

Abstract
The brain endocannabinoid system is a potential target for the treatment of psychiatric and metabolic conditions. Here, a novel CB1 receptor antagonist (ABD459) was synthesized and assayed for pharmacological efficacy in vitro and for modulation of food consumption, vigilance staging and cortical electroencephalography in the mouse. ABD459 completely displaced the CB1 agonist CP99540 at a Ki of 8.6 nmol/l, and did not affect basal, but antagonized CP55940-induced GTPγS binding with a KB of 7.7 nmol/l. Acute ABD459 (3–20 mg/kg) reliably inhibited food consumption in nonfasted mice, without affecting motor activity. Active food seeking was reduced for 5–6 h postdrug, with no rebound after washout. Epidural recording of electroencephalogram confirmed that ABD459 (3 mg/kg) robustly reduced rapid eye movement (REM) sleep, with no alterations of wakefulness or non-REM sleep. Effects were strongest during 3 h postdrug, followed by a progressive washout period. The CB1 antagonist AM251 (3mg/kg) and agonist WIN-55,212-2 (WIN-2: 3 mg/kg) also reduced REM, but variously affected other vigilance stages. WIN-2 caused a global suppression of normalized spectral power. AM251 and ABD459 lowered delta power and increased power in the theta band in the hippocampus, but not the prefrontal cortex. The neutral antagonist ABD459 thus showed a specific role of endocannabinoid release in attention and arousal, possibly through modulation of cholinergic activity.
Keywords: agonist WIN-2, antagonist AM251, cannabinoids, cannabinoid receptor 1 receptors, electroencephalogram, endocannabinoid, mouse, neutral antagonist ABD459, nonrapid eye movement sleep, rapid eye movement sleep
Introduction
It is widely known that Δ9-tetrahydrocannabinol (Δ9-THC), the active ingredient of marijuana, affects a multitude of neuronal elements, with consequences on the regulation of food intake and circadian rhythms (Pivik et al., 1972; Riedel et al., 2009), and endocannabinoids are powerful modulators of these behaviours. Consequently, regulation of endocannabinoid targets may produce beneficial outcomes and be of therapeutic relevance in eating/sleeping disorders. Here, we investigated a novel antagonist, which is devoid of inverse agonism on cannabinoid receptor 1 (CB1), and explored its hypophagic and sleep-reducing properties.
The most common mechanistic description of inverse agonism is based on the premise that G-protein coupled receptors such as CB1 receptors exist in at least two conformational states: an inactive R and an active R* state (Pertwee, 2005). In this model, agonists have higher affinity for the R* state and shift the equilibrium towards R*, resulting in G-protein activation and an increase in GDP/GTP exchange. In contrast, inverse agonists bind preferentially to the R state, resulting in a decrease in constitutive activity. CB1 antagonists such as SR141716A (rimonabant) and AM251 are also known to be CB1 receptor inverse agonists. It has been shown that a hydrogen bond formed between a lysine residue (Lys192) and the oxygen of the amide in compounds such as rimonabant is pivotal for inverse agonism to occur (Lange et al., 2005; Hurst et al., 2006). The hydrogen bond formed stabilizes a salt bridge between the lysine and an adjacent aspartate residue. This salt bridge is formed because of the presence of a pronounced kink in the receptor helix found only in the inactive state of the receptor, thereby stabilizing this inactive state and increasing its proportion relative to its active state (Hurst et al., 2002; Lange et al., 2005). Mutation at this site removes the inverse agonist properties of rimonabant, but allows it to continue to behave as an antagonist (Pan et al., 1998). On this basis, we synthesized a ketone derivative of rimonabant, which we hypothesized would behave as a CB1 receptor neutral antagonist, ABD459 (Fig. 1a).
Fig 1.

(a) Structure of ABD459 and (b) binding affinity in an [3H]CP55940 displacement essay in mouse brain membranes. (c) We found no inhibition of [35S]GTPγS binding as a measure of inverse efficacy to mouse brain membranes relative to SR141716A (SR141). (d) There was a clear antagonism for CB1 measured in a [35S]GTPγS binding assay in mouse brain membranes for the agonist, CP55940. **P<0.01; ***P <0.001.
The relevance of the endocannabinoid system for nutrition and energy balance has been confirmed over the last decade [for a recent review, see: André and Gonthier (2010)], and there is a strong contribution of central CB1 receptors towards these effects. Overall, endocannabinoid levels increase during periods of fasting and are reduced during satiety. Consequently, CB1 agonists exert hyperphagic effects, whereas antagonists are known to reduce food intake in fasted and nonfasted subjects (Cota et al., 2006; Riedel et al. 2009). Although rimonabant progressed clinically because of its anorexic properties, it was eventually withdrawn owing to unacceptable side-effects that precluded its use (Engeli, 2012). CB1 antagonists devoid of inverse agonism appear to show a much more acceptable pharmacological profile and yet exert hypophagic properties (Hodge et al., 2008; Cluny et al., 2011). We thus first explored the anorexigenicity of ABD459 in mice fed a normal diet.
There is now accumulating evidence that cerebral blood flow differs not only during stages of hunger and satiety but also between normal weight and eating disorders (Gautier et al., 2000; Del Parigi et al., 2002). Particularly striking are abnormal reductions in resting state activity in prefrontal, paralimbic and temporal brain regions in underweight and obese subjects (Babiloni et al., 2011). Typically, satiety coincided with decreased delta band (1–5 Hz) spectral power, whereas theta (5–9 Hz) and early alpha (9–10 Hz) power increased (Hoffman and Polich, 1998). In addition, diurnal vigilance patterns are modulated by food availability, such that starvation coincides with heightened wakefulness and overall sleep reduction, increasing energy expenditure (Yamanaka et al., 2003; Koban et al., 2008), and obesity increases sleep (Laposky et al., 2006). However, this relationship is controversial and it remains unclear whether the nutritional stage determines global brain activity and sleep abnormalities, or vice versa (Jauregui-Lobera, 2012). Ideally, treatment to normalize (increase/reduce) food intake should mimic mental states of hunger/satiety, but not otherwise interfere with vigilance.
Related to this issue is the long-standing notion that sleep and brain activity in the lower frequency bands are important for memory formation (Platt and Riedel, 2011), and that central CB1 receptors have a role to play in cognitive processing (Riedel and Davies, 2005; Rubino and Parolaro, 2011). This pertains not only to short-term memory (Goonawardena et al., 2010a, 2010b, 2011a, 2011b) but also to the consolidation process underlying long-term memory formation (Clarke et al., 2008; Robinson et al., 2008, 2010). Memory consolidation, however, is critically dependent on the occurrence of regular sleep patterns (Brankack et al., 2009; Platt and Riedel, 2011). Indeed, Δ9-THC increases sleep in both humans and animals (Pivik et al., 1972; Freemon et al., 1974; Feinberg et al., 1975, 1976; Buonamici et al., 1982; Freemon, 1982), and these effects are mimicked by activation of the endogenous cannabinoid, arachidonoyl ethanolamide (anandamide) (Murillo-Rodriguez et al., 1998), and prevented by the CB1 receptor antagonist/inverse agonist rimonabant (Santucci et al., 1996). Sleep regulation is therefore likely mediated by the activation of central CB1 receptors (Devane et al., 1992; Howlett, 1995), but studies utilizing rimonabant have limited power because of its CB1 antagonism on the one hand (McMahon and Koek, 2007; Sokal et al., 2008) and its inverse agonism on the other (Landsman et al., 1997; Sim-Selley et al., 2001). Hence, it is difficult to attribute the wake-promoting properties of rimonabant to the blockade of endocannabinoid activity. To this end, the evaluation of a neutral antagonist, such as ABD459 described here, would be highly instructive.
Methods
Chemistry of ABD459
ABD459 was synthesized from the previously reported 5-(4-bromophenyl)-1-(4-chlorophenyl)-4-methyl-1H-pyrazole-3-carbonyl chloride (Lan et al., 1999). The acid chloride was coupled with N, O-dimethyl hydroxylamine in dichloromethane/pyridine to yield 5-(4-bromophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxylic acid methoxy-methyl-amide. This Weinreb amide was dissolved in THF and reacted at 0°C with a Grignard prepared from 4-bromoanisole and magnesium in dry THF, to yield 5-(4-bromophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazol-3-yl]-4-methoxyphenyl-methanone (ABD459) as a white solid following purification by column chromatography.
Radioligand binding assays
Mouse brain membrane preparation
Whole brains from adult male MF1 mice were suspended in centrifugation buffer (320 mmol/l sucrose, 2 mmol/l EDTA, 5 mmol/l MgCl2) and the tissues were homogenized using an Ultra-Turrex homogenizer (Fisher Scientific, Loughborough, UK). Tissue homogenates were centrifuged at 1600g for 10 min and the resulting supernatant was collected. This pellet was resuspended in centrifugation buffer, centrifuged as before and the supernatant was collected. Supernatants were combined before being subjected to further centrifugation at 28 000g for 20 min. The supernatant was discarded and the pellet was resuspended in buffer A (50 mmol/l Tris, 2 mmol/l EDTA, 5 mmol/l MgCl2 at pH 7.0) and incubated at 37°C for 10 min. Following incubation, the suspension was centrifuged for 20 min at 23 000g. After resuspending the pellet in buffer A, the suspension was incubated for 40 min at room temperature before a final centrifugation for 15 min at 11 000g. This pellet was resuspended in buffer B (50 mmol/l Tris, 1 mmol/l EDTA, 3 mmol/l MgCl2) and the final protein concentration determined using the Bio-Rad Dc kit (Bio-Rad, Hemel Hemstead, UK) was 1 mg/ml. All centrifugation procedures were carried out at 4°C. Prepared brain membranes were stored at −80°C and defrosted on the day of the experiment.
