). Jagged/ DLL ECDs, like those of Notch, have a modular extracellular domain architecture consisting of a region termed module at the N-terminus of Notch ligands (MNNL) domain followed by a Delta/Serrate/Lag-2 (DSL) domain, and either 6 (DLL3), 8 (DLL1 and DLL4), or 16 (Jagged1 and Jagged2) EGF repeats (19, 23). Alanine scanning mutagenesis studies have identified conserved residue Phe207 of the Jagged1 DSL domain and residues Leu468, Asp469, and Ile477 of Notch1 EGF12 as critical to receptor-ligand interactions (18, 24).
Structural information about Notch-Delta/Jagged interactions is absent largely due to several complicating factors that have precluded the reconstitution of Notch receptor-ligand complexes for structural studies. Notch possesses an intrinsically low affinity for its ligands (25, 26). Furthermore, optimal ligand binding requires proper modification by O-glycosyltransferases in the recombinant Notch fragments used for structural analysis (13). Finally, the calcium binding by several Notch EGFs is important for folding and ligand engagement (5, 18, 27). To overcome these obstacles and capture a stable complex for structure determination, we used in vitro evolution to engineer mutations in DLL4 stability and enhance affinity for Notch1.
Affinity maturation of DLL4
We targeted the interaction between Notch1 and DLL4 for affinity maturation because they represent the mammalian receptor-ligand pair with the highest intrinsic affinity (21). As a platform for in vitro evolution, we expressed DLL4 comprising the predicted receptor-binding region spanning from the N terminus to EGF2, plus EGFs 3 to 5, on the surface of yeast as a fusion to cell wall protein Aga2 (Fig. 1A). DLL4 was displayed, rather than Notch1, because yeast do not encode for the O-glycosylation enzymes required for modification of Notch EGFs. To isolate higher-affinity DLL4 variants, we generated a mutant library of DLL4(N-EGF5) using unbiased error-prone polymerase chain reaction and performed several rounds of selection on magnetic beads with decreasing concentrations of Notch1(1–14) tetramers or monomers. Wild-type DLL4 did not bind Notch1(1–14) (Fig. 1A) but gradually increased in affinity after each round of selection (Fig. 1B). To further enhance DLL4-Notch1 affinity, the selectants were pooled and subjected to a second cycle of mutagenesis. Clones isolated from the first- and second-generation libraries were sequenced and revealed a series of consensus mutations in only the MNNL and DSL domains (Fig. 1C and fig. S1).
Fig. 1. In vitro evolution of Notch1-DLL4 interactions with yeast surface display.
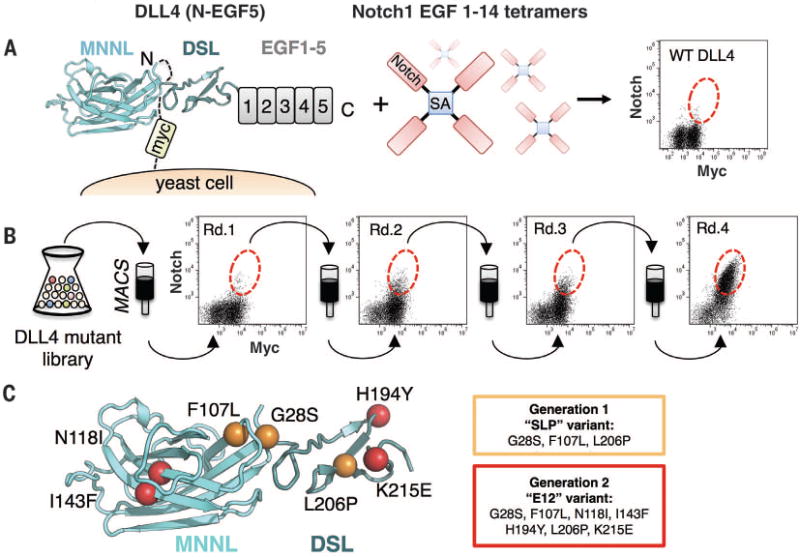
(A) Schematic representation of the yeast-displayed DLL4(N-EGF5)construct used for mutant library generation, stained with Notch1(1–14) tetramers. The flow cytometry dot plot shows double staining of DLL4 (N-EGF5) with 50 nM Notch1 tetramer and antibody to c-Myc. (B) Four rounds of selection were performed on magnetic-activated cell sorting (MACS) columns using a mutant library of DLL4(N-EGF5). Yeast isolated from selection rounds 1 to 4 (Rd.1 to Rd.4) were each double stained with 50 nM Notch1(1-14) tetramer and antibody to c-Myc. Dot plots for each round are displayed, with enriched populations circled with a red dotted line. (C) Affinity-enhancing mutants isolated from generation 1 and generation 2 libraries are mapped onto the surface of MNNL/DSL domains from the DLL4 crystal structure. The first generation SLP mutations are depicted as orange spheres. The second generation E12 variant includes the SLP mutations, plus four additional mutations (colored red).
