

Abstract
The MyD88 signaling pathway operates in multiple cell types downstream of Toll-like receptors (TLRs) and IL-1 receptor (IL-1R) family members. To investigate the role of the MyD88 pathway in CD4+ T cell responses, we have generated mice carrying a T cell-specific deletion of MyD88 (MyD88T-KO mice). Using these mice in conjunction with mice carrying an IL-1R-deficient T cell compartment, we show that is required to overcome suppression of the TH1 response by regulatory T cells (Tregs). The ablation of MyD88 specifically in Tregs had no effect on the CD4+ T cell response, suggesting that IL-1 acts on naïve CD4+ T cells instead of on Tregs themselves. Together, these findings demonstrate that TLR-induced IL-1 renders naïve CD4+ T cells refractory to Treg mediated suppression in order to allow their differentiation into TH1 cells. In addition, we show that IL-1 is also important for the generation of functional CD4+ memory T cells.
Introduction
The innate instruction of adaptive immunity is controlled on multiple levels. The activation of pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) in dendritic cells (DCs) leads to their maturation, upregulation of costimulatory molecules, and secretion of proinflammatory cytokines. This activation program provides a critical layer in the discrimination between self and non-self and is essential for the activation of T cell responses (Iwasaki and Medzhitov, 2011; Schenten and Medzhitov, 2011). Despite the progress in the general understanding of the underlying rules that govern the interaction between DCs and cognate CD4+ T cells following TLR activation, the specific roles of individual TLR-induced cytokines and T cell-specific TLR signals in shaping CD4+ T cell responses remain incompletely understood.
CD4+ T cells express several TLRs, although the precise patterns of TLR expression in particular CD4+ T cell subsets are still subject to debate (Cairns et al., 2006; Caramalho et al., 2003; Fukata et al., 2008; Gelman et al., 2004; Gonzalez-Navajas et al., 2010; Kabelitz, 2007). Multiple studies have demonstrated various effects of TLR stimulation in T cells. For example, the stimulation of CD4+ T cells with TLR9 agonists causes enhanced proliferation, survival, and secretion of IL-2 (Gelman et al., 2004). Interestingly, these triggers induce MyD88, the essential signaling adaptor of most TLRs and IL-1 family receptors, to activate both NF-κB and PI3K (Gelman et al., 2006). The former pathway is thought to provide survival signals, while the latter pathway seems to induce IL-2 production and proliferation. Likewise, T cell-specific TLR2 activation can enhance the generation of TH17 responses (Reynolds et al., 2010). Some TLRs also appear to influence naturally-occurring CD4+ CD25+ Tregs directly by dampening their suppressive capabilities, in part by lowering the expression levels of FoxP3, the transcription factor that is critical for the development and function of this T cell lineage (LaRosa et al., 2007) (Liu et al., 2006; Sutmuller et al., 2006). Thus, TLRs seem to modulate both CD4+ effector T cell and Treg responses simultaneously in order to promote the generation of CD4+ T cell responses.