Equilibrium binding assays
Binding assays were performed with the CB1 receptor agonist, [3H]CP55940 (0.7 nmol/l), and the CB1 receptor antagonist, [3H]SR141716A (1.2 nmol/l), 1 mg/ml BSA and 50 mmol/l Tris buffer containing 0.1 mmol/l EDTA and 0.5 mmol/l MgCl2 (pH 7.4), total assay volume 500 μl. Binding was initiated by the addition of mouse brain membranes (30 μg). Assays were carried out at 37°C for 60 min before termination by the addition of ice-cold wash buffer (50 mmol/l Tris buffer, 1 mg/ml BSA) and vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester, Gaithersburg, Maryland, USA) and Whatman GF/B glass-fibre filters that had been soaked in wash buffer at 4°C for 24 h. Each reaction tube was washed five times with a 4 ml aliquot of buffer. The filters were oven-dried for 60 min and then placed in 5 ml of scintillation fluid (Ultima Gold XR; PerkinElmer, Buckinghamshire, UK), and radioactivity was quantified by liquid scintillation spectrometry. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 μmol/l of the corresponding unlabelled ligand and was 70–80% of the total binding.
Values have been expressed as means and variability as SEM or as 95% confidence limits (95% CI). The concentration of CP55940 that produced a 50% displacement of radioligand from specific binding sites (IC50 value) was calculated using GraphPad Prism 4 (Graphpad Software Inc., San Diego, California, USA). Its dissociation constant (Ki value) was calculated using the equation of Cheng and Prusoff (1973).
[35S]GTPγS binding assay
Mouse brain membranes (5 μg protein) were pre-incubated for 30 min at 30°C with adenosine deaminase (0.5 U/ml). The membranes were then incubated with the agonist with vehicle or modulator for 60 min at 30°C in assay buffer (50 mmol/l Tris-HCl; 50 mmol/l Tris-Base; 5 mmol/l MgCl2; 1 mmol/l EDTA; 100 mmol/l NaCl; 1 mmol/l DTT; 0.1% BSA) in the presence of 0.1 nmol/l [35S]GTPγS and 30 μmol/l GDP, in a final volume of 500 μl. Binding was initiated by the addition of [35S]GTPγS. Nonspecific binding was measured in the presence of 30 μmol/l GTPγS. The reaction was terminated by rapid vacuum filtration (50 mmol/l Tris-HCl; 50 mmol/l Tris-Base; 0.1% BSA) using a 24-well sampling manifold (Cell harvester; Brandel, Gaithersburg, Maryland, USA) and GF/B filters (Whatman, Maidstone, UK) that had been soaked in buffer (50 mmol/l Tris-HCl; 50 mmol/l Tris-Base; 0.1% BSA) for at least 24 h. Each reaction tube was washed six times with a 1.2 ml aliquot of ice-cold wash buffer. The filters were oven-dried for at least 60 min and then placed in 5 ml of scintillation fluid (Ultima Gold XR; Packard). Radioactivity was quantified by liquid scintillation spectrometry.
EC50 and maximal effects (Emax) and the SEM or 95% CI of these values were calculated by nonlinear regression analysis using the equation for a sigmoidal concentration–response curve (GraphPad Prism). KB values for antagonism of LTB4 were calculated by substituting a single concentration ratio value into the equation (x−1) = B/KB, where x (the ‘concentration ratio’) is the concentration of agonist that produced a particular size of effect in the presence of antagonist at a concentration, B, divided by the concentration of agonist that produced an identical effect in the absence of antagonist (Tallarida et al., 1979).
Food intake and feeding orientated behaviour
Subjects
Thirty-two C57Bl/6 mice (Harlan, Derby, UK) were used to determine the effects of ABD459 on activity, food intake and feeding-orientated behaviour. Before the start of testing, mice were group housed (10 animals per cage) and subjected to a 12 h light/dark cycle (lights off at 19:00 h) with temperature maintained at 23 ± 2°C and relative humidity of 40–60%. All experiments followed the guidelines on the ethical use of animals from the European Communities Council Directive of 24 November 1986 (86/609/EEC) and UK Home Office regulations (Scientific Procedures Act 1986).
Apparatus
Home cage activity and feeding-orientated behaviour were measured using PhenoTyper (Noldus, Wageningen, the Netherlands) cages containing video-based observation software (Ethovision 3.1; Noldus IT) for long-term continuous monitoring of activity in mice, following the protocol detailed in the study by Riedel et al. (2009). Briefly, testing was conducted with mice singly housed and constant lighting, temperature and feeding regimes as detailed above. From the video tracks, the following dependent variables were recorded and processed: (i) total ambulatory activity in the arena pooled into hourly bins during experimental days; (ii) time spent in the food zone (area proximal to the feeder) and (iii) time spent in the water zone (area proximal to the water bottle) as indices for eating and drinking activities. These parameters were also monitored continuously and analysed in hourly bins as well as averaged for light and dark periods. They were complemented by daily determination of body weights, and overall 24 h food consumption and water intake (conducted between 10.00 and 11.00 h).
Behavioural testing
Following habituation for 3–4 days in the PhenoTyper, animals were matched for body weight and assigned to treatment groups of ABD459 (3, 10 and 20 mg/kg) or vehicle (triethylene glycol and PBS; 50 : 50 vol/vol) (n = 8 per group). Compounds were injected intraperitoneally at a volume of 0.1 ml/10 g body weight at 17.00 h during the light phase of the circadian cycle. Animals were returned into PhenoTypers and locomotor activity was recorded for another 48 h.
Data analysis
All data are presented as group mean ± SEs and reliability tested using the PC-based statistics package Prism 4.01 (Graphpad Software Inc.). Two-way repeated-measures analyses of variance (ANOVAs) were carried out using drug-treatment as a between-subjects factor and time as a within-subjects factor; one-way ANOVAs and t-tests were performed to contrast specific groups, with α set to a P value less than 0.05.
Multichannel electroencephalogram
Subjects
Twenty-four C57Bl/6 wild-type mice (Harlan) weighing 30–40 g were used. All housing and behavioural procedures were identical to the ones described above.
Surgery
Implantation of surface electrodes was performed as described previously (Jyoti et al., 2010). In brief, anaesthesia was induced with 3% isoflurane in medical-grade oxygen and, after head shaving and stereotaxic fixation (Stoelting, Kiel, Wisconsin, USA), maintained on 1–1.5% isoflurane. The skull was exposed and co-ordinates were determined from the Bregma. Burr holes were drilled for insertion of gold-plated surface screw electrodes at the following co-ordinates: medial prefrontal cortex (2 mm AP, 0.1 mm L), parietal cortex overlaying both left and right hippocampi (2 mm AP, ±1.5 mm L). Surface recordings at this position are dominated by coherent hippocampal discharges (Megevand et al., 2008; Jyoti et al., 2010) and will thus be referred to as the hippocampal recording site. Additional reference/ground electrodes were placed over parietal and occipital regions; all were assembled into a seven-pin head stage and anchored to the skull by Durelon dental cement and tissue glue. Postsurgical care included saline replacement (0.5 ml intraperitoneally) and 0.01 ml analgesic (Temgesic, subcutaneously). All animals were allowed at least 7–10 days of recovery before experiments commenced, during which time every effort was made to minimize stress and potential suffering.
Drug groups
ABD459, the full CB1 receptor agonist WIN-55,212-2 (WIN-2), and the antagonist/inverse agonist AM251 (both from Tocris, Bristol, UK) were dissolved in vehicle (triethylene glycol and PBS; 50 : 50 vol/vol). All animals were divided into four treatment groups (n=6 per group) and were administered either vehicle, ABD459 (3.0 mg/kg), AM251 (3.0 mg/kg) or WIN-2 (3.0 mg/kg) intra-peritoneally at midday (12.00 h) during the sleep phase of the cycle and electroencephalographic (EEG) recordings commenced for a total duration of 6 h (until 18:30 h).
Apparatus and analyses of sleep recordings
Wireless Neurologgers (New Behavior, Zurich, Switzerland) were attached to the head stage to register the EEG activity from freely behaving mice at three channels, with a sampling rate of 200 Hz. Neural activity was band pass filtered (0.1 Hz high-pass–70 Hz low-pass) at an expected input range of ±500 μV. A built-in accelerometer recorded all movement activity. Data were downloaded offline to a PC, using USB plug-in docking stations, transformed with Matlab 7 (The MathWorks Inc., Natick, Massachusetts, USA) and imported into SleepSign (Kissei Comtec Co. Ltd, Nagano, Japan) for vigilance staging and extrapolation of spectral power (for details, see Jyoti et al., 2010; Goonawardena et al., 2011c). Vigilance stages [wakefulness, nonrapid eye movement (NREM) and rapid eye movement (REM) sleep] of 4-s epochs/bins were identified on the basis of combined fast Fourier transform (FFT; delta/theta ratio from hippocampal EEGs) and accelerometer activity (body movement). Automated staging was followed by visual inspection and corrections excluding any movement-related artefacts from spectral analyses. FFTs were finally calculated for each epoch with a resolution of 0.77 Hz, Hamming window smoothed and averaged. EEG power spectra (1–20 Hz) for each vigilance state were normalized relative to the absolute peak power and averaged for each drug group for the hippocampus and the prefrontal cortex (spectral bands: delta: 0.5–5 Hz, theta: 5–9 Hz, alpha: 9–14 Hz and beta 14–20 Hz). Sleep scoring and all power spectral analyses were carried out by a single examiner unaware of the treatment group.
