

Abstract
The present study investigated the effect of silibinin, the principal potential anti-inflammatory flavonoid contained in silymarin, a mixture of flavonolignans extracted from Silybum marianum seeds, on palmitate-induced insulin resistance in C2C12 myotubes and its potential molecular mechanisms. Silibinin prevented the decrease of insulin-stimulated 2-NBDG (2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose) uptake and the downregulation of glutamate transporter type 4 (GLUT4) translocation in C2C12 myotubes induced by palmitate. Meanwhile, silibinin suppressed the palmitate-induced decrease of insulin-stimulated Akt Ser473 phosphorylation, which was reversed by wortmannin, a specific inhibitor of phosphatidylinositol-3-kinase (PI3K). We also found that palmitate downregulated insulin-stimulated Tyr632 phosphorylation of insulin receptor substrate 1 (IRS-1) and up-regulated IRS-1 Ser307 phosphorylation. These effects were rebalanced by silibinin. Considering several serine/threonine kinases reported to phosphorylate IRS-1 at Ser307, treatment with silibinin downregulated the phosphorylation of both c-Jun N-terminal kinase (JNK) and nuclear factor-κB kinase β (IKKβ), which was increased by palmitate in C2C12 myotubes mediating inflammatory status, whereas the phosphorylation of PKC-θ was not significantly modulated by silibinin. Collectively, the results indicated that silibinin prevented inhibition of the IRS-1/PI3K/Akt pathway, thus ameliorating palmitate-induced insulin resistance in C2C12 myotubes.
Keywords: Silibinin, Insulin resistance, Palmitate, C2C12
Introduction
Metabolic detuning has been reported to be involved in obesity, dyslipidemia, diabetes mellitus, and hypertension, all of which characterize metabolic syndrome and are closely associated with insulin resistance. In recent decades, the sustained increases in obesity and metabolic syndrome that have occurred worldwide have resulted in greater interest in the cellular events related to insulin resistance and in how to prevent and treat such resistance (1).
Skeletal muscle is the primary site of glucose uptake, disposal, and storage, accounting for approximately 75% of the entire body's glucose uptake under insulin stimulation (2). Increased plasma free fatty acid (FFA) levels are observed in the above-mentioned diseases, and a growing body of evidence indicates that FFA levels play a central role in the pathophysiology of skeletal muscle insulin resistance (3,4). It has been proposed that several mechanisms account for the inhibition of insulin signaling by saturated fatty acids, including the activation of various serine/threonine kinases, such as protein kinase C isoforms (PKCs), nuclear factor-κB kinase β (IKKβ), c-Jun N-terminal kinase (JNK), and p38 MAP kinase (5). These kinases are activated in high-fat diet-induced or saturated fatty acid-induced insulin resistance and have been reported to catalyze the phosphorylation of serine residues in insulin receptor substrate 1 (IRS-1), leading to a reduction in the phosphorylation of tyrosine residues of IRS-1 and in the activity of downstream signaling pathways activated by insulin (6,7).
Silibinin, the principal flavonoid contained in silymarin, a mixture of flavonolignans extracted from Silybum marianum seeds, is widely used to treat a variety of liver ailments (8), such as nonalcoholic fatty liver disease, which is a chronic metabolic disorder related to a puzzling crosstalk between liver, muscle, and adipose tissue regarding FFA utilization (9). The therapeutic effect of silibinin on insulin resistance has been reported in both clinical studies (10,11) and experimental liver injury models (12-14). However, whether and how silibinin can improve insulin resistance in skeletal muscle cells induced by FFA remains to be elucidated.
Materials and Methods
Reagents
The mouse C2C12 myoblast cell line was obtained from American Type Culture Collection (ATCC, USA). HG-DMEM (Dulbecco's modified Eagle's medium with high glucose) was from GIBCO™ (USA). Fetal bovine serum (FBS) and horse serum were purchased from Hyclone (USA). Insulin, fatty acid-free bovine serum albumin (BSA), palmitate, silibinin, cytochalasin B, and wortmannin were from Sigma (USA). We obtained 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG) from Invitrogen (USA). IRS, phospho-IRS-1 (Thr632), phospho-IRS-1 (ser307), Akt, phospho-Akt (Ser473), phospho-JNK, phospho-IKKβ, and phospho-PKC-θ antibodies were purchased from Cell Signaling Technology (USA).