We expressed recombinant forms of wild-type DLL4 and engineered DLL4 variants in insect cells and measured their binding affinities for Notch1 by surface plasmon resonance. Wild-type DLL4(N-EGF2) bound Notch1(1–14), Notch1 (10–14), and Notch1(11–13) with dissociation constant (Kd) values of 12.7 μM, 8.6 μM, and 7.5 μM, respectively (Fig. 2A). The similar DLL4 affinity for Notch1(11–13) and Notch1(1–14) is consistent with previous studies suggesting that EGFs 11 and 12 constitute the major binding determinants (18, 20). We next determined the affinity of a variant containing SLP consensus mutations (G28S, F107L, and L206P) named DLL4SLP, and the highest-affinity variant from the second-generation library, named DLL4E12, containing the three SLP mutations plus four additional substitutions (N118I, I143F, H194Y, and K215E) (Fig. 1C). Minimal binding regions of DLL4SLP(N-EGF1) or DLL4SLP(N-EGF2) bound our Notch1 constructs with a Kd of ∼260 to 540 nM. DLL4E12(N-EGF2) interacted with an even lower Kd range of ∼56 to 68 nM (Fig. 2A), a 125- to 225-fold increase in affinity relative to wild-type DLL4(N-EGF2) (Fig. 2A). The kinetics of the Notch1-DLL4 interactions suggests that affinity-enhancing mutations have slowed the dissociation rate of the complex (fig. S2).
Fig. 2. Notch1 binding and activation by high-affinity DLL4 variants.
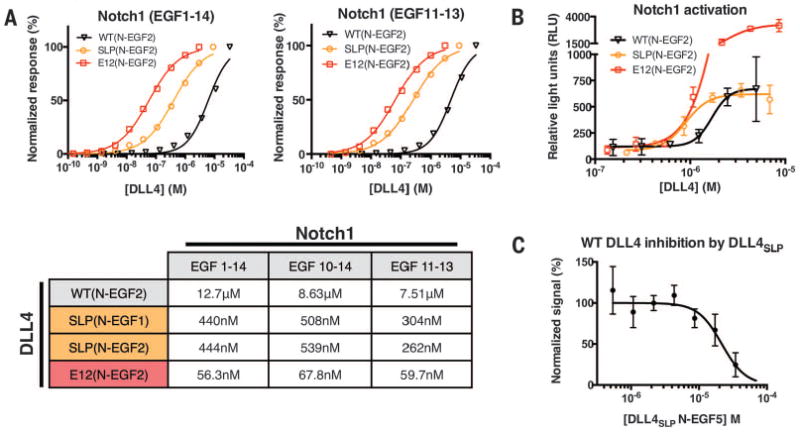
(A) Binding isotherms were obtained by surface plasmon resonance. Wild-type DLL4(N-EGF2), generation 1 DLL4SLP(N-EGF1 and N-EGF2) and generation 2 DLL4E12(N-EGF2) constructs were flowed over immobilized Notch1 EGF1–14, 10–14, or 11–13. Curves were fit to a 1:1 binding model to obtain the Kd values indicated in the table. (B) Notch1 activation was measured in a luciferase assay using cell lines expressing Notch1 and stably integrated with a luciferase gene under control of a CSL-driven promotor. Plates (96-well) were coated with serial dilutions of wild-type and high-affinity DLL4 variants. High-affinity mutants have increased potency relative to wild type. (C) Soluble DLL4SLP (N-EGF5) inhibited Notch1 activation by immobilized, wild-type DLL4(N-EGF5) in the luciferase assay.
Notch1 activation by DLL4 variants
To ensure that the affinity-enhancing mutations in DLL4 did not diminish function, we assayed the signaling activity of the high-affinity DLL4 variants. We performed a luciferase assay using a stable cell-line expressing Notch1 and a luciferase reporter gene under control of the Notch-driven CBF1, Suppressor of Hairless, Lag-1 (CSL) promoter. Cells incubated with DLL4SLP(N-EGF2) gave a similar maximal response (Emax) as wild-type DLL4(N-EGF2) (Fig. 2B), but there was a marked left-shift in the dose-response curve for DLL4SLP(N-EGF2) as a result of its higher affinity. The highest-affinity variant, DLL4E12(N-EGF2), yielded a five-fold increase in Emax relative to wild-type DLL4 and DLL4SLP proteins (Fig. 2B). Performing activation assays with longer DLL4(N-EGF5) constructs increased the Emax of wild-type DLL4 and the SLP variant, suggesting that EGF3-5 may provide the low and intermediate affinity ligands with sufficient length to access the cellular Notch1 receptors in the plate-based format (fig. S3) (21). We also assessed the ability of the first generation SLP variant to compete with immobilized wild-type DLL4 for Notch1 binding. The soluble DLL4SLP(N-EGF5) effectively inhibited Notch1 activation (Fig. 2C). Thus, the affinity-enhanced DLL4 variants remain functional in activating Notch signaling.
Crystallization of the Notch1-DLL4 complex
We attempted to make Notch1-DLL4 complexes with a variety of different combinations of DLL4 affinity-matured constructs and Notch1. DLL4SLP(N-EGF2) formed biochemically stable complexes with Notch1(11-13) (fig. S4). Heterogeneous N-linked glycans were enzymatically trimmed in DLL4SLP(N-EGF2) by sensitizing the insect cells with a mannosidase inhibitor, kifunensine, followed by treatment with endoglycosidase F1 (Endo F1). This strategy digests all but the proximal N-acetyl glucosamine (Nag) from asparagine residues without affecting O-linked sugars. Crystals of Notch1(11-13) bound to the glycan-shaved DLL4SLP(N-EGF2) diffracted to a resolution of 3.4 Å (table S1). Crystals of a complex containing a shorter DLL4SLP(N-EGF1) construct were obtained by the same strategy and diffracted to a resolution of 2.3 Å (table S1). We determined both structures by molecular replacement using models of unliganded Notch1(11–13) and Jagged1 (18,19). The electron density maps obtained from the high-resolution Notch1(11–13)–DLL4SLP(N-EGF1) structure were used for analysis of atomic contacts, whereas the lower-resolution Notch1(11–13)–DLL4SLP(N-EGF2) structure allowed us to model the additional EGF2 domain.