Statistical analysis
Statistical significance of all vigilance stage parameters (i.e. total time; average length of wakefulness, REM and NREM events; latencies to first NREM and REM episodes) was assessed using one-way ANOVA, followed by post-hoc analysis (unpaired t-tests, two tailed) for comparisons between different treatment groups using Prism, version 5.0 (GraphPad Software Inc.). In addition, two-way ANOVAs, with treatment and time as factors, assessed the effects of each treatment on time spent in each vigilance stage across the 6 h recording period, followed by individual comparisons between treatments of interest. For EEG spectral power, a two-way factorial ANOVA was carried out using group (drug treatment) and frequencies as discriminators. Post-hoc planned unpaired comparisons and frequency specific analyses were carried out on preselected frequency bands to determine effects between drug groups. All data are expressed as group mean ± SE and α was set to a P value less than 0.05. Only significant results are presented.
Results
In-vitro pharmacology of ABD459
In equilibrium binding assays, ABD459 completely displaced [3H]CP55940 with a Ki value of 8.61 nmol/l (95% CI: 4.23–17.5 nmol/l; n=4; Fig. 1b). In [35S]GTPγS binding assays, ABD459 had no effect on basal binding, in contrast to rimonabant, which significantly reduced basal [35S]GTPγS binding at concentrations of 100 nmol/l, 1 μmol/l and 10 μmol/l (n=6; Fig. 1c). Furthermore, ABD459 produced a significant antagonism of CP55940 stimulated [35S]GTPγS binding with a KB value of 7.7 nmol/l (n = 4; Fig. 1d).
ABD459 has hypophagic properties
Administration of ABD459 dose-dependently reduced body weight during the night cycle following treatment (Fig. 2a). The overall ANOVA confirmed a significant difference between drug doses [F(3,30) = 3.23; P < 0.05] and both 10 and 20 mg/kg, but not the 3 mg/kg group, showed significant effects in post-hoc t-tests (P's ≤ 0.05). This overall weight loss corresponded with reduced food intake [F(3,30) = 6.55; P < 0.002] (Fig. 2b) in both 3 and 10 mg groups (P's< 0.01). At the same time, ABD 459 did not affect water intake at any dose (F< 1) (Fig. 2c).
Fig 2.

Hypophagic effects of acute administration of ABD459. (a) Reduced body weight was evident 12 h following ABD459 at doses of 10 and 20 mg/kg, but not at 3 mg/kg. (b) At the same time, ABD459-treated mice showed a decrease in food intake (3 and 10 mg/kg) compared with the vehicle group, while not showing any change in the overall consumption of water (c). Means± SEM; *, **, ***P < 0.05. Ctrl, control; NS, nonsignificant.
ABD459 effects on feeding-orientated behaviour and activity
Independent of dose, ABD459 effects on feeding-orientated behaviour in a home cage system confirmed decreased time spent in the food zone in the hours following drug treatment (Fig. 3a) and thus led to a significant treatment-by-time interaction term [F(192,1728) =1.32; P <0.005]. This recovered in the latter part of the night and animals returned to normal visit regimes. The overall drug action was clearly visible during the initial 5 h after injection [Fig. 3b: main effect of treatment; F(3,108) =6.72; P< 0.005] with all ABD459 doses significantly different from vehicle (F's>8.5; P's < 0.01). As there was no difference in food zone visits in the following 7 h (Fig. 3c; F<1), this suggests a washout period of about 6 h. At the same time, a failure to observe a rebound in food zone visits seems to indicate that the overall weight loss and lowering of food intake detailed in Fig. 2 arose from the first 5–6 h after treatment.
Fig 3.

Feeding-related behavioural changes following single dose ABD459 in a home-cage-like environment. (a) Circadian activity profile of visits to the food hopper during 3 days. There was a typical enhancement in feeding activity during night hours (horizontal bars). Drug injection took place 1–2 h before night onset on day 2. Although there was no difference in activity during pretreatment and post-treatment nights, fewer entries into the food zone were observed in the hours postdrug. (b) Activity during the initial 5 h after treatment clearly indicates a reduction in the time spent in the food zone following ABD459 (3 mg/kg) exposure in all drug groups. By contrast, there was no activity difference during the rest of the night once the drug had been washed out (c). (d) All groups spent similar amounts of time eating in the first nocturnal period of observation (pre). Compared with this baseline value, only controls remained stably active in eating, but activity was significantly reduced in the dark period when animals were under ABD459 treatment. Note that the effects were similar for all doses. (e) Total activity in PhenoTyper home cages, monitored as the distance moved, was not different between drug groups at any stage of testing. Heightened activity was observed during nocturnal periods of wakefulness under all conditions, but drug injection did not affect global activity. (f) Correlational analysis of food intake and time spent in food zone in initial 5 h following treatment. Increased time in the food zone corresponded with a higher food intake, validating time in the food zone as a proxy for hunger and food intake in the absence of automatic recording of food intake. Means± SEM. *P < 0.05. Ctrl, control.
As a follow-up, we next pooled the overall time spent in the food zone and compared the group performance during matching hours on the nights before and during drug treatment (Fig. 3d). Apart from main effects of treatment and time (F's > 4, P's < 0.005), the drug effect was reflected in the significant interaction term [F(3,56) = 2.89; P<0.05], which provides compelling evidence that vehicle controls did not alter performance between days (P>0.2; t-test), but ABD459 3 and 10 mg/kg groups significantly reduced their visiting times to the food zone by about 35% (all P's < 0.005). However, 20 mg/kg marginally failed to attain significance (P = 0.07) when comparing predosing and postdosing periods.
Overall weight loss and lowering of food intake would be readily explained in terms of heightened locomotor activity. Consequently, we explored the overall ambulatory activity in the PhenoTyper over 3 days, including predrug and postdrug periods (Fig. 3e). Clearly, there were normal circadian rhythms in all drug groups including pronounced activity increases during nocturnal periods. This time effect was reliable [F(64,1728) = 23.95; P< 0.001], but no significant interaction or effect of treatment was observed, confirming that gross locomotion was not affected by ABD459 at any dose.
To confirm that time in the food zone during the 5-h post-treatment period is a valid proxy for food consumption, we correlated the two parameters (Fig. 3f); a positive correlation (r=0.44; P<0.01) corroborates our prediction of equality of measures so that increased time in food zone compellingly reflects higher food consumption.
Effects of ABD459 on vigilance states in comparison with WIN-2 and AM251
A multitude of effects on vigilance states occurred following the administration of cannabinoids, as summarized in Figs 4 and 5. They reflect the pharmacological properties of each compound injected at 12:00 h and contrast the full CB1 agonist WIN-2 (3 mg/kg) with the antagonist/inverse agonist AM251 (3 mg/kg) as well as the neutral antagonist ABD459 (3 mg/kg). Most prominent is the overall reduction in REM sleep induced consistently by all cannabinoids (Fig. 4a, f and g). As is clear from the hypnogram (Fig. 4a), REM sleep was absent during 4 h postdrug in all cannabinoid groups, but slowly recovered thereafter (see also Fig. 4g). Consequently, there was a significant main effect of treatment for the total time spent in REM sleep for the 6 postdrug recording hours [F(3,23) = 7.36, P<0.01], with all cannabinoids lowering REM sleep (all t's > 2.9, P's<0.01; Fig. 4f) and significant overall main effects of treatment [F(3,220) = 7.36, P<0.01] and time [F(11,220) = 15.24, P<0.001] in the factorial two-way analysis. A post-hoc planned comparison of each cannabinoid group with vehicle confirmed significant reductions in REM time for all treatments (all F's > 8.90, all P's < 0.05; Fig. 4g).
Fig 4.