Members of the IL-1 family of cytokines are known to control several aspects of T cell responses directly (Dinarello, 2009; Sims and Smith, 2010). In recent years, IL-1 has received much attention because of its involvement in the differentiation of TH17 cells. These cells express high levels of the IL-1 receptor (IL-1R) and several studies have suggested that IL-1 enhances the differentiation of naïve CD4+ T cells into TH17 cells in vitro (Acosta-Rodriguez et al., 2007; Chung et al., 2009; Kryczek et al., 2007; Wilson et al., 2007). IL-1 signaling in CD4+ T cells is also important for the induction of TH17 cells in vivo, which is illustrated by the observation that CD4+ T cells deficient in IL-1 signaling fail to induce the TH17-dependent diseases experimental autoimmune encephalomyelitis (EAE) and colitis (Chung et al., 2009; Fukata et al., 2008; Sutton et al., 2006; Tomita et al., 2008). In addition to its role in TH17 biology, however, it has been suggested that IL-1 plays an important role in the generation of both primary as well as secondary TH1 responses (Ben-Sasson et al., 2009; Sims and Smith, 2010). The expression level of the IL-1R on naive and TH1 cells is still unclear as some studies detect the receptor in these cells, while others find no evidence for this (Chung et al., 2009; Guo et al., 2009; Taylor-Robinson and Phillips, 1994). Nonetheless, it seems clear that IL-1 signaling contributes to the generation of TH1 responses. For example, it has been suggested to promote the upregulation of the IL-2 receptor α (CD25), prevent apoptosis, and enhance the TH1 expansion (Ben-Sasson et al., 2009; O’Neill, 2008). Moreover, bone marrow chimeras harboring IL-1R-deficient T cells have reduced numbers of TH1 cells during the course of EAE (Ben-Sasson et al., 2009; Chung et al., 2009). Therefore, IL-1 plays a role in the generation of both TH1 and TH17 responses. In contrast, the IL-1-related cytokine IL-18 seems to be more exclusively linked to the generation of TH1 responses as TH1 cells express high amounts of the IL-18R in a T-bet-dependent fashion. In this regard, the function of IL-18 in TH1 cells may parallel that of IL-1 and IL-33 in TH17 and TH2 cells, respectively, namely to reinforce or stabilize CD4+ T cell lineage commitments (Guo et al., 2009). Thus, members of the IL-1 family appear to be involved in both the activation of CD4+ T cells as well as maintaining the subsequent lineage commitment decision. In addition to its effects on the development of specific CD4+ T effector subsets, IL-1 signaling also appears to regulate the interaction between effector T cells and Tregs. Tregs express the IL-1R, even though the roles of IL-1 on Treg function are not clearly understood (Chaudhry et al., 2009; Mercer et al.). Nonetheless, IL-1 may influence the function of Tregs directly by blocking the suppressive function of Tregs (O’Sullivan et al., 2006). Tregs are also thought to deprive CD4+ effector T cells of IL-1 (Chaudhry et al., 2009). Moreover, IL-1 has been implicated enabling the conversion of induced Tregs into TH17 cells (Chung et al., 2009).
Despite this progress in the understanding of TLR and IL-1 biology, the T cell-specific function of these signals are still incompletely understood, in part due to the pleiotropic nature of these triggers. In this study, we sought to investigate the T cell-specific roles of TLRs, IL-1 and IL-18 on the generation of CD4+ T cell responses in vivo. Here, we report on the generation and analysis of mice carrying a T cell-specific ablation of MyD88, the essential signaling adaptor of most TLRs and receptors of the IL-1 family. Our study identifies a novel function for IL-1 in rendering CD4+ T cells refractory to Treg-mediated suppression. In addition, we show that IL-1 is also necessary to generate functional TH1 memory cells.
Results
T cell-specific MyD88 signaling is required for the induction of both TH1 and TH17 responses
In order to investigate the T cell-specific function of signals induced by TLRs and IL-1 family members, we generated mice that ablate MyD88 specifically in the T cell lineage. We flanked exons 3–5 of the MyD88 gene with loxP sites to allow its deletion by Cre-mediated recombination. These exons encode the essential TIR domain of MyD88. Moreover, splicing from exon 2 to exon 6 results in a frame-shift mutation. The targeting strategy is outlined in Supplementary Figure S1A. After the identification of correctly targeted embryonic stem (ES) cells (Figure S1B, C) and successful germline transmission, we intercrossed the resulting MyD88FL/+ mice with CD4-cre mice in order to obtain MyD88FL/FL; CD4-cre mice. These mice, which we call MyD88T-KO mice, ablated MyD88 in all T cells (Figure S1D).