Cell culture and treatments
Myoblast C2C12 cells were maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere with 5% CO2 at 37°C. Cells were reseeded at a density of 2×104 cells/mL. After 48 h (∼80% confluence), the medium was switched to HG-DMEM with 2% (v/v) horse serum and replaced every other day. Experiments were initiated on day 5 when differentiation was complete. Silibinin was added at various concentrations 2 h prior to the experiments. Palmitate treatment (0.75 mM) of myotubes was carried out during the last 16 h of silibinin treatment.
Palmitate solution preparation
BSA-bound palmitate was prepared according to a previously described procedure (15), with some modifications. Palmitate was dissolved in 0.1 M NaOH to a concentration of 75 mM by heating at 70°C in a shaking water bath, and the solution was then diluted with 10% FFA-free BSA-DMEM at a stock solution of 5 mM at 55°C in a shaking water bath. After filtration (0.45-μm pore size membrane filter), this solution was stored at -20°C and used within 2 weeks. Stored stock solution was heated for 15 min at 55°C and then cooled to room temperature before use. The same concentration of NaOH mixed with 10% FFA-free BSA was used as a control.
2-NBDG uptake
Glucose uptake was measured by adding the fluorescent D-glucose analog 2-NBDG as a tracer to the culture medium. Immediately after treatments with silibinin and BSA-bound palmitate, cells plated in 24- or 96-well plates were incubated with or without insulin (100 nM) for 15 min before 2-NDBG was added at a final concentration of 50 μM; incubation was then continued for a further 20 min. After incubation, free 2-NBDG was washed out 3 times, and fluorescence densities in cell monolayers were measured with a fluorescence microplate reader (Molecular Devices, USA) set at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. The protein concentration of each sample was determined by the Bradford method. Results were normalized by mg of total protein. Nonspecific 2-NBDG uptake was measured in the presence of 20 µM cytochalasin B and subtracted from the total 2-NBDG uptake.
Subcellular fractionation
Cells were collected in ice-cold phosphate-buffered saline (PBS), washed twice with the same buffer, suspended in 100 µL cold sample preparation buffer, sonicated 4-5 times for 10 s each, and centrifuged at 100,000 g for 60 min at 4°C. The resulting pellet was resuspended in 100 µL homogenization buffer to which was added Triton X-100 (final concentration 0.5%) and incubated on ice for 1 h to extract soluble membrane proteins. Samples were centrifuged again at 100,000 g for 1 h at 4°C to remove insoluble membrane components. The resultant supernatant was kept as the plasma membrane fraction. Protein concentrations in the plasma membrane fraction were determined using the Bradford method.
Immunoblotting
Immediately after treatments, the media were aspirated, and the cells were washed twice in ice-cold PBS and lysed in 100 µL lysis buffer. The samples were then briefly sonicated, heated for 5 min at 95°C, and centrifuged at 14,000 g for 5 min. Protein concentrations in the supernatants were determined using the Bradford method. The supernatants were diluted to the same protein concentration, electrophoresed on sodium dodecyl sulfate-polyacrylamide (8%) gels (SDS-PAGE), and transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore, USA). The blots were incubated overnight at room temperature with primary antibodies and then washed 3 times in Tris-buffered saline/0.1% Tween 20 prior to 1 h incubation with horseradish peroxidase-conjugated secondary antibodies at room temperature. Bound antibodies were detected using an enhanced chemiluminescence system (Amersham Pharmacia Biotech, UK) and measured by densitometry using a ChemiDoc XRS digital imaging system and the MultiAnalyst software from Bio-Rad Laboratories (USA).
Statistical analysis
Data are reported as means±SD, and statistical comparisons between groups were carried out using Student's t-test or one-way analysis of variance (ANOVA). P<0.05 was considered statistically significant.