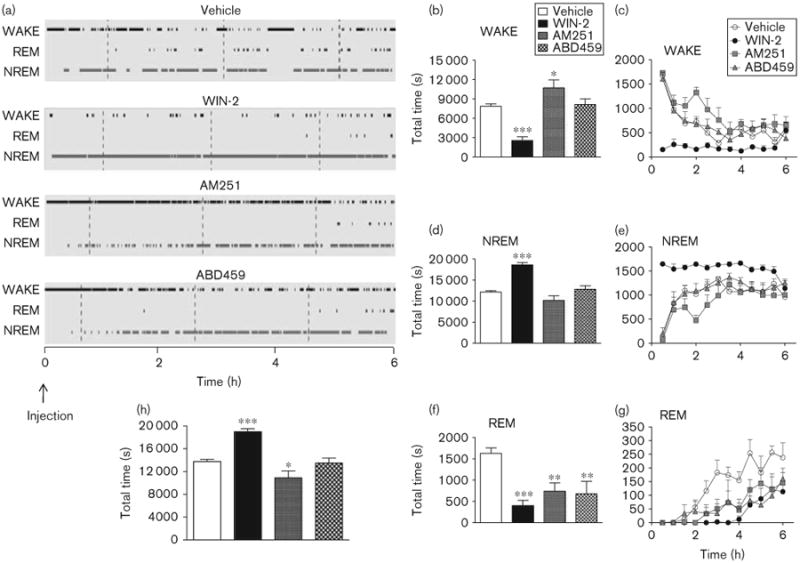
Effects of cannabinoids on vigilance parameters. Means±SEM. *P < 0.05; **P < 0.01; ***P < 0.001 for paired comparisons relative to vehicle treatment. (a) Sample hypnograms of representative individuals across the 6-h EEG recording period showing the amount of WAKEfulness, NREM and REM sleep following vehicle, WIN-2 (3 mg/kg), AM251 (3 mg/kg) and ABD459 (3 mg/kg) injection. The time of injection (12:00 h) was recorded as 0. Note the loss of REM sleep across the first 4 h following the treatment with all cannabinoids. Total time spent in WAKE (b), NREM (d), REM (f) and total sleep (h) following pharmacological treatments. Also, time course of vigilance staging broken down into 30 min bins and plotted against the 6-h recording time for WAKE (c), NREM (e) and REM sleep (g). ABD459, AM251 and WIN-2 significantly reduced REM sleep throughout the recording period. In addition, AM251 enhanced WAKE, whereas WIN-2 decreased it. WIN-2 also significantly enhanced NREM sleep. ABD459 had little effect on WAKE and NREM sleep. EEG, electroencephalogram; NREM, nonrapid eye movement; REM, rapid eye movement.
Fig 5.

Cannabinoid effects on vigilance parameters. (a) Overall sleep composition (% of NREM vs. REM). Controls showed about 12% REM sleep, cannabinoid treatment significantly reduced this amount and there was a near total lack of REM in the WIN-2 group. Latency to first NREM (b) and first REM (c) episodes was significantly altered by WIN-2 (3 mg/kg) and AM251 (3 mg/kg), but not ABD459 (3 mg/kg). Bout lengths of WAKE (d), NREM (e) and REM sleep (f) episodes were altered by WIN-2 in all vigilance stages. As for overall time in REM, bout lengths were reliably shorter when animals were under the influence of cannabinoid. Means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 for paired comparisons relative to vehicle treatment. NREM, nonrapid eye movement; REM, rapid eye movement.
For wakefulness and NREM, however, effects were more variable. Although WIN-2-treated mice presented with significantly reduced wakefulness [Fig. 4a–c; t= 8.12, P< 0.001 as post-hoc to overall ANOVA: F(3,23)= 16.97, P<0.001], wakefulness was enhanced in the AM251 groups (t=2.22, P<0.05) and remained unaffected by ABD459. The reduction in wakefulness in the WIN-2 group was evident throughout the recording period [main effect of WIN-2: F(1,110)=65.92, P< 0.001, and time epoch: F(11,110) =7.00, P <0.001; Fig. 4c]. All other groups were highly active during the initial part of the recording period, possibly because of handling and drug administration, but settled within 2–3 h. AM251-treated animals in particular were more awake during this habituation period [effect of AM251: F(1,110)=4.9; P<0.05; effect of time: F(11,110) =14.7; P<0.001; Fig. 4c].
For the total time spent in NREM sleep, we also observed overall changes [F(3,23) = 22.41, P < 0.001; Fig. 4d] and WIN-2 reliably increased NREM sleep (t=22.41, P<0.001), whereas ABD459 and AM251 exerted no effect. This was readily obvious from the hypnogram (Fig. 4a), but also from the time course of NREM sleep (Fig. 4e), for which an overall factorial ANOVA also yielded a treatment × time epoch interaction [F(33,220) = 3.56, P< 0.001] because of a continuously high amount of NREM sleep in the WIN-2 group [F(1,110) = 97.05, P< 0.001]. Moreover, there was a significant main effect of treatment for the total time spent sleeping (NREM + REM) for the 6 h postdrug [F(3,23)= 17.08, P<0.001; Fig. 4h]. Post-hoc planned comparison of each cannabinoid group with vehicle confirmed significant increases in total sleep for the WIN-2 group (t= 8.12, P<0.001) and reductions for the AM251 group (t= 2.23, P<0.05), with the ABD459 group showing no change.
It follows from these data that the overall sleep composition is altered by cannabinoid treatments (ABD459, WIN-2 and AM251; Fig. 5a). Keeping in mind the fact that recordings took place during the rest/sleep period of the mice, it is not surprising that controls slept for 88.2% of time. However, this was considerably increased in the ABD459, WIN-2 and AM251 groups to 95.1, 97.9 and 93.4%, respectively.
These alterations were further paralleled by changes in latency to first sleep (NREM and REM) episodes (Fig. 5b and c). Overall main effects of treatment were significant for latency to first NREM [F(3,23) = 12.39, P < 0.001] and first REM episodes [F(3,23) = 7.06, P < 0.01]. WIN-2 significantly reduced the time taken to first NREM episode (t =7.20, P < 0.001) such that animals readily fell asleep, thereby increasing the latency to first REM episode (t =6.70, P< 0.001). Neither antagonist affected initiation of NREM sleep, but AM251 delayed onset of REM (t=2.06, P<0.05). This difference in the pharmacological profile may arise from the inverse agonism shown by AM251. Data from ABD459, however, seem to indicate that endogenous cannabinoid release may not contribute towards sleep onset.
Changes in vigilance patterns also affected individual bouts of events (Fig. 5d–f). Significant main effects of treatment were observed on average bout lengths of wake [F(3,23) =6.10, P< 0.01; Fig. 5d], NREM [F(3,23)= 22.98, P <0.001; Fig. 5e] and REM [F(3,23) = 3.92, P<0.05; Fig. 5f] states, and all cannabinoids shortened REM episodes significantly (WIN-2: t= 3.86, P< 0.01; AM251: t=1.87, P<0.05; ABD459: t=2.05, P<0.05). Most dramatic were alterations caused by WIN-2, such that not only the overall total time of wake (Fig. 4b) but also the bout length was reduced (t=3.45, P<0.01; Fig. 5d), whereas both NREM time and event length were markedly increased (t= 4.90, P<0.001; Fig. 5e). These results strongly suggest that the endocannabinoid system may have a more complex involvement in modulating REM sleep as opposed to wakefulness and NREM sleep.
An important additional observation, selective for the WIN-2 group, pertained to the quality of the EEG recording trace during the NREM periods. Compared with vehicle subjects (Fig. 6a and c), WIN-2 traces presented with smaller amplitudes and desynchronized frequency patterns (Fig. 6b). Intriguingly, these changes occurred specifically in the first 3 h after treatment and recovered thereafter [F(64,256) = 22, P<0.01; Fig. 6d]. Although this reflects an overall reduction in excitatory drive in the brain, it also indicates the time course of drug efficacy and washout of WIN-2.
Fig 6.
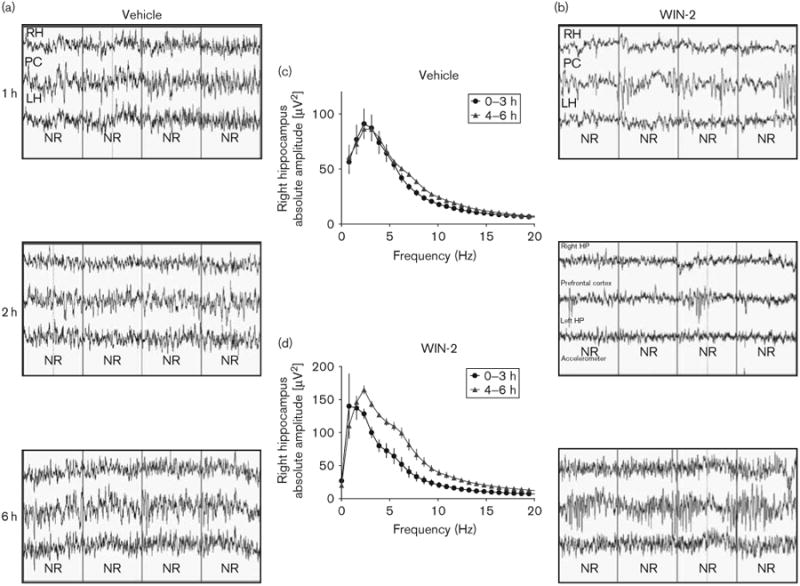
Acute effects of WIN-2 on EEG recordings during NREM sleep and their recovery. Comparison of raw EEG traces recorded at 1, 2 and 6 h after treatment for vehicle (a) and WIN-2 (3 mg/kg) (b) groups. Each recording shows traces from the right hippocampus (RH; top), the prefrontal cortex (PC; middle) and the left hippocampus (LH; bottom). The reduction in amplitude and power was obvious for the first 3 h, following which recovery set in. Comparison of quantitative EEG of absolute spectral power yielded no difference between the first and the second 3 h block for the vehicle group (c), but significantly reduced power during the 0–3 h block in comparison with the 4–6 h block for the WIN-2 group (d), and affected all frequency bands (0–20 Hz; right hippocampus). The absolute amplitudes are represented as mean± SEM. EEG, electroencephalogram; NREM, nonrapid eye movement.