Architecture of the Notch1-DLL4 complex
In the complex of Notch1(11–13) and DLL4SLP(N-EGF2), the two proteins form a colinear, antiparallel interaction across a narrow interface that buries ∼1100 Å2 of protein and glycan surface area (Fig. 3A). This antiparallel binding orientation is compatible with either a trans cell-cell contact to initiate signaling or a self-inhibited cis interaction, as has also been proposed (Fig. 3B) (28,29). Notch1 EGFs 11 to 13 assemble into a ∼90 Å rod stabilized by three disulfide bonds per EGF and by Ca2+ ions coordinated at the inter-EGF junctions. DLL4SLP(N-EGF2) MNNL (C2-like), DSL, EGF1, and EGF2 domains stretch 120 Å lengthwise. Notch1 ligand-binding residues localize to a continuous surface of EGF 11 and 12, providing structural validation for long-standing cellular, genetic, and biochemical studies implicating this region in Notch activation (18, 20, 24, 30). Although Notch EGF12 and the Delta/Jagged DSL domain are widely regarded as the central interacting nodes, the complex structure indicates that these regions are offset such that EGF11 lies opposite the DSL and EGF12 engages a disulfide-tethered loop at the base of the MNNL (Fig. 3A). Calcium ions did not contribute directly to the interface in the Notch1-DLL4 structure but are coordinated by residues at the N terminus of EGF11 and at the EGF11-12/EGF12-13 junctions and potentiate the interaction by rigidifying the DLL4-binding platform (Fig. 3A) (31). Although it has been proposed that Notch activation requires calcium-dependent phospholipid binding by ligand MNNL domains, DLL4 was not bound to any calcium ions, which would suggest that this feature is not essential for signal transduction by all ligands (19).
Fig. 3. Structure of the Notch1(11–13)– DLL4SLP(N-EGF2) complex.
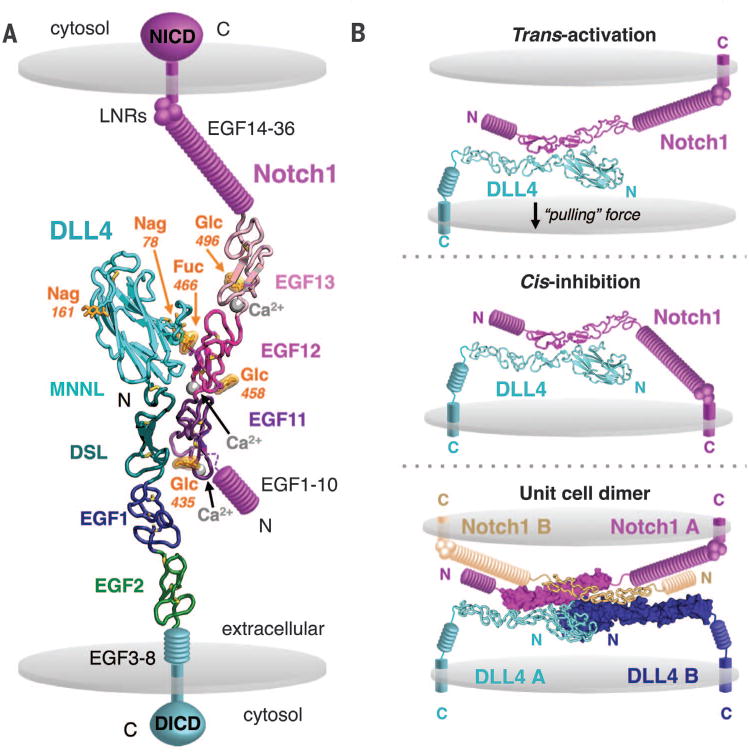
(A) The structure of Notch1(11–13) bound to DLL4SLP(N-EGF2) is shown in ribbon representation. Calcium ions are represented as gray spheres, Notch1 O-glycans are highlighted in orange, and cell membranes are depicted as gray discs. The remaining domains of the Notch1 or DLL4 receptors are displayed schematically in magenta or teal, respectively. DICD, DLL4 intracellular domain. (B) The Notch1-DLL4 crystal structure is modeled as the interaction is predicted to occur in cellular contexts. Notch is trans-activated upon binding to ligands on adjacent cells or cis-inhibited upon binding to ligands on the same cell. In one model for trans-activation, mechanical pulling force resulting from endocytosis of ligand by adjacent cells activates Notch. The 2:2 dimer observed in the crystal asymmetric unit is also displayed as it may occur between two cells. The same dimer organization was observed in crystals of Notch1(11–13)–DLL4SLP(N-EGF1) and is further described in fig. S5.
The Notch1-DLL4 complex forms an anti-parallel 2:2 dimer in the asymmetric unit of the crystal (fig. S5A). The same dimer organization was present in crystals of both different Notch1-DLL4 complexes (fig. S5B). Although such a dimer would be feasible to form between two cells, it is unclear whether the dimer observed in the crystal lattices represents a physiologically relevant assembly (Fig. 3B). Multi-angle light-scattering analyses of Notch1(11–13) and DLL4SLP(N-2EGF) indicated that the proteins formed a 1:1 complex in solution (fig. S6). It is possible that the high local concentration of proteins in the two-dimensional environment of cell membranes may drive oligomerization in vivo, but this remains to be shown experimentally.