Results
Silibinin prevented the palmitate-induced decrease of insulin-stimulated glucose
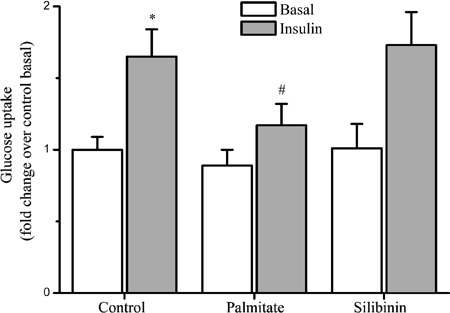
The effects of silibinin and palmitate on glucose uptake activity were tested in differentiated C2C12 myotubes. We found that 0.75 mM palmitate decreased 2-NBDG uptake in insulin-stimulated C2C12 myotubes by 29% (P<0.05), while the effect of palmitate was not significant in naive myotubes. Silibinin treatment did not significantly affect basal or insulin-mediated glucose uptake (Figure 1).
Figure 1. Effects of chronic exposure of C2C12 skeletal muscle cells to silibinin or palmitate on glucose uptake. Myotubes were incubated for 16 h in growth medium supplemented with 100 μg/mL silibinin or 0.75 mM palmitate, and then left untreated or stimulated with 100 nM insulin, followed by assessment of 2-NBDG uptake as described in Methods. Data are reported as means±SD of 4 determinations. *P<0.05, compared to control basal values; #P<0.05, compared to control group treated with acute insulin (Student's t-test).
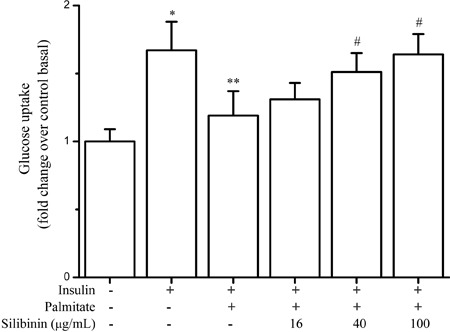
To examine whether silibinin affected the insulin-mediated glucose uptake in palmitate-induced insulin-resistant cells, various concentrations of silibinin were added 2 h prior to the addition of palmitate. As shown in Figure 2, the decrease of 2-NBDG uptake was prevented by silibinin in a dose-dependent manner. Compared with the 0.75 mM palmitate group, insulin-stimulated glucose uptake was improved by 10%, 27% (P<0.05), and 38% (P<0.05) in the presence of 16, 40, and 100 μg/mL silibinin, respectively.
Figure 2. Effect of silibinin on insulin-stimulated glucose uptake in palmitate-treated C2C12 myotubes. C2C12 myotubes were incubated with palmitate (0.75 mM) or silibinin + palmitate, then stimulated with insulin (100 nM), followed by assay for 2-NBDG uptake as described in Methods. Data are reported as means±SD of 4 determinations. *P<0.05, compared to control basal values; **P<0.05, compared to insulin control values; #P<0.05, compared to palmitate group (Student's t-test).
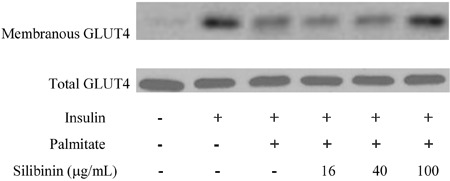
Downregulation of glutamate transporter type 4 translocation induced by palmitate was prevented by silibinin
To elucidate the mechanism by which silibinin prevented the decrease of insulin-stimulated glucose uptake induced by palmitate, we measured glutamate transporter type 4 (GLUT4) expression and translocation. As shown in Figure 3, total GLUT4 protein content was non-significantly reduced in palmitate-treated cells. Although GLUT4 levels in the plasma membrane fraction were lowered significantly by palmitate, this decrease was prevented by silibinin.
Figure 3. Effects of silibinin on insulin-stimulated GLUT4 translocation in palmitate-treated C2C12 myotubes. C2C12 myotubes were incubated with palmitate (0.75 mM) or silibinin + palmitate as described in Methods. Before harvesting, the cells were incubated in the presence or absence of 100 nM insulin for 30 min and lysed for Western blotting. Figures are representative of 3 independent experiments.