Endocannabinoids contribute towards activation at lower frequency bands
FFT of EEG spectra over all frequency bands and vigilance stages are summarized in Fig. 7. Again, the 6 h long recording was pooled into two 3 h periods because of specific drug effects in the WIN-2 group. For clarity, analysis concentrates on alterations produced by ABD459, WIN-2 and AM251 relative to vehicle treatment, and statistically significant anomalies are shown below each frequency band. Overall, it is evident that cannabinoids exerted stronger effects on spectral power in the hippocampus compared with the prefrontal cortex. This was particularly evident for WIN-2, which caused a leftward shift in power to lower frequencies in the hippocampus than in the prefrontal cortex (Fig. 7a, c, g, j). Significant reductions occurred in both the alpha [F(1,60) = 20.88, P<0.01] and the beta frequency bands [F(1,70) = 11.57, P<0.01] in the hippocampus (Fig. 7g and j), whereas prefrontal effects were more subtle and involved modulations within delta, alpha and beta frequency bands during wakefulness (all F's > 2.6; P's < 0.05; Fig. 7a and c). For NREM sleep, WIN-2 again caused a prominent leftward shift of spectral power in the hippocampus, such that delta band power was enhanced, but power in all higher frequency bands was reduced (Fig. 7b, e, h, k, all F's > 2.8, P's < 0.05). Stronger alterations were noted for the first 3 h after treatment and prefrontal power was not affected by WIN-2 during hours 4–6 (Fig. 7e).
Fig 7.

Normalized EEG power spectra recorded from electrodes positioned above the prefrontal cortex (PC; a–f) and the parietal cortex/dorsal (right) hippocampus (RH; g–l) following systemic treatment with vehicle, WIN-2 (3 mg/kg), AM251 (3 mg/kg) or ABD459 (3 mg/kg). Vigilance stages of WAKEfulness, NREM and REM sleep were isolated and FFT power spectra were computed for 0–3 h and 4–6 h after treatment. The frequency bands that were used for analysis included delta (0–5 Hz), theta (5–9 Hz), alpha (9–14 Hz) and beta (14–20 Hz). Note that the low number of REM episodes from 0–3 h after cannabinoid treatment precluded a meaningful analysis of spectral power; only the vehicle group is presented (c, i). Significant main effects of drug are shown in black in the respective frequency band, whereas significant drug×frequency interactions within a specific frequency band are indicated in grey. The normalized power for all data points (0.77 Hz increments from 0 to 20 Hz) is represented as mean±SEM. EEG, electroencephalogram; NREM, nonrapid eye movement; REM, rapid eye movement.
Too few REM episodes were observed during the first hours after injection to allow determination of spectral power. Only typical spectra for vehicle controls are shown (Fig. 7c and i). At 4–6 h after treatment, WIN-2 produced significant reductions in both theta [F(1,40) = 6.18, P<0.05] and alpha [F(6,60)= 11.56, P<0.001] power in the hippocampus (Fig. 7l), whereas it enhanced alpha [F(6,60) = 4.63, P< 0.001] and reduced delta power [F(5,50) = 5.75, P<0.001] in the prefrontal cortex (Fig. 7f).
Assessment of the contribution of endogenous CB1 activation was attempted using the antagonist/inverse agonist AM251 and the neutral antagonist ABD459. We hypothesized that alterations observed after AM251 and not verified by ABD459 are most likely because of the inverse agonism of AM251. In this latter category are drug effects observed for prefrontal recordings, which were all very small. These are shown in Fig. 7a–f, but not considered here. Of potential interest are alterations in hippocampal recordings for which there was a simultaneous reduction in delta and increase in theta power during wakefulness, and a lowering of delta power during NREM sleep for both antagonists (Fig. 7g, h, i, k; F's > 2.; P's < 0.05). The co-occurrence of these effects in both antagonist groups compellingly suggests that tonic CB1 activation significantly contributes towards power in the lower EEG bands. REM sleep changes could only be determined for the second recording period, for which both AM251 and ABD459 also reduced delta power (Fig. 7l), but this was only significant for the inverse agonist [F(5,50) = 2.59, P<0.05]. Other significant effects observed in the AM251 group included the reduction of beta power during NREM sleep (Fig. 7h and k; F's > 2.6, P's < 0.05) independent of the time of recording. This was reminiscent of the lowering in beta observed for WIN-2, leading to the interpretation that this AM251 effect was because of inverse agonism by the drug. This was further supported by the fact that ABD459 exerted no effect on beta power.
Discussion
ABD459 is a neutral antagonist for CB1 receptors
The chemical removal of the amide group from rimonabant has been used previously as a method of preventing inverse agonism: for example, VCHSR (Hurst et al., 2002) and 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-3-hexyl-1H-1,2,4-triazole (Jagerovic et al., 2004). Unfortunately, many of these compounds expressed significantly reduced binding to CB1 receptors. Therefore, it appears that the amide group should be replaced by a moiety capable of acting as a hydrogen-bond acceptor so not to completely lose this binding interaction. A number of relevant derivatives with bioisosteric substitutes of the amide group by sulphonamide (Srivastava et al., 2008), oxadiazole (Lee et al., 2008) and imidazol-4-thione (Wu et al., 2009) have been attempted; however, these derivatives either lost binding affinity for CB1 or retained inverse agonism. In terms of size and spatial requirements, a ketone is an excellent replacement for an amide, but any functional consequences of a small, partially negative charge residing on the oxygen of the ketone are difficult to foresee. We proposed that a ketone would still bind to the receptor, and possibly to be less efficacious in stabilizing the salt bridge in the inactive form of the receptor; this may weaken inverse agonistic properties. Consequently, a number of ketone derivatives were synthesized, based on rimonabant, and replacing the N-aminopiperidine moiety with an aryl or a cycloalkyl ring, either directly linked to the carbonyl or containing a methylene spacing group. Most of these were indeed neutral antagonists and a number had excellent binding affinities (Ki) and antagonist potencies (KB) of less than 10 nmol/l; from these we selected ABD459 for further study. In-vitro pharmacology experiments presented here show that ABD459 binds with high affinity to the CB1 receptor and behaves as a competitive antagonist. In contrast to rimonabant, there was no inverse agonism on basal CB1 signalling.
Cannabinoids and food intake
The involvement of the endocannabinoid system in hunger and satiety (see Engeli 2012; Kang and Park, 2012 for a review) has been exploited medically in the treatment of anorexia and obesity, cachexia and nausea (Kirkham and Williams, 2001; Verty et al., 2011). Rimonabant did enter the clinic as a licensed medicine for a short time, but was withdrawn because of side-effects (Jones, 2008; Sam et al., 2011). There is nevertheless general acceptance that inhibition of CB1 receptors may reduce food intake and this has been confirmed in multiple behavioural models (Riedel et al., 2009; Engeli, 2012). Indeed, centrally and peripherally active neutral CB1 antagonists such as AM4113 and AM6545, which expressed poor brain penetration, also suppressed food intake at doses between 2 and 50 mg/kg (Chambers et al., 2007; Sink et al., 2008a, 2008b; Cluny et al., 2010; Randall et al., 2010). This occurred in nondeprived mice and both drugs were ineffective in CB1 receptor knockout mice, suggesting that block of endocannabinoid activity is hypophagic. Similarly, we also observed acute weight reduction and lowering of food intake with ABD459 at doses between 3 and 20 mg in mice on a normal diet, and this is in the same dose range as determined previously for AM251 (Riedel et al. 2009). Importantly, food intake and time in the food zone of the home cage correlated positively, suggesting that a genuine reduction in hunger led to a decrease in visits to the food hopper. Our method of observation furthermore offers a sensitive proxy for the length of the drug effect; fewer visits to the food zone were registered during the first 5 h postdrug, but not during the rest of the dark period. This period was somewhat longer for ABD459 than for AM251 (3 h) and also did not result in rebound after washout. We have not explored here whether ABD459 is also devoid of the negative pharmacological effects inherent in AM251 or rimonabant, such as nausea, vomiting, compulsory scratching or grooming syndrome. However, given its similar pharmacological profile, it seems unlikely that ABD459 differs considerably from AM4113 in this respect (Chambers et al., 2007; Sink et al., 2008a, 2008b), and our home cage observation data confirm that there was no overall reduction in ambulatory locomotion, which would have been expected in case of repeated obsessive episodes. Compulsory syndromes to rimonabant, however, are mechanistically dissociable from anorectic responses (Wright and Rodgers, 2013).
Unexplored, to date, are the actions of neutral antagonists on withdrawal of palatable foods, which typically induces a negative emotional state and the inverse agonist/antagonist rimonabant is able to precipitate through a block of the endocannabinoid tone in the amygdala (Blasio et al., 2013). Our neutral antagonist ABD459 may thus exert fewer negative effects than rimonabant.
Cannabinoid effects on vigilance stages
Both the wake-promoting properties of the CB1 antagonist rimonabant (Santucci et al., 1996) and the sleep-enhancing effects of agonists such as anandamide (Murillo-Rodriguez et al., 1998, 2001) and indirect endocannabinoid elevation by VDM-11 in rats (Murillo-Rodriguez et al., 2008), after systemic or local intracranial administration, have confirmed the modulation of vigilance by the endocannabinoid system. However, Δ9-THC and anandamide are partial CB1 agonists and we only recently determined the effects of the full agonists WIN-2 on vigilance in rats (Goonawardena et al., 2011c). This was extended here to mice and we applied wireless microchip-based recording devices (Neurologger) enabling free movement of subjects to determine the pharmaco-EEG. Particular weight was given to the sleep period and recordings concentrated on the daytime. As only rimonabant has been assessed as a selective CB1 antagonist so far, contrasting putative inverse agonistic side-effects with a neutral antagonist, such as ABD459, would differentiate against rimonabant. ABD459 was also compared with the full agonist WIN-2. We focused on endocannabinoid function and did not seek to titrate the antagonists with the agonist WIN-2.