The Notch1-DLL4 interaction is partitioned across two interfaces: a “site 1” between the DLL4 MNNL domain and Notch1 EGF12, and a “site 2” between the DLL4 DSL domain and Notch1 EGF11 (Fig. 4A and table S2). With respect to site 1, mutagenesis studies of Notch1-EGF12 defined a patch of residues—Leu468, Asp469, and Ile477—localized on a single face of EGF12 as critical for Jagged1 recognition, and O-glycosylation of residue Thr466 on this same face enhances binding to DLL1, DLL4, and Jagged1 (13, 24). Remarkably, the Notch1(11–13)–DLL4SLP(N-EGF1) structure indicates that the Notch1 Thr466 O-fucose modification centrally anchors the DLL4-binding interface, essentially acting as a surrogate amino acid (Fig. 4A). O-Fuc466 buries more than 80% of its total surface area between Notch1 EGF12 and the surface of the MNNL, where it participates in a network of glycan-protein contacts (Fig. 4, A and B). The MNNL domain presents a fucose-binding platform formed by interstrand loops stabilized by the Cys61-Cys74 disulfide bond. The platform accommodates the O-Fuc466 ring atop the planar side chains of DLL4 His64 and Tyr65 (Fig. 4A). A hydroxyl group of O-Fuc466 points downward toward the MNNL platform and hydrogen bonds (H-bonds) to the main-chain carbonyl of Tyr65 (Fig. 4A). In addition to the Thr466 fucose, several Notch1 residues that were previously implicated in Jagged1-binding (Leu468, Asp469, and Ile477) are buried at the MNNL interface (24). Leu468 and Ile477 occupy a hydrophobic cleft of DLL4 composed of His64, Phe76, and Phe109, whereas Asp469 accepts H-bonds from the side-chain hydroxyl and backbone amine of Thr110 (Fig. 4, A and B).
Fig. 4. Molecular determinants and conservation of Notch1-DLL4 interactions.
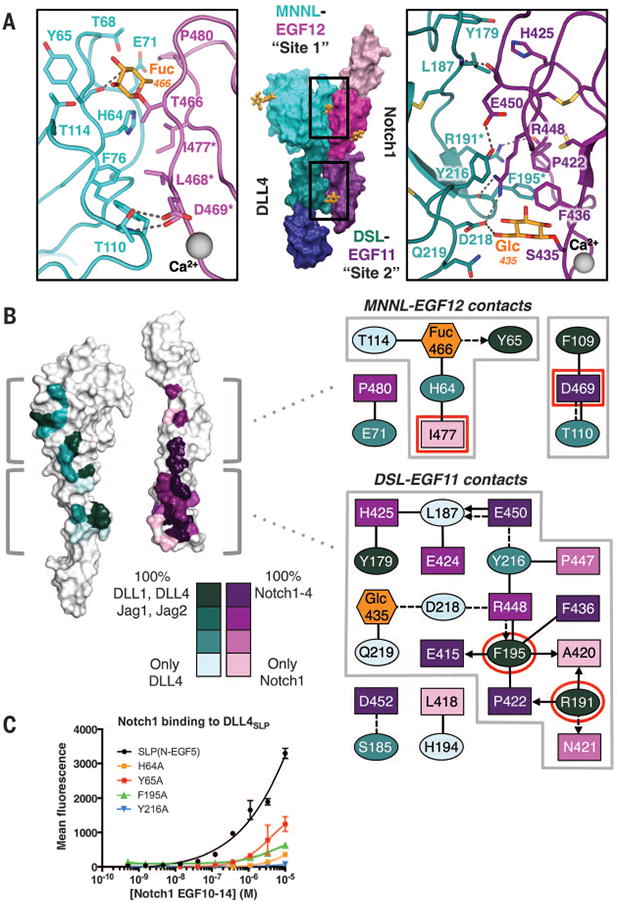
(A) The Notch1(11–13)–DLL4SLP(N-EGF1) structure is displayed in surface representation with individual domains colored as in Fig. 3A. Site 1 and site 2 interacting regions of Notch1 and DLL4 are outlined in black boxes. In the zoomed panels, dashed gray lines represent H-bonds or salt bridges. (B) White surface representations of Notch1 and DLL4 represent an “open-book” view of the contact residues. Conservation of contact residues among mammalian paralogs is indicated by a color gradient painted onto the structures. Jag1, Jagged1. Jag2, Jagged2. The panels to the right serve as an interface contact map. Ovals represent DLL4 residues, rectangles represent Notch1 residues, and hexagons represent O-glycans. Each residue is colored according to conservation, and glycans are colored orange. Dashed lines connecting residues are H-bonds or salt bridges, and solid lines are VDW contacts. Interactions between side chains and backbones are represented as arrows pointing toward the backbone. Red outlines indicate residues that, when mutated, result in null binding or dysfunction in developmental assays (18, 24). (C) DLL4SLP(N-EGF5)and DLL4SLP (N-EGF5) containing H64A, Y65A, F195A, or Y216A mutations were expressed on yeast and evaluated for binding to biotinylated Notch1(10–14). Binding is indicated as mean fluorescence intensity from SA-647 secondary staining.