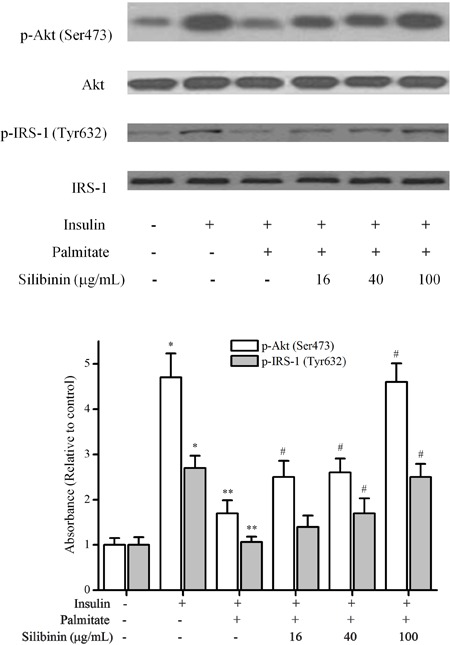
Silibinin prevented IRS-1/PI3K/Akt pathway inhibition
To determine whether silibinin improved palmitate-induced insulin resistance in C2C12 myotubes through the canonical insulin signaling pathway, we examined IRS-1 and Akt phosphorylation. Palmitate markedly decreased the insulin-stimulated Ser473 phosphorylation of Akt and Tyr632 phosphorylation of IRS-1, which was prevented by 16, 40, and 100 μg/mL silibinin treatment (Figure 4).
Figure 4. Effects of silibinin on the palmitate-inhibited insulin signaling pathway in C2C12 myotubes. C2C12 myotubes were incubated with palmitate (0.75 mM) or silibinin + palmitate as described in Methods. Before harvesting, the cells were incubated in the presence or absence of 100 nM insulin for 30 min and lysed for Western blotting. Data are reported as means±SD of 4 determinations. *P<0.05, compared to control group; **P<0.05, compared to insulin group; #P<0.05, compared to palmitate group (Student's t-test).
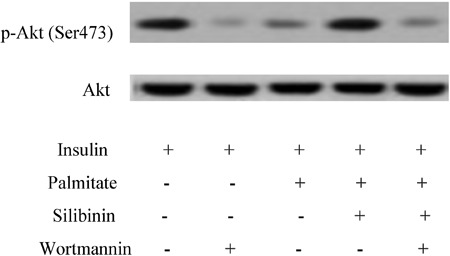
As shown in Figure 5, 50 nM wortmannin, a specific inhibitor of phosphatidylinositol-3-kinase (PI3K), suppressed the increase of Akt phosphorylation induced by 100 μg/mL silibinin in insulin-resistant C2C12 myotubes, which indicated that the effect of silibinin upon the inhibition of Akt phosphorylation by palmitate is PI3K-dependent.
Figure 5. The increase of Akt phosphorylation by silibinin in insulin-resistant C2C12 myotubes was suppressed by 50 nM wortmannin, a specific inhibitor of phosphatidylinositol-3-kinase (PI3K). Differentiated C2C12 skeletal muscle cells were incubated with palmitate (0.75 mM) and/or silibinin (100 μg/mL) in the presence or absence of wortmannin (50 nM), and then stimulated with insulin (100 nM) before cells were harvested for Western blotting. Figures are representative of 3 independent experiments.
Meanwhile, we found that 0.75 mM palmitate upregulated IRS-1 Ser307 phosphorylation in the presence of insulin; this up-regulation was significantly reduced by 100 μg/mL silibinin (Figure 6).
Figure 6. Silibinin modulated the phosphorylation of IRS-1 Ser307, JNK, IKKβ but not PKC-θ in skeletal muscle cells. Differentiated C2C12 skeletal muscle cells were incubated with palmitate (0.75 mM) or silibinin (100 μg/mL) + palmitate, then stimulated with insulin (100 nM) before cells were harvested for Western blotting. Data are reported as means±SD of 4 determinations. *P<0.05, compared to insulin group; #P<0.05, compared to palmitate group in the presence of insulin (Student's t-test).