Sleep vigilance state analyses confirmed that all three cannabinoids, ABD459, AM251 and WIN-2, disrupted normal sleep by markedly reducing REM sleep. These alterations were brought about by suppressing REM sleep within the 6h recording period. The WIN-2-induced alterations in the sleep–wake architecture corroborate previous studies that have shown considerable increases in the total amount and bout length of NREM sleep at the expense of wakefulness following cannabinoid (anandamide and Δ9-THC) administration in rats (Buonamici et al., 1982; Murillo-Rodriguez et al., 1998, 2001, 2008) and humans (see Schierenbeck et al., 2008 for a review). This increase in sleep was efficiently blocked by rimonabant (Murillo-Rodriguez et al., 2001, 2008), suggesting a role of CB1 receptors in this effect. However, recent work from our laboratory found both CB1-dependent and CB1-independent actions of WIN-2 in the presence of AM251 in rats (Goonawardena et al., 2011c). What the CB1-independent component reflects is unclear at present, but the strong suppression of REM sleep may be because of a lowering of the cholinergic tone after WIN-2, an action that is consistent with behavioural data (Goonawardena et al., 2010a; Robinson et al., 2010) and also with the notion that REM sleep is dependent on high cholinergic activity (Platt and Riedel, 2011 for review). By contrast, REM sleep reductions observed after AM251, compound 64 and ABD459 treatment are unlikely to be explained by cholinergic mechanisms, but may reflect genuine inhibition of CB1-dependent modulation of GABAergic activity in sleep-relevant brain centres such as the lateral hypothalamus and brainstem (Saper et al., 2001; Gottesmann, 2002; Blanco-Centurion et al., 2006; Jacobson et al., 2011).
In terms of endocannabinoid function, rimonabant reduced NREM and increased wakefulness (Santucci et al., 1996). Although we did not repeat these experiments, AM251 appears to express a somewhat different pharmacology and we could not detect a lowering of NREM sleep in either rats (Goonawardena et al., 2011a, 2011b, 2011c) or mice (this study), and there was also no change in sleep bout length. However, there was a reliable increase in wakefulness after rimonabant (Santucci et al., 1996) or AM251 (this study) limited to 1–3 h postdrug. As this effect was not observed in the ABD459 group, it appears to be because of the inverse agonism inherent to the rimonabant and AM251 (Pertwee, 2005), and seems to suggest the progressive washout of drug during 3 h, following which there was a normalization of vigilance staging. Therefore, we distinguished between the first 3 h of drug action and a later period when considerable washout had appeared. Overall, ABD459 did not affect any of the parameters examined for wakefulness or NREM sleep, suggesting that endocannabinoids may not play a critical role in these stages.
Endocannabinoids have little effect on EEG spectral power
The global reduction (i.e. leftward shift) in normalized spectral power following WIN-2 treatment during wakefulness suggests a lowering of neuronal synchrony in hippocampal–cortical projections (Robbe et al., 2006; Goonawardena et al., 2011a). At the same time, the loss in alpha (9–14 Hz) power during wakefulness may explain why performance in working/short-term memory paradigms (Hampson and Deadwyler, 1999; Hampson et al., 2003; Goonawardena et al., 2010a, 2010b) is compromised and hippocampal cell ensemble firing during task-specific events (i.e. encoding) is disrupted. A similar leftward shift in spectral power in the hippocampus, especially in theta and alpha frequency bands, may explain the observed decrease in REM and significant increase in NREM sleep. Both lowered alpha during wakefulness and heightened delta during NREM sleep are characteristic of overweight or obese humans and mice (Laposky et al., 2006; Babiloni et al., 2011), and in keeping with the notion that WIN-2-induced weight gain reproduces EEG anomalies typical for these conditions.
Similarly, a global reduction in spectral power was also observed in rats treated with THC and CP55940, but not with other synthetic cannabinoid agonists (Kucewicz et al., 2011; Uchiyama et al., 2012). However, the exact vigilance stage was not determined in their recordings, making it difficult to draw firm conclusions from these data. A lowering of rhythmic activity is consistent with an overall reduction of attention or arousal and could be explained by a desynchronization of prefrontal–hippocampal network activity (Kucewicz et al., 2011), a reduction in amplitude of auditory event-related P300 (D'Souza et al., 2012) and an action mediated by CB1 receptors (Goonawardena et al., 2011c).
Administration of antagonists was more selective and our interest focused on similarities between AM251 and ABD459 as they may reflect genuine CB1-mediated endocannabinoid actions. Indeed, subtle differences in spectral power following the administration of inverse agonists and neutral agonists for benzodiazepine-binding sites on the GABA receptor have long been known (Santucci et al., 1989). Here, alterations in spectral power, which were observed throughout the recording period and appeared independent of washout, mainly occurred in the hippocampus and presented as a lowering of delta power during NREM sleep and wakefulness and as an increase in spectral power of theta activity during wakefulness. Such a lowering of spectral power has also been reported during NREM sleep for rimonabant (Santucci et al., 1996) and compound 64 (Jacobson et al., 2011) in rats. However, the fact that rimonabant was not efficient during wakefulness may yet again point towards different pharmacological properties compared with AM251. Intriguingly, the modulation of spectral power by ABD459 appears to be independent of effects on vigilance stages (Fig. 4). Nevertheless, changes in spectral power map onto the neuronal anomalies observed during weight loss (Chowdhury et al., 2003) and ABD459 may be favourably therapeutic over AM251 or rimonabant as it did not affect wakefulness or NREM sleep.
Conclusion
Here, we introduce a novel neutral CB1 receptor antagonist ABD459. In keeping with the reference compound AM251, ABD459 also exerted hypophagic properties in nonfasted mice and led to changes in global EEG power similar to alterations found in underweight subjects. That no changes in wakefulness and NREM sleep were observed in the ABD459 group further underlines the enhanced potential clinical utility of the drug over existing antagonists/inverse agonists for the treatment of obesity.
Acknowledgments
This study was supported by NIDA/NIH grants DA08549 to R.E.H.; DA03672 (to R.G.P and R.R.), DA03934 to R.R. and MRC to G.R.
Footnotes
Conflicts of interest: There are no conflicts of interest.