Although site 2 is dominated by protein-protein contacts, there is an unexpected interaction between an O-glycan attached to Ser435 of Notch1 and DLL4 (21). DLL4 DSL engages Notch EGF11 with several residues at the apices of disulfide-knotted loops linking Cys188-Cys200 and Cys208-Cys217 (Fig. 4A). Phe195 and Tyr216 protrude from these loops to form a clamp sandwiching the side chain of Notch1 Arg448, which serves as the major linchpin for DLL4 engagement. The Notch1 Arg448 side chain extends deeply into a DSL groove, burying ∼100 Å2 of surface area and forming a salt bridge with the carboxylate head group of DLL4 Asp218, an H-bond with the backbone carbonyl of Phe195, and engaging in aliphatic interactions with Tyr216 (Fig. 4, A and B). The O-Glc modification Ser435 of Notch1 interacts with DLL4 by donating an H-bond to the carboxylate head group of DLL4 Asp218 and participating in a van der Waals (VDW) interaction with the side chain of Gln219 (Fig. 4, A and B). We modeled a β1-glucose at this position because recent mass spectroscopy data classified the sugar as a hexose, although we cannot rule out the possibility of other O-linked hexoses such as man-nose or galactose (21). The role of the Ser435 modification is much less clear than that of the well-characterized O-Fuc modification of Thr466. Ser435 is only present in Notch1 and Notch4, is part of neither a Poglut nor a Pofut1 consensus motif, and there is no strong evidence for another functionally important O-glycosyltransferase that directly modifies Notch receptors. Additional contacts that stabilize site 2 include the benzene ring of DLL4 Phe195 filling a hydrophobic pocket of EGF11, where it participates in a triad of VDW interactions with Pro422 and Phe436 of Notch1 (Fig. 4, A and B).
To assess the relative importance of key interface residues to binding, we mutated MNNL fucose-binding platform residues His64 and Tyr65 and DSL residues Phe195 and Tyr216 to alanine and analyzed cell-surface capture of Notch1(10–14) using a fluorescence-based assay. Mutation of Y216A completely abolished binding to Notch1, whereas H64A, Y65A, and F195A substantially diminished Notch1(10–14) interactions relative to DLL4SLP(N-EGF5) (Fig. 4C).
Location of affinity-enhancing mutations
We examined the structural context of our engineered mutations to better understand the molecular basis for affinity enhancement. Surprisingly, none of the three DLL4 SLP substitutions directly contacted Notch1 (fig. S7A). G28S and F107L mutations are in close spatial proximity within the MNNL and probably strengthen the interaction by stabilizing the MNNL-DSL hinge or the adjacent EGF12-contacting residues Phe109 and Thr110 (fig. S7A). The L206P mutation lies at the center of a DSL beta strand, where it may indirectly enhance binding (fig. S7A). Modeling of the additional mutations in our second-generation E12 construct revealed that only the H194Y and K215E substitutions occur at DLL4 contact positions. Thus, it is unlikely that the evolved mutations have introduced differences in the manner that Notch1 binds to wild-type DLL4.
Conservation of contact residues
The four mammalian Notch receptor homologs (Notch1 to 4) signal in response to engagement by four different ligands (Jagged1, Jagged2, DLL1, and DLL4). We performed a conservation analysis to assess whether the Notch1(11–13)–DLL4SLP(N-EGF1) complex structure is broadly representative of a conserved Notch receptor-ligand interaction mode (Fig. 4B and fig. S8). Along the EGF11 interface, discontinuous Notch1 residues Glu415, Pro422, Phe436, Glu450, and Asp452 collectively form a DSL-binding surface that is conserved in Notch1 to 4 (Fig. 4B). The complementary DLL4 DSL surface includes three conserved DLL4 residues: Tyr179, Arg191, and Phe195. The importance of Arg191 and Phe195 to the interaction is supported by the deleterious functional effects of mutating the equivalent positions in Jagged1 in the Drosophila melanogaster ligand Serrate (Fig. 4B) (18). Aligning the sequence of Notch ligands to divergent family member DLL3 revealed that only 6 out of 15 Notch1-binding residues are conserved in DLL3 (fig. S8). Because DLL3 has been reported not to bind or activate Notch, it was omitted from our structural analysis of interface conservation (32). EGF12 interface contacts are more divergent than those of EGF11: Only the calcium-coordinating Asp469 is identical in all four Notch receptors (Fig. 4B and fig. S8). The functionally relevant, O-fucose–bearing Thr466 is encoded by Notch1, Notch2, and Notch4 (fig. S8). On the MNNL side, the interacting Phe109 and a fucose-binding Tyr65 residue are conserved (Fig. 4B). Collectively, we propose that the EGF11-DSL interface is the conserved focal point for ligand binding and the more variable EGF12-MNNL interface facilitates ligand pleiotropy. The limited sequence identity at the MNNL interface may then contribute to diverse surface chemistry important for Fringe-mediated tuning of ligand specificity (discussed below). Taken together, our analyses suggest that the general binding mode observed in the Notch1(11–13)–DLL4SLP(N-EGF1) structure is consistent among Notch receptor and ligand paralogs.