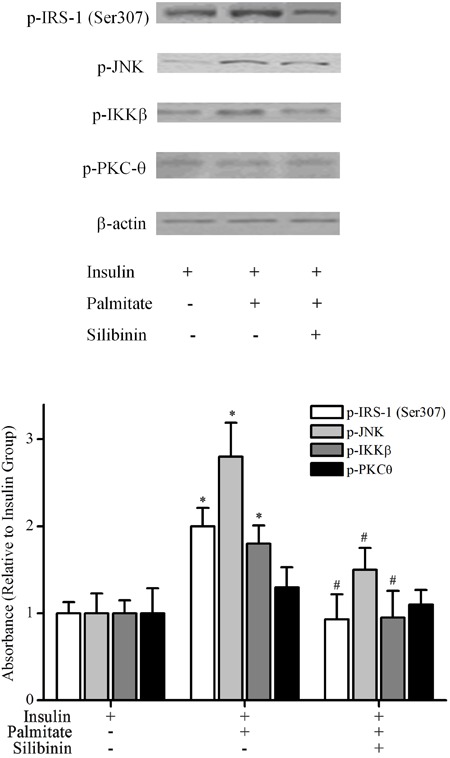
Silibinin treatment modulated the phosphorylation of JNK and IKKβ but not PKC-θ in palmitate-treated C2C12 myotubes
Treatment with silibinin downregulated the phosphorylation of JNK and IKKβ, both of which were increased by 0.75 mM palmitate in C2C12 myotubes. The phosphorylation of PKC-θ was not significantly modulated by palmitate, either alone or combined with 100 μg/mL silibinin (Figure 6).
Discussion
Insulin resistance is a common pathological state found in the metabolic syndrome associated with obesity and type 2 diabetes mellitus, in which target tissues fail to respond properly to physiologic insulin levels (5,7). Skeletal muscle is the major tissue in which a decrease of insulin-mediated glucose uptake is one of the earliest abnormalities indicating insulin resistance. It was suggested that circulating fatty acids significantly increased in obesity and that associated diseases might play an important role in the development of insulin resistance in skeletal muscle (16). In the present study, insulin-stimulated 2-NBDG uptake and GLUT4 translocation in insulin-resistant C2C12 myotubes induced by palmitate was investigated to elucidate the potential effect and mechanism of silibinin on these processes. Our results showed that silibinin (between 16 and 100 μg/mL) was not toxic to C2C12 myotubes, using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay (data not shown), and it prevented the decrease in insulin-stimulated glucose uptake and downregulation of GLUT4 translocation in insulin-resistant C2C12 myotubes induced by palmitate. However, other researchers have reported that silibinin inhibited glucose uptake by directly interacting with GLUT transporters in 3T3-L1 adipocytes and Chinese hamster ovary (CHO) cells (17). On the other hand, contrary results were reported for several other substances, such as genistein, quercetin, and green tea (18). Genistein was reported to suppress insulin-mediated glucose uptake in adipocytes (19) and to promote glucose uptake and GLUT4 translocation in L6 myotubes (20). Quercetin was also reported to inhibit glucose uptake in isolated rat adipocytes (21) but to prevent the downregulation of glucose uptake in skeletal muscle cells (22). These inconsistent and seemingly contrasting results may be due to the use of different cells and tissues. Both the increases of glucose uptake in skeletal muscle and the decreases in adipose tissue are attractive targets for the prevention of diabetes mellitus and obesity (18).
Insulin stimulates the canonical IRS-PI3K-Akt pathway under physiological conditions, inactivates Akt substrate 160 (AS160), promotes GLUT4 translocation to the membrane from inner vesicules and consequently stimulates glucose uptake (23). At the molecular level, decreased insulin-stimulated glucose uptake is connected to reduced tyrosine phosphorylation of IRS-1 and PI3K activation in insulin-resistant states (7). The immunoblot results in the present study suggested that silibinin improved insulin resistance in C2C12 myotubes by preventing the inhibition of the insulin signaling pathway, including Tyr632 tyrosine phosphorylation of IRS1, PI3K activation, and Ser473 phosphorylation of Akt.
IRS1 contains pleckstrin homology and phosphotyrosine domains, which provide a docking site for PI3K when phosphorylated, and it plays a critical role in the insulin signaling pathway. It has been postulated for several years that serine phosphorylation of IRS1 is involved in the desensitization of the action of insulin, due to poor balance between “positive” IRS1 tyrosine phosphorylation and “negative” serine phosphorylation (7). Our results showed that the upregulation of IRS-1 Ser307 phosphorylation in C2C12 myotubes induced by 0.75 mM palmitate was significantly reduced by 100 μg/mL silibinin.