References
- André A, Gonthier MP. The endocannabinoid system: its roles in energy balance and potential as a target for obesity treatment. Int J Biochem Cell Biol. 2010;42:1788–1801. doi: 10.1016/j.biocel.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Babiloni C, Marzano N, Lizio R, Valenzano A, Triggiani AI, Petito A, et al. Resting state cortical electroencephalographic rhythms in subjects with normal and abnormal body weight. Neuroimage. 2011;58:698–707. doi: 10.1016/j.neuroimage.2011.05.080. [DOI] [PubMed] [Google Scholar]
- Blanco-Centurion C, Xu M, Murillo-Rodriguez E, Gerashchenko D, Shiromani AM, Salin-Pascual RJ, et al. Adenosine and sleep homeostasis in the basal forebrain. J Neurosci. 2006;26:8092–8100. doi: 10.1523/JNEUROSCI.2181-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasio A, Iemolo A, Sabino V, Petrosino S, Steardo L, Rice KC, et al. Rimonabant precipitates anxiety in rats withdrawn from palatable food: role of the central amygdala. Neuropsychopharmacology. 2013;38:2498–2507. doi: 10.1038/npp.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankack J, Platt B, Riedel G. Sleep and hippocampus: do we search for the right things? Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:806–812. doi: 10.1016/j.pnpbp.2009.03.027. [DOI] [PubMed] [Google Scholar]
- Buonamici M, Young GA, Khazan N. Effects of acute delta 9-THC administration on EEG and EEG power spectra in the rat. Neuropharmacology. 1982;21:825–829. doi: 10.1016/0028-3908(82)90071-5. [DOI] [PubMed] [Google Scholar]
- Chambers AP, Vemuri VK, Peng Y, Wood JT, Olszewska T, Pittman QJ, et al. A neutral CB1 receptor antagonist reduces weight gain in rat. Am J Physiol Regul Integr Comp Physiol. 2007;293:R2185–R2193. doi: 10.1152/ajpregu.00663.2007. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chowdhury U, Gordon I, Lask B, Watkins B, Watt H, Christie D. Early-onset anorexia nervosa: is there evidence of limbic system imbalance? Int J Eat Disord. 2003;33:388–396. doi: 10.1002/eat.10155. [DOI] [PubMed] [Google Scholar]
- Clarke JR, Rossato JI, Monteiro S, Bevilaqua LR, Izquierdo I, Cammarota M. Posttraining activation of CB1 cannabinoid receptors in the CA1 region of the dorsal hippocampus impairs object recognition long-term memory. Neurobiol Learn Mem. 2008;90:374–381. doi: 10.1016/j.nlm.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol. 2010;161:629–642. doi: 10.1111/j.1476-5381.2010.00908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluny NL, Chambers AP, Vemuri VK, Wood JT, Eller LK, Freni C, et al. The neutral cannabinoid CB1 receptor antagonist AM4113 regulates body weight through changes in energy intake in the rat. Pharmacol Biochem Behav. 2011;97:537–543. doi: 10.1016/j.pbb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Tschöp MH, Horvath TL, Levine AS. Cannabinoids, opioids and eating behavior: the molecular face of hedonism? Brain Res Rev. 2006;51:85–107. doi: 10.1016/j.brainresrev.2005.10.004. [DOI] [PubMed] [Google Scholar]
- D'Souza DC, Fridberg DJ, Skosnik PD, Williams A, Roach B, Singh N, et al. Dose-related modulation of event-related potentials to novel and target stimuli by intravenous Δ9-THC in humans. Neuropsychopharmacology. 2012;37:1632–1646. doi: 10.1038/npp.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Parigi A, Gautier JF, Chen K, Salbe AD, Ravussin E, Reiman E, Tataranni PA. Neuroimaging and obesity: mapping the brain responses to hunger and satiation in humans using positron emission tomography. Ann NY Acad Sci. 2002;967:389–397. [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Engeli S. Central and peripheral cannabinoid receptors as therapeutic targets in the control of food intake and body weight. Handb Exp Pharmacol. 2012;209:357–381. doi: 10.1007/978-3-642-24716-3_17. [DOI] [PubMed] [Google Scholar]
- Feinberg I, Jones R, Walker JM, Cavness C, March J. Effects of high dosage delta-9-tetrahydrocannabinol on sleep patterns in man. Clin Pharmacol Ther. 1975;17:458–466. doi: 10.1002/cpt1975174458. [DOI] [PubMed] [Google Scholar]
- Feinberg I, Jones R, Walker J, Cavness C, Floyd T. Effects of marijuana extract and tetrahydrocannabinol on electroencephalographic sleep patterns. Clin Pharmacol Ther. 1976;19:782–794. doi: 10.1002/cpt1976196782. [DOI] [PubMed] [Google Scholar]
- Freemon FR. The effect of chronically administered delta-9-tetra-hydrocannabinol upon the polygraphically monitored sleep of normal volunteers. Drug Alcohol Depend. 1982;10:345–353. doi: 10.1016/0376-8716(82)90036-9. [DOI] [PubMed] [Google Scholar]
- Freemon FR, Salinas-Garcia RF, Ward JW. Sleep patterns in a patient with a brain stem infarction involving the raphe nucleus. Electroencephalogr Clin Neurophysiol. 1974;36:657–660. doi: 10.1016/0013-4694(74)90232-6. [DOI] [PubMed] [Google Scholar]
- Gautier JF, Chen K, Salbe AD, Bandy D, Pratley RE, Heiman M, et al. Differential brain responses to satiation in obese and lean men. Diabetes. 2000;49:838–846. doi: 10.2337/diabetes.49.5.838. [DOI] [PubMed] [Google Scholar]
- Goonawardena AV, Robinson L, Hampson RE, Riedel G. Cannabinoid and cholinergic systems interact during performance of a short-term memory task in the rat. Learn Mem. 2010a;17:502–511. doi: 10.1101/lm.1893710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonawardena AV, Robinson L, Riedel G, Hampson RE. Recruitment of hippocampal neurons to encode behavioral events in the rat: alterations in cognitive demand and cannabinoid exposure. Hippocampus. 2010b;20:1083–1094. doi: 10.1002/hipo.20706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonawardena AV, Riedel G, Hampson RE. Cannabinoids alter-spontaneous firing, bursting, and cell synchrony of hippocampal principal cells. Hippocampus. 2011a;21:520–531. doi: 10.1002/hipo.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonawardena AV, Sesay J, Sexton CA, Riedel G, Hampson RE. Pharmacological elevation of anandamide impairs short-term memory by altering the neurophysiology in the hippocampus. Neuropharmacology. 2011b;61(5–6):1016–1025. doi: 10.1016/j.neuropharm.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goonawardena AV, Plano A, Robinson L, Platt B, Hampson RE, Riedel G. A pilot study into the effects of CB1 cannabinoid receptor agonist, WIN55,212-2 or the antagonist/inverse agonist AM251 on sleep in rats. Sleep disorders. 2011c;2011:178469. doi: 10.1155/2011/178469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesmann C. GABA mechanisms and sleep. Neuroscience. 2002;111:231–239. doi: 10.1016/s0306-4522(02)00034-9. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Deadwyler SA. Cannabinoids, hippocampal function and memory. Life Sci. 1999;65:715–723. doi: 10.1016/s0024-3205(99)00294-5. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Simeral JD, Kelly EJ, Deadwyler SA. Tolerance to the memory disruptive effects of cannabinoids involves adaptation by hippocampal neurons. Hippocampus. 2003;13:543–556. doi: 10.1002/hipo.10081. [DOI] [PubMed] [Google Scholar]
- Hodge J, Bow JP, Plyler KS, Vemuri VK, Wisniecki A, Salamone JD, et al. The cannabinoid CB1 receptor inverse agonist AM 251 and antagonist AM 4113 produce similar effects on the behavioral satiety sequence in rats. Behav Brain Res. 2008;193:298–305. doi: 10.1016/j.bbr.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LD, Polich J. EEG, ERPs and food consumption. Biol Psychol. 1998;48:139–151. doi: 10.1016/s0301-0511(98)00010-6. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- Hurst DP, Lynch DL, Barnett-Norris J, Hyatt SM, Seltzman HH, Zhong M, et al. N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) interaction with LYS 3.28(192) is crucial for its inverse agonism at the cannabinoid CB1 receptor. Mol Pharmacol. 2002;62:1274–1287. doi: 10.1124/mol.62.6.1274. [DOI] [PubMed] [Google Scholar]
- Hurst D, Umejiego U, Lynch D, Seltzman H, Hyatt S, Roche M, et al. Biarylpyrazole inverse agonists at the cannabinoid CB1 receptor: importance of the C-3 carboxamide oxygen/lysine3.28(192) interaction. J Med Chem. 2006;49:5969–5987. doi: 10.1021/jm060446b. [DOI] [PubMed] [Google Scholar]
- Jacobson LH, Commerford SR, Gerber SP, Chen YA, Dardik B, Chaperon F, et al. Characterization of a novel, brain-penetrating CB1 receptor inverse agonist: metabolic profile in diet-induced obese models and aspects of central activity. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:565–581. doi: 10.1007/s00210-011-0686-y. [DOI] [PubMed] [Google Scholar]
- Jagerovic N, Hernandez-Folgado L, Alkorta I, Goya P, Navarro M, Serrano A, et al. Discovery of 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-3-hexyl-1h-1,2,4-triazole, a novel in vivo cannabinoid antagonist containing a 1,2,4-triazole motif. J Med Chem. 2004;47:2939–2942. doi: 10.1021/jm031099y. [DOI] [PubMed] [Google Scholar]
- Jáuregui-Lobera I. Electroencephalography in eating disorders. Neuropsychiatric Dis Treat. 2012;8:1–11. doi: 10.2147/NDT.S27302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. End of the line for cannabinoid receptor 1 as an anti-obesity target? Nat Rev Drug Discov. 2008;7:961–962. doi: 10.1038/nrd2775. [DOI] [PubMed] [Google Scholar]
- Jyoti A, Plano A, Riedel G, Platt B. EEG, activity, and sleep architecture in a transgenic AbetaPPSWE/PSEN1A246E Alzheimer's disease mouse. J Alzheimers Dis. 2010;22:873–887. doi: 10.3233/JAD-2010-100879. [DOI] [PubMed] [Google Scholar]
- Kang JG, Park CY. Anti-obesity drugs: a review about their effects and safety. Diabetes Metab J. 2012;36:13–25. doi: 10.4093/dmj.2012.36.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham TC, Williams CM. Endogenous cannabinoids and appetite. Nutr Res Rev. 2001;14:65–86. doi: 10.1079/NRR200118. [DOI] [PubMed] [Google Scholar]