We immunized MyD88T-KO and control mice with Ovalbumin (OVA) in the presence of LPS using incomplete Freund’s adjuvant (IFA) as a carrier and measured the ensuing CD4+ T cell response. To this end, we isolated CD4+ T cells from the draining lymph nodes 7 days after immunization, at which point the majority of cells displayed a phenotype of CXCR5+ PD-1+ T follicular helper cells (TFH cells) (Supplementary Figure S2), and restimulated the cells with OVA in the presence of irradiated splenocytes as APCs in vitro. Wild-type CD4+ T cells proliferated robustly and secreted both IFN-γ and IL-17. In contrast, the proliferative response of CD4+ T cells from MyD88T-KO mice was reduced and the cells were significantly impaired in their ability to secrete both IFN-γ and IL-17 (Figure 1A and 1B, Supplementary Figure S9A and S9B). As dying cells, such as irradiated splenocytes, might have been a source of IL-1 in the T cell cultures and thus might have affected the experimental outcome during the in vitro phase of the assay, we controlled for the presence of IL-1 in the cultures. While we could readily detect IL-1α and IL-1β in cultured macrophages after stimulation with LPS and ATP, we failed to do so in the T cell assays after re-stimulation with OVA, even in the presence of a 4-fold higher number of irradiated splenocytes (Supplementary Figure S3A). Indeed, CD4+ T cells from MyD88T-KO mice also failed to expand and secrete IFN-γ after restimulation in the presence of irradiated APCs from Caspase-1-deficient mice, which are impaired in their ability to secrete IL-1 (Supplementary Figure S3B and S3C). These results suggest that IL-1 released by dying APCs after in vitro stimulation with antigen is not responsible for the defects observed in MyD88-deficient CD4+ T cells. The impairment of the CD4+ T cell response in MyD88T-KO mice is therefore due to defective IL-1 signaling in vivo.
Figure 1. Impaired CD4+ T cells response in MyD88T-KO mice.

(A) Proliferation of CD4+ T cells from MyD88T-KO mice and wild-type controls. CD4+ T cells were isolated from the draining lymph nodes 7 days after immunization in the feet with OVA + LPS in IFA and re-stimulated with OVA in the presence of irradiated splenocytes. Proliferation was measured 3 days later by 3H-thymidine incorporation. A representative of 3 experiments is shown. (B). Cytokine secretion of CD4+ T cells following restimulation with OVA. (C). TH1 response in MyD88T-KO mice and wild-type controls after immunization with 2W peptide + LPS in IFA and restimulation with 2W peptide. (D). Frequency of 2W:I-Ab+ CD4+ T cells in MyD88T-KO and wild-type control mice T cells 7 days after footpad immunization with 2W peptide + LPS in IFA. (E). Cytokine production of 2W:I-Ab+ CD4+ T cells in MyD88T-KO and wild-type control mice after restimulation with an αCD3e antibody. (F). Absolute numbers of all cells, CD4+ T cells, and 2W:I-Ab+ CD4+ T cells in the draining lymph nodes of MyD88T-KO and wild-type controls immunized with 2W peptide + LPS in IFA 7 days earlier.
In order to assess whether the activation of other TLRs results in a similar defect of the CD4+ T cell response in MyD88T-KO mice, we also immunized mice with OVA plus either polyIC, triggering TLR3, or CpG DNA, triggering TLR9, as adjuvant, using IFA as a carrier. Compared to wild-type controls, CD4+ T cells of MyD88T-KO mice failed to expand properly after restimulation and secreted only limited amounts of IFN-γ also under these additional stimuli (Supplementary Figure S4A–B). Moreover, the CD4+ T cell response was also defective in MyD88T-KO mice following immunization with OVA in the more complex Complete Freund’s Adjuvant (CFA) (Supplementary Figure S4C). Next, we asked whether the impairment of the CD4+ T cell response was caused by a defect in the generation or function of the cells. We therefore immunized the mice with 2W peptide in the presence of LPS and IFA, as this system allows for the tracking of the antigen-specific CD4+ T cells with 2W:I-Ab tetramers (Moon et al., 2007). Upon immunization with 2W peptide, the TH1 response in MyD88T-KO mice was also impaired following restimulation with 2W peptide, suggesting that the choice of antigen does not influence the outcome of the T cell response as measured by restimulation (Figure 1C). At the peak of the response, activated CD44+ CD4+ T cells specific for the 2W peptide comprised approximately 1.3 % of all CD4+ T cells in the draining lymph nodes of wild-type controls. Surprisingly, the frequency of these cells was similar in MyD88T-KO mice (Figure 1D). While the cellular composition of the lymph nodes did not change after immunization (Supplementary Figure S5A–D), the number of cells in the draining lymph nodes of MyD88T-KO mice was reduced compared to that of MyD88WT mice, leading to a reduction in the absolute number of 2W:I-Ab-positive CD4+ T cells in MyD88T-KO mice, which may have been caused by a failure of the antigen-specific T cells to secrete inflammatory cytokines (Figure 1E).