Notch1 O-linked glycans
O-glucosylation and O-fucosylation of Notch1 are strictly required for Notch signaling (11,12). Notch1 EGF11 to 13 contains two Poglut glucosylation consensus sequences at Ser458 and Ser496, one Pofut1 fucosylation sequence Thr466, and a Ser435 hexose of unknown origin (Fig. 5A) (21). We observed clear electron density for the proximal O-glycan at all four predicted positions (Fig. 5B). Neither O-Glc458 nor O-Glc496 contacted DLL4 in the complex structure; thus, their effect on signaling activity would appear to be indirect (Fig. 3A). O-Glc458 lies flat against the surface of EGF12, opposite the DLL4-binding face, where it shields hydrophobic Pro460 and Phe474 side chains from solvent exposure. Glc496 similarly covers the Pro494 and Phe512 residues at equivalent positions in EGF13. As opposed to O-fucose, which directly influences ligand binding, Poglut-mediated Notch O-glucose modifications enhance signaling by increasing Notch receptor susceptibility to proteolytic processing (11,33). From a structural perspective, O-glucose may then facilitate efficient proteolysis by protecting hydrophobic surfaces from aggregation as Notch clusters on the cell surface. The newly identified O-glycan modification of S435 does not occur at a known O-glycosyltransferase consensus motif but is a salient feature of the structure, given its position at the DLL4 interface (Figs. 3A and 4A). Elongation of O-Glc435 has the potential to regulate Notch by enhancing or inhibiting interaction with the opposing DSL domain.
Fig. 5. Basis of affinity tuning by O-fucose modification.
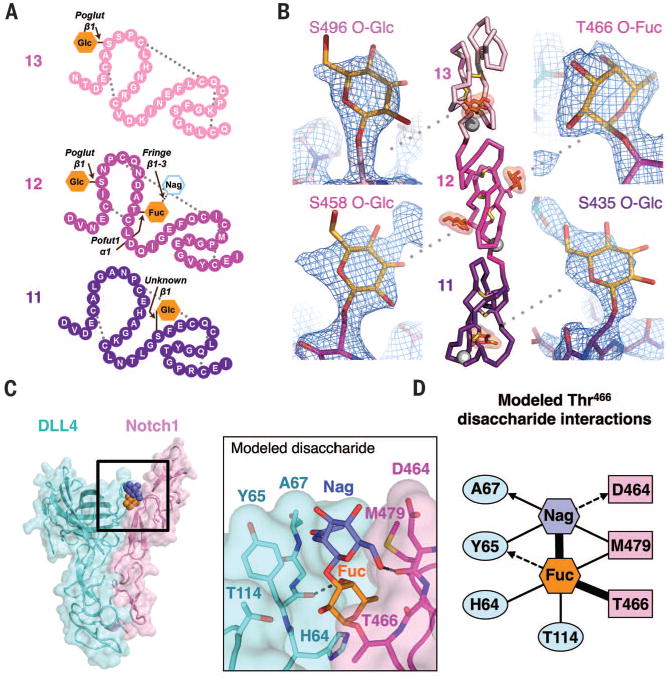
(A) Schematic diagram of Notch1(11–13) with amino acids depicted as colored spheres and disulfide connectivity as dotted gray lines. The O-linked glycans observed in the structure are shown as filled orange hexagons. The Thr466 O-Fuc is elongated by Fringe enzymes to form a disaccharide, represented as an outlined hexagon. Enzymes responsible for each modification, along with the glycosidic linkages, are shown above each O-glycan. (B) Ribbon diagram of Notch1(11–13) indicating the position of disulfide bonds (yellow sticks), Ca2+ ions (gray spheres), and O-linked glycans (orange highlighted sticks). Simulated annealing composite omit map electron density surrounding each of the four O-glycans is shown in zoomed panels as blue mesh and contoured at 1.5 σ. (C) The Notch1 Thr466 disaccharide is modeled as spheres in the context of a Notch1-DLL4 complex and are colored as in (A). The zoomed panel depicts Thr466 disaccharide interactions with protein side chains, with H-bonds represented as dashed gray lines. (D) Contact map describing interactions between the modeled disaccharide and Notch1-DLL4. Sugars, Notch1, and DLL4 residues are colored corresponding to the panels in (C). Thick black lines represent covalent bonds, thin black lines are VDW interactions, and dashed black lines are H-bonds.
Modeling Thr466 fucose elongation
Regulation of ligand specificity by Fringe enzymes is a complex process that involves the coordinated elongation of several Notch1 O-fucose moieties (34). Recent biophysical studies have begun to deconvolve this process by determining that O-fucosylation of Thr466 of Notch1 substantially enhances binding to Jagged1/DLL1 and moderately enhances binding to DLL4 (13). Affinity was further increased in all cases when a β1-3 Nag was added to O-Fuc466 by Fringe enzymes. To probe the molecular basis for Fringe-mediated affinity enhancement, we generated a model of the naturally occurring Thr466 O-linked disaccharide based on the single O-fucosylation we see in the structure (Fig. 5C). The modeled disaccharide tunnels outward through a crevice at the Notch1-DLL4 interface and adopts a similar conformation to that observed in an unliganded, disaccharide-modified Notch1(11–13) structure (13). The elongated Nag glycan appears to facilitate interactions by generating a network of H-bonds and VDW contacts with both Notch1 and DLL4 and acting as a molecular bridge (Fig. 5D). The modeled Nag is sandwiched between hydrophobic side chains presented by Tyr65 of DLL4 and Met479 of Notch1 and donates a H-bond to the backbone carbonyl of Asp464 of Notch1 (Fig. 5C). It is clear from our model that elongated O-glycans are capable of providing substantial additional energetic contributions to Notch-ligand interfaces.