- Koban M, Sita LV, Le WW, Hoffman GE. Sleep deprivation of rats: the hyperphagic response is real. Sleep. 2008;31:927–933. [PMC free article] [PubMed] [Google Scholar]
- Kucewicz MT, Tricklebank MD, Bogacz R, Jones MW. Dysfunctional prefrontal cortical network activity and interactions following cannabinoid receptor activation. J Neurosci. 2011;31:15560–15568. doi: 10.1523/JNEUROSCI.2970-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, et al. Structure-activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- Landsman RS, Burkey TH, Consroe P, Roeske WR, Yamamura HI. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur J Pharmacol. 1997;334:R1–R2. doi: 10.1016/s0014-2999(97)01160-6. [DOI] [PubMed] [Google Scholar]
- Lange C, Luque I, Hervás M, Ruiz-Sanz J, Mateo PL, De la Rosa MA. Role of the surface charges D72 and K8 in the function and structural stability of the cytochrome c from Nostoc sp. PCC 7119. FEBS J. 2005;272:3317–3327. doi: 10.1111/j.1742-4658.2005.04747.x. [DOI] [PubMed] [Google Scholar]
- Laposky AD, Shelton J, Bass J, Dugovic C, Perrino N, Turek FW. Altered sleep regulation in leptin-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;290:R894–R903. doi: 10.1152/ajpregu.00304.2005. [DOI] [PubMed] [Google Scholar]
- Lee SH, Seo HJ, Lee SH, Jung ME, Park JH, Park HJ, et al. Biarylpyrazolyl oxadiazole as potent, selective, orally bioavailable cannabinoid-1 receptor antagonists for the treatment of obesity. J Med Chem. 2008;51:7216–7233. doi: 10.1021/jm800843r. [DOI] [PubMed] [Google Scholar]
- McMahon LR, Koek W. Differences in the relative potency of SR 141716A and AM 251 as antagonists of various in vivo effects of cannabinoid agonists in C57BL/6J mice. Eur J Pharmacol. 2007;569:70–76. doi: 10.1016/j.ejphar.2007.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megevand P, Quairiaux C, Lascano AM, Kiss JZ, Michel CM. A mouse model for studying large-scale neuronal networks using EEG mapping techniques. Neuroimage. 2008;42:591–602. doi: 10.1016/j.neuroimage.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Sánchez-Alavez M, Navarro L, Martinez-González D, Drucker-Colín R, Prospéro-García O. Anandamide modulates sleep and memory in rats. Brain Res. 1998;812:270–274. doi: 10.1016/s0006-8993(98)00969-x. [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Cabeza R, Mendez-Díaz M, Navarro L, Prospéro-García O. Anandamide-induced sleep is blocked by SR141716A, a CB1 receptor antagonist and by U73122, a phospholipase C inhibitor. Neuroreport. 2001;12:2131–2136. doi: 10.1097/00001756-200107200-00018. [DOI] [PubMed] [Google Scholar]
- Murillo-Rodríguez E, Millán-Aldaco D, Di Marzo V, Drucker-Colín R. The anandamide membrane transporter inhibitor, VDM-11, modulates sleep and c-Fos expression in the rat brain. Neuroscience. 2008;157:1–11. doi: 10.1016/j.neuroscience.2008.08.056. [DOI] [PubMed] [Google Scholar]
- Pan X, Ikeda SR, Lewis DL. SR 141716A acts as an inverse agonist to increase neuronal voltage-dependent Ca2 + currents by reversal of tonic CB1 cannabinoid receptor activity. Mol Pharmacol. 1998;54:1064–1072. doi: 10.1124/mol.54.6.1064. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Pivik RT, Zarcone V, Dement WC, Hollister LE. Delta-9-tetra-hydrocannabinol and synhexyl: effects on human sleep patterns. Clin Pharmacol Ther. 1972;13:426–435. doi: 10.1002/cpt1972133426. [DOI] [PubMed] [Google Scholar]
- Platt B, Riedel G. The cholinergic system, EEG and sleep. Behav Brain Res. 2011;221:499–504. doi: 10.1016/j.bbr.2011.01.017. [DOI] [PubMed] [Google Scholar]
- Randall PA, Vemuri VK, Segovia KN, Torres EF, Hosmer S, Nunes EJ, et al. The novel cannabinoid CB1 antagonist AM6545 suppresses food intake and food-reinforced behavior. Pharmacol Biochem Behav. 2010;97:179–184. doi: 10.1016/j.pbb.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel G, Davies SN. Cannabinoid function in learning, memory and plasticity. Handb Exp Pharmacol. 2005;168:445–477. doi: 10.1007/3-540-26573-2_15. [DOI] [PubMed] [Google Scholar]
- Riedel G, Fadda P, McKillop-Smith S, Pertwee RG, Platt B, Robinson L. Synthetic and plant-derived cannabinoid receptor antagonists show hypophagic properties in fasted and non-fasted mice. Br J Pharmacol. 2009;156:1154–1166. doi: 10.1111/j.1476-5381.2008.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Montgomery SM, Thome A, Rueda-Orozco PE, McNaughton BL, Buzsaki G. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat Neurosci. 2006;9:1526–1533. doi: 10.1038/nn1801. [DOI] [PubMed] [Google Scholar]
- Robinson L, McKillop-Smith S, Ross NL, Pertwee RG, Hampson RE, Platt B, Riedel G. Hippocampal endocannabinoids inhibit spatial learning and limit spatial memory in rats. Psychopharmacol. 2008;198:551–563. doi: 10.1007/s00213-007-1012-8. [DOI] [PubMed] [Google Scholar]
- Robinson L, Goonawardena AV, Pertwee RG, Hampson RE, Platt B, Riedel G. WIN55,212-2 induced deficits in spatial learning are mediated by cholinergic hypofunction. Behav Brain Res. 2010;208:584–592. doi: 10.1016/j.bbr.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino T, Parolaro D. Sexually dimorphic effects of cannabinoid compounds on emotion and cognition. Front Behav Neurosci. 2011;5:64. doi: 10.3389/fnbeh.2011.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam AH, Salem V, Ghatei MA. Rimonabant: from RIO to Ban. J Obesity. 2011;2011:432607. doi: 10.1155/2011/432607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santucci V, Fournier M, Worms P, Keane P, Biziere K. Cerebral-activating (EEG) properties of two inverse agonists and of an antagonist at the benzodiazepine receptor in the rat. Naunyn-Schmiedeberg's Arch Pharmacol. 1989;340:93–100. doi: 10.1007/BF00169213. [DOI] [PubMed] [Google Scholar]
- Santucci V, Storme JJ, Soubrie P, Le FG. Arousal-enhancing properties of the CB1 cannabinoid receptor antagonist SR 141716A in rats as assessed by electroencephalographic spectral and sleep-waking cycle analysis. Life Sci. 1996;58:L103–L110. doi: 10.1016/0024-3205(95)02319-4. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–731. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Schierenbeck T, Riemann D, Berger M, Hornyak M. Effect of illicit recreational drugs upon sleep: cocaine, ecstasy and marijuana. Sleep Med Rev. 2008;12:381–389. doi: 10.1016/j.smrv.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ, Brunk LK, Selley DE. Inhibitory effects of SR141716A on G-protein activation in rat brain. Eur J Pharmacol. 2001;414:135–143. doi: 10.1016/s0014-2999(01)00784-1. [DOI] [PubMed] [Google Scholar]
- Sink KS, McLaughlin PJ, Wood JA, Brown C, Fan P, Vemuri VK, et al. The novel cannabinoid CB(1) receptor neutral antagonist AM4113 suppresses food intake and food-reinforced behavior but does not induce signs of nausea in rats. Neuropsychopharmacology. 2008a;43:946–955. doi: 10.1038/sj.npp.1301476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sink KS, Vemuri VK, Olszewska T, Makriyannis A, Salamone JD. Cannabinoid CB1 antagonists and dopamine antagonists produce different effects on a task involving response allocation and effort-related choice in food-seeking behavior. Psychopharmacology (Berl) 2008b;196:565–574. doi: 10.1007/s00213-007-0988-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal DM, Benetti C, Girlanda E, Large CH. The CB1 receptor antagonist, SR141716A, prevents high-frequency stimulation-induced reduction of feedback inhibition in the rat dentate gyrus following perforant path stimulation in vivo. Brain Res. 2008;1223:50–58. doi: 10.1016/j.brainres.2008.05.065. [DOI] [PubMed] [Google Scholar]
- Srivastava BK, Soni R, Joharapurkar A, Sairam KV, Patel JZ, Goswami A, et al. Bioisosteric replacement of dihydropyrazole of 4S-(-)-3-(4-chlorophenyl)-N-methyl-N′-[(4-chlorophenyl)-sulfonyl]-4-phenyl -4,5-dihydro-1H-pyrazole-1-caboxamidine (SLV-319) a potent CB1 receptor antagonist by imidazole and oxazole. Bioorg Med Chem Lett. 2008;18:963–968. doi: 10.1016/j.bmcl.2007.12.036. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, Cowan A, Adler MW. pA2 and receptor differentiation: a statistical analysis of competitive antagonism. Life Sci. 1979;25:637–654. doi: 10.1016/0024-3205(79)90505-8. [DOI] [PubMed] [Google Scholar]
- Uchiyama N, Kikura-Hanajiri R, Matsumoto N, Huang ZL, Goda Y, Urade Y. Effects of synthetic cannabinoids on electroencephalogram power spectra in rats. Forensic Sci Int. 2012;215:179–183. doi: 10.1016/j.forsciint.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Verty ANA, Evetts MJ, Crouch GJ, McGregor IS, Stefanidis A, Oldfield BJ. The cannabinoid receptor agonist THC attenuates weight loss in a rodent model of activity-based anorexia. Neuropsychopharmacology. 2011;36:1349–1358. doi: 10.1038/npp.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright FL, Rodgers RJ. Low dose naloxone attenuates the pruritic but not anorectic response to rimonabant in male rats. Psychopharmacology (Berl) 2013;226:415–431. doi: 10.1007/s00213-012-2916-5. [DOI] [PubMed] [Google Scholar]
- Wu CH, Hung MS, Song JS, Yeh TK, Chou MC, Chu CM, et al. Discovery of 2-[5-(4-chloro-phenyl)-1-(2,4-dichloro-phenyl)-4-ethyl-1H-pyrazol-3-yl]-1,5,5-trimethyl-1,5-dihydro-imidazol-4-thione (BPR-890) via an active metabolite. A novel, potent and selective cannabinoid-1 receptor inverse agonist with high antiobesity efficacy in DIO mice. J Med Chem. 2009;52:4496–4510. doi: 10.1021/jm900471u. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, et al. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]