Consistent with normal frequency of antigen-specific CD4+ T cells in MyD88T-KO mice, these cells incorporated BrdU into their DNA as efficiently as wild-type controls, implying that the clonal expansion of antigen-specific CD4+ T cells is not impaired in MyD88T-KO mice (Figure 2A). CD4+ T cells from these mice also did not display an increased tendency to undergo apoptosis (Figure 2B). However, antigen-specific 2W+ CD4+ T cells of MyD88T-KO mice expressed lower amounts of T-bet, the lineage-defining transcription factor of TH1 cells, when compared to wild-type control cells (Figure 2C). Together, our findings therefore demonstrate that T cell-specific MyD88 signaling is required for the generation of functionally competent TH1 cells, even though the initial expansion of the antigen-specific CD4+ T cells is not compromised.
Figure 2. Proliferation, viability, and differentation of 2W:IA(b)+ CD4+ T cells from MyD88T-KO and wild-type control mice.

(A) Proliferation of antigen-specific 2W:I-A(b)+ CD4+ T cells in the draining lymph nodes of MyD88T-KO and wild-type control mice between day 4–7 after immunization with 2W peptide + LPS in IFA as measured by incorporation of BrdU. Left panel: A representative experiment is shown. Right panel: Statistical representation of all experiments normalized to the level of BrdU incorporation of 2W:I-A(b)+ CD4+ T cells of MyD88WT mice. Each dot represents one mouse. (B) Frequency of apoptotic antigen-specific 2W:I-A(b)+ CD4+ T cells in the draining lymph nodes of MyD88T-KO and wild-type control mice on day 7 after immunization with 2W peptide + LPS in IFA, measured by staining for active caspase-3. Shown is the mean ± standard deviation. (C) T-bet expression by qPCR in immunized CD4+ T cells in the draining lymph nodes of MyD88T-KO and wild-type control mice. A representative experiment using the pooled samples from 5–10 mice per genotype is shown.
Impaired CD4+ T cell response in MyD88T-KO mice is caused by defective IL-1 signaling
MyD88 relays signals from most TLRs and also from members of the IL-1 family. In order to distinguish between these two types of signals, we first analyzed the regulation of the receptors of the IL-1 family on antigen-specific T cells 7 days after immunization with 2W peptide and LPS in IFA. We detected an upregulation of the IL-1R in antigen-specific CD4+ T cells from both MyD88T-KO mice and wild-type controls compared to naïve CD4+ T cells. In contrast, the IL-18R was downregulated in all CD4+ T cells from both MyD88T-KO mice and wild-type controls when compared to naïve CD4+ T cells (Figure 3A and 3B).
Figure 3. Impaired CD4+ T cells response in MyD88T-KO mice is caused by defective IL-1 signaling.