The central chemical and structural role of an O-linked glycan in mediating a receptor-ligand interaction, essentially functionalizing the Notch protein with an expanded capacity for molecular recognition beyond the 20 naturally occurring amino acids, is unusual. It is intriguing that two other key receptor-ligand systems that act as developmental checkpoints in mammals, Wnt/Frizzled and sonic hedgehog (Shh)/patched, also rely on posttranslational modifications for function. Wnt is lipidated, and Shh is cholesterol modified (17, 35). It is possible that these primordial systems took advantage of the biosynthetic modification pathways (e.g., Pofut and fringe) to achieve fine regulation of signaling in a way that would be difficult to achieve through nonpolymorphic amino acid contacts alone. By linking molecular recognition to the O-glycan heterogeneity, the Notch-ligand interaction can be tuned to reflect the developmental and metabolic state of the cell.
Trans-versus cis-interaction
Canonical Notch signaling involves trans-activation by ligands expressed on adjacent cells, though cis-inhibition of Notch by ligands expressed on the same cell is a potential regulatory mechanism (28, 29). The antiparallel orientation observed in our structure could mediate both trans- and cis-interactions because of the interdomain flexibility of regions of Notch receptors and ligands (Fig. 3B) (36, 37). That the same binding site could mediate trans-interactions between opposing cells and cis-interactions on the same cell has been observed in several immune receptors (38).
Comparison of Notch1-DLL4 complex with unliganded structures
Notch activation involves ligand-induced conformational changes that render Notch susceptible to proteolytic cleavage. To assess whether conformational changes occur in the interacting regions upon binding, we compared the individual Notch and DLL4 subunits of the complex structure with unliganded structures of Notch1(11–13) and Jagged1. Unliganded Notch1(11–13) superimposed onto the backbone and side chains of complexed Notch1(11–13) with a root mean square deviation of 1.44 Å (fig. S9A), indicating that the protein does not undergo major conformational changes upon binding to DLL4 (18). Because there is no available structure of DLL4 alone, we compared Notch1-complexed DLL4SLP to the unliganded structure of homolog Jagged1. The DSL, EGF1, and EGF2 of DLL4 are rotated ∼25° about the MNNL domain relative to their position in Jagged1 (fig. S9B), suggesting that ligands may flex at an MNNL-DSL “hinge” to engage Notch. Given that the Notch-interacting residues of DLL4 are well conserved (Fig. 4B and fig. S8) and that many of our affinity-enhancing mutations were at noncontact positions, it is possible that the lower affinity of ligands Jagged1 and DLL1 are due to differences in domain orientations about the MNNL-DSL linker.
AGS disease mutations
AGS is an inherited developmental disease that is frequently caused by mutations in Jagged1 (10). We mapped AGS-associated Jagged1 missense mutations onto their equivalent DLL4 positions in the Notch1(11–13)–DLL4SLP(N-EGF2) complex to clarify their effect on receptor binding (fig. S7B) (39). The majority of AGS mutations are predicted to disrupt disulfide bond formation, perturb the hydrophobic core of the MNNL domain, or interfere with inter-EGF packing. However, mutations at conserved Thr110 and Tyr179 residues would directly affect binding, whereas other mutations at Gly28, Pro112, and Val151 positions could indirectly influence interactions by distorting the MNNL-DSL linker (fig. S7B). Notably, one AGS patient had a Jagged1 mutation analogous to the DLL4 G28S mutation isolated from our affinity maturation experiments, raising the possibility that the mutation disrupts Notch binding in the context of Jagged1 (fig. S7B) (40).
Implications for therapeutic targeting of the Notch pathway
The distinct pathologies linked to Notch receptors and ligands necessitates the development of more precisely targeted therapies. For example, Notch1 mutations are associated with leukemia and aortic valve disease, Notch3 mutations with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and Jagged1/Notch2 mutations with AGS (39,41–44). Physiological processes can also be differentially regulated by ligands, evidenced by the opposing roles of Jagged1 and DLL4 in angiogenesis and the role of the Notch1-DLL4 axis in lymphatic development and the lymphatic system (45, 46). Certain advantages of receptor-specific therapeutics have begun to emerge in studies describing the inhibition of tumor growth by Notch1-antagonist antibodies and the control of graft-versus-host disease by DLL1/4-antagonist antibodies (47,48). The Notch1(11–13)–DLL4SLP(N-EGF1) structure provides new insights toward the development of receptor- and ligand-specific drugs. Antibodies against Notch EGF11 and 12 reportedly have limited effectiveness in cellular assays; based on the structure, this is likely due to occlusion of relevant protein epitopes by the Ser435 O-glucose and Thr466 O-fucose moieties (49). More effective therapeutic targeting of EGF11 and 12 may be achieved with engineered high-affinity ligands such as those presented here, which have co-evolved with Notch receptors to accommodate O-glycan binding.
Acknowledgments
We thank A. Velasco, D. Waghray, and J. Spangler for assistance. We also thank the staff of the Advanced Photon Source for support and access to the 23-ID-D beamline; the staff of the Advanced Light Source for support and access to the 8.2.2 Howard Hughes Medical Institute (HHMI) beamline; and the staff of the Stanford Synchrotron Radiation Lightsource, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract DE-AC02-76SF00515. This work was supported by NIH-1RO1-GM097015, Ludwig Cancer Foundation, and the HHMI (K.C.G.). V.C.L. was supported by a Cancer Research Institute postdoctoral fellowship and an NIH-Immunology postdoctoral training grant. M.V.N. was supported by the Stanford Discovery Innovation fund, and N.W.P. was supported by an HHMI fellowship of the Helen Hey Whitney Foundation. K.C.G. and V.L. are inventors on a patent applied for by Stanford University describing high-affinity Notch ligands. Coordinates and structure factors of 1egf DLL4 and 2egf DLL4 have been deposited in the Protein Data Bank with accession codes 4XL1 and 4XLW.