Several serine/threonine kinases including JNK, IKKβ, and PKC-θ have been reported to phosphorylate IRS1 at Ser307 and to inhibit its function, which represents a mechanistic link between FFA and insulin resistance. In our skeletal muscle cell model of palmitate-induced insulin resistance, PKC-θ phosphorylation was not markedly modulated, which is consistent with the report that diacylglycerol derived from saturated fatty acid appears to be a poor activator of PKC, whereas that produced from polyunsaturated fatty acid is a much stronger stimulus (24). Conversely, the decrease in insulin-mediated IRS1 tyrosine phosphorylation by FFA was linked to increased PKC-θ activity (25); PKC-θ inactivation prevented defects in insulin signaling and glucose uptake in skeletal muscle (26). It has also been documented that PKC-θ negatively regulates IRS1 in 3T3-L1 adipocytes (27). The inconsistency is probably due to the use of different cell types and variations between in vivo and in vitro studies, but this requires further study.
In our study, the phosphorylation of both JNK and IKKβ was increased by 0.75 mM palmitate in C2C12 myotubes, and this was downregulated by 100 μg/mL silibinin. This is consistent with previous studies that reported that activities of JNK and IKKβ are increased in diabetic and obese mice or by saturated fatty acids (28,29) and that these increases are associated with marked inhibition of the action of insulin, due to the phosphorylation of serine residues on IRS-1 and the consequent inhibition of phosphorylation of tyrosine residues (5,30). However, it needs to be further elucidated how silibinin downregulates the phosphorylation of JNK and IKKβ and decreases a palmitate-induced inflammatory response in C2C12 myotubes.
In conclusion, the present results provide important evidence for the role of silibinin in the prevention of palmitate-induced insulin resistance and inhibition of the IRS-1/PI3K/Akt pathway in skeletal muscle cells. Potential mechanisms of these actions include downregulation of JNK and IKKβ phosphorylation, thus causing a rebalance between “positive” IRS-1 tyrosine phosphorylation and “negative” serine phosphorylation.
Acknowledgments
This research was supported by the Doctoral Fund of the Ministry of Education of China (#20093317120003), the Key Program of School Foundation of Zhejiang University of Technology (#2012XZ004), and the Natural Science Foundation of Zhejiang Province (#Y14H310011). It was also generously supported by the Chinese Key Laboratory for Biomedical Engineering of the Ministry of Education.
Footnotes
First published online.
References
- 1.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith AG, Muscat GE. Skeletal muscle and nuclear hormone receptors: implications for cardiovascular and metabolic disease. Int J Biochem Cell Biol. 2005;37:2047–2063. doi: 10.1016/j.biocel.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Silveira LR, Fiamoncini J, Hirabara SM, Procopio J, Cambiaghi TD, Pinheiro CH, et al. Updating the effects of fatty acids on skeletal muscle. J Cell Physiol. 2008;217:1–12. doi: 10.1002/jcp.21514. [DOI] [PubMed] [Google Scholar]
- 4.Bloch-Damti A, Bashan N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid Redox Signal. 2005;7:1553–1567. doi: 10.1089/ars.2005.7.1553. [DOI] [PubMed] [Google Scholar]
- 5.Martins AR, Nachbar RT, Gorjao R, Vinolo MA, Festuccia WT, Lambertucci RH, et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis. 2012;11:30. doi: 10.1186/1476-511X-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7:1040–1052. doi: 10.1089/ars.2005.7.1040. [DOI] [PubMed] [Google Scholar]