References
- 1.Artavanis-Tsakonas S, Rand MD, Lake RJ. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 2.Milner LA, Bigas A. Blood. 1999;93:2431–2448. [PubMed] [Google Scholar]
- 3.Nyfeler Y, et al. EMBO J. 2005;24:3504–3515. doi: 10.1038/sj.emboj.7600816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radtke F, MacDonald HR, Tacchini-Cottier F. Nat Rev Immunol. 2013;13:427–437. doi: 10.1038/nri3445. [DOI] [PubMed] [Google Scholar]
- 5.Fleming RJ. Cell Dev Biol. 1998;9:599–607. doi: 10.1006/scdb.1998.0260. [DOI] [PubMed] [Google Scholar]
- 6.Struhl G, Greenwald I. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 7.De Strooper B, et al. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 8.Brou C, et al. Mol Cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 9.Gordon WR, et al. Nat Struct Mol Biol. 2007;14:295–300. doi: 10.1038/nsmb1227. [DOI] [PubMed] [Google Scholar]
- 10.Hansson EM, Lendahl U, Chapman G. Semin Cancer Biol. 2004;14:320–328. doi: 10.1016/j.semcancer.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 11.Acar M, et al. Cell. 2008;132:247–258. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okajima T, Irvine KD. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- 13.Taylor P, et al. Proc Natl Acad Sci USA. 2014;111:7290–7295. doi: 10.1073/pnas.1319683111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hicks C, et al. Nat Cell Biol. 2000;2:515–520. doi: 10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- 15.Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Moloney DJ, Stanley P. Proc Natl Acad Sci USA. 2001;98:13716–13721. doi: 10.1073/pnas.241398098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Science. 2012;337:59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cordle J, et al. Nat Struct Mol Biol. 2008;15:849–857. doi: 10.1038/nsmb.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chillakuri CR, et al. Cell Reports. 2013;5:861–867. doi: 10.1016/j.celrep.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rebay I, et al. Cell. 1991;67:687–699. doi: 10.1016/0092-8674(91)90064-6. [DOI] [PubMed] [Google Scholar]
- 21.Andrawes MB, et al. J Biol Chem. 2013;288:25477–25489. doi: 10.1074/jbc.M113.454850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto S, et al. Science. 2012;338:1229–1232. doi: 10.1126/science.1228745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Souza B, Miyamoto A, Weinmaster G. Oncogene. 2008;27:5148–5167. doi: 10.1038/onc.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whiteman P, et al. J Biol Chem. 2013;288:7305–7312. doi: 10.1074/jbc.M112.428854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hicks C, et al. J Neurosci Res. 2002;68:655–667. doi: 10.1002/jnr.10263. [DOI] [PubMed] [Google Scholar]
- 26.Nichols JT, et al. J Cell Biol. 2007;176:445–458. doi: 10.1083/jcb.200609014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fehon RG, et al. Cell. 1990;61:523–534. doi: 10.1016/0092-8674(90)90534-l. [DOI] [PubMed] [Google Scholar]
- 28.de Celis JF, Bray S. Development. 1997;124:3241–3251. doi: 10.1242/dev.124.17.3241. [DOI] [PubMed] [Google Scholar]
- 29.Sprinzak D, et al. Nature. 2010;465:86–90. doi: 10.1038/nature08959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordle J, et al. J Biol Chem. 2008;283:11785–11793. doi: 10.1074/jbc.M708424200. [DOI] [PubMed] [Google Scholar]
- 31.Hambleton S, et al. Structure. 2004;12:2173–2183. doi: 10.1016/j.str.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Ladi E, et al. J Cell Biol. 2005;170:983–992. doi: 10.1083/jcb.200503113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rana NA, Haltiwanger RS. Curr Opin Struct Biol. 2011;21:583–589. doi: 10.1016/j.sbi.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rampal R, Arboleda-Velasquez JF, Nita-Lazar A, Kosik KS, Haltiwanger RS. J Biol Chem. 2005;280:32133–32140. doi: 10.1074/jbc.M506104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter JA, Young KE, Beachy PA. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 36.Xu A, Lei L, Irvine KD. J Biol Chem. 2005;280:30158–30165. doi: 10.1074/jbc.M505569200. [DOI] [PubMed] [Google Scholar]
- 37.Morgan WD, et al. J Mol Biol. 1999;289:113–122. doi: 10.1006/jmbi.1999.2753. [DOI] [PubMed] [Google Scholar]
- 38.Held W, Mariuzza RA. Nat Rev Immunol. 2008;8:269–278. doi: 10.1038/nri2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spinner NB, et al. Hum Mutat. 2001;17:18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 40.Warthen DM, et al. Hum Mutat. 2006;27:436–443. doi: 10.1002/humu.20310. [DOI] [PubMed] [Google Scholar]
- 41.Weng AP, et al. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 42.Garg V, et al. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 43.Joutel A, et al. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 44.McDaniell R, et al. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benedito R, et al. Cell. 2009;137:1124–1135. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 46.Niessen K, et al. Blood. 2011;118:1989–1997. doi: 10.1182/blood-2010-11-319129. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y, et al. Nature. 2010;464:1052–1057. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 48.Tran IT, et al. J Clin Invest. 2013;123:1590–1604. doi: 10.1172/JCI65477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falk R, et al. Methods. 2012;58:69–78. doi: 10.1016/j.ymeth.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]