- 7.Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Loguercio C, Festi D. Silybin and the liver: from basic research to clinical practice. World J Gastroenterol. 2011;17:2288–2301. doi: 10.3748/wjg.v17.i18.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trauner M, Arrese M, Wagner M. Fatty liver and lipotoxicity. Biochim Biophys Acta. 2010;1801:299–310. doi: 10.1016/j.bbalip.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Federico A, Trappoliere M, Tuccillo C, de Sio I, Di Leva A, Del Vecchio Blanco C, et al. A new silybin-vitamin E-phospholipid complex improves insulin resistance and liver damage in patients with non-alcoholic fatty liver disease: preliminary observations. Gut. 2006;55:901–902. doi: 10.1136/gut.2006.091967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andreone P, Brisc M, Chiaramonte M, Federico A, Floreani A, Freni MA, et al. Silybin conjugated with phosphatidylcholine and vitamin E improves liver damage in patients with NAFLD: the results of a randomized multicentre double-blind vs placebo trial. J Hepatol. 2011;54((Suppl 1)):S330–S331. [Google Scholar]
- 12.Yao J, Zhi M, Minhu C. Effect of silybin on high-fat-induced fatty liver in rats. Braz J Med Biol Res. 2011;44:652–659. doi: 10.1590/S0100-879X2011007500083. [DOI] [PubMed] [Google Scholar]
- 13.Salamone F, Galvano F, Marino GA, Paternostro C, Tibullo D, Bucchieri F, et al. Silibinin improves hepatic and myocardial injury in mice with nonalcoholic steatohepatitis. Dig Liver Dis. 2012;44:334–342. doi: 10.1016/j.dld.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Salamone F, Galvano F, Cappello F, Mangiameli A, Barbagallo I, Li Volti G. Silibinin modulates lipid homeostasis and inhibits nuclear factor kappa B activation in experimental nonalcoholic steatohepatitis. Transl Res. 2012;159:477–486. doi: 10.1016/j.trsl.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Cousin SP, Hugl SR, Wrede CE, Kajio H, Myers MG, Jr, Rhodes CJ. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology. 2001;142:229–240. doi: 10.1210/endo.142.1.7863. [DOI] [PubMed] [Google Scholar]
- 16.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhan T, Digel M, Kuch EM, Stremmel W, Fullekrug J. Silybin and dehydrosilybin decrease glucose uptake by inhibiting GLUT proteins. J Cell Biochem. 2011;112:849–859. doi: 10.1002/jcb.22984. [DOI] [PubMed] [Google Scholar]
- 18.Ashida H, Furuyashiki T, Nagayasu H, Bessho H, Sakakibara H, Hashimoto T, et al. Anti-obesity actions of green tea: possible involvements in modulation of the glucose uptake system and suppression of the adipogenesis-related transcription factors. Biofactors. 2004;22:135–140. doi: 10.1002/biof.5520220126. [DOI] [PubMed] [Google Scholar]
- 19.Bazuine M, van den Broek PJ, Maassen JA. Genistein directly inhibits GLUT4-mediated glucose uptake in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2005;326:511–514. doi: 10.1016/j.bbrc.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 20.Ha BG, Nagaoka M, Yonezawa T, Tanabe R, Woo JT, Kato H, et al. Regulatory mechanism for the stimulatory action of genistein on glucose uptake in vitro and in vivo . J Nutr Biochem. 2012;23:501–509. doi: 10.1016/j.jnutbio.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 21.Strobel P, Allard C, Perez-Acle T, Calderon R, Aldunate R, Leighton F. Myricetin, quercetin and catechin-gallate inhibit glucose uptake in isolated rat adipocytes. Biochem J. 2005;386:471–478. doi: 10.1042/BJ20040703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anhe GF, Okamoto MM, Kinote A, Sollon C, Lellis-Santos C, Anhe FF, et al. Quercetin decreases inflammatory response and increases insulin action in skeletal muscle of ob/ob mice and in L6 myotubes. Eur J Pharmacol. 2012;689:285–293. doi: 10.1016/j.ejphar.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 24.MohammadTaghvaei N, Taheripak G, Taghikhani M, Meshkani R. Palmitate-induced PTP1B expression is mediated by ceramide-JNK and nuclear factor kappaB (NF-kappaB) activation. Cell Signal. 2012;24:1964–1970. doi: 10.1016/j.cellsig.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 26.Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest. 2004;114:823–827. doi: 10.1172/JCI200422230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2024–2034. doi: 10.1210/me.2003-0383. [DOI] [PubMed] [Google Scholar]
- 28.Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes. 2008;32((Suppl 7)):S52–S54. doi: 10.1038/ijo.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 30.Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. 2006;103:16454–16459. doi: 10.1073/pnas.0607626103. [DOI] [PMC free article] [PubMed] [Google Scholar]