(A, B) Expression of the receptors for IL-1 (A) and IL-18 (B) in CD4+ T cells in the draining lymph nodes of immunized MyD88T-KO and wild-type control mice as measured by qPCR. The expression levels were normalized to the level of naïve CD4+ T cells. A representative experiment using the pooled samples from 5–10 mice per genotype is shown. (C, D) CD4+ T cell response of Rag2-deficient mice reconstituted with a 3:1 mix of bone marrow from TCRβ-deficient mice and IL1RKO, IL18KO, or TLR2KO; TLR4KO mice, respectively. CD4+ T cells were restimulated with OVA in the presence of irradiated splenocytes 7 days after the immunzation of the bone marrow chimeras in the feet with OVA + LPS in IFA. The proliferation was measured 3 days later by 3H-thymidine incorporation (C) and the secretion of IFNγ and IL-17 of the restimulated CD4+ T cells was measured by ELISA (D). A epresentative experiment is shown. (E, F). CD4+ T cell response of mixed bone marrow chimeras harboring bone marrow of IL1RKO, IL18KO, or wild-type mice mixed with bone marrow of TCRβKO mice in a ratio of 1:3 after footpad immunization with OVA + LPS in IFA. A representative experiment is shown.
The regulation of the IL-1R implicated IL-1 signaling as an important event in the generation of the CD4+ T cell response, but did not rule out an involvement of TLR signaling. We therefore generated mixed bone marrow chimeras, in which the T cell compartment consisted of T cells derived from TLR2- and TLR4-deficient compound mutant mice, IL-1R-deficient mice, or wild-type mice. We immunized these mice with OVA and LPS in IFA and analyzed the ensuing T cell response. Chimeric mice harboring a wild-type or a TLR2 and TLR4-deficient T cell compartment mounted a robust CD4+ T cell response. In contrast, proliferation and cytokine secretion of IL-1R-deficient CD4+ T cells was significantly impaired (Figure 3C–D and Supplementary Figure S6). While CD4+ T cells had down-regulated the IL18R expression 7 days after immunization, it still remained possible that IL-18 signaling plays an important role in the early stages of the T cell response. Thus, we also compared the CD4+ T cell response to immunization with OVA + LPS in IFA in bone marrow chimeras that harbored either IL-1R- or IL-18R- deficient T cells. As before, chimeras with an IL-1R-deficient T cell compartment failed to mount a robust response. In contrast, chimeras carrying a IL-18R-deficient T cell compartment mounted a CD4+ T cell response similar to that of wild-type controls (Figure 3E–F and Supplementary Figure 6), suggesting that IL-18 signaling in CD4+ T cells is not essential for the generation of a CD4+ T cell response upon immunization with OVA + LPS in IFA.
We obtained similar results for bone marrow chimeras that carried either a TLR9 or IL-1R-deficient T cell compartment, which we had immunized with OVA and CpG DNA in IFA (Supplementary Figure S7A–B and Supplementary Figure 8). We noticed in some, but not all experiments, a moderate contribution of TLR9 signaling to the response, consistent with the suggested role of T cell-intrinsic TLR9 signaling in the generation of CD4 T cell responses (Gelman et al., 2004). However, IL-1 signaling was also required under these conditions. Thus, our results collectively demonstrate that the defective CD4+ T cell response in MyD88T-KO mice is caused by the inability of the T cells to recognize IL-1 signals, whereas TLR-driven signals play a minor role.
IL-1 signaling is required to render CD4+ T cells refractory to Treg-mediated suppression
We had previously shown that transient depletion of regulatory T cells (Tregs) in complete MyD88-deficient mice is able to restore the TH1 response (Pasare and Medzhitov, 2004). However, neither the molecular signals nor the cell type responsible for this phenotype were known. In light of our current results, we asked whether IL-1 signaling is influencing the interplay between the naive and effector CD4+ T cell population and the Treg population. We therefore first analyzed the CD4+ T cell response in the absence of Tregs. To this end, we depleted CD25+ Tregsfrom MyD88T-KO and control mice using a monoclonal αCD25 antibody three days prior to immunization with OVA and LPS in IFA. We verified the successful Treg depletion on the day of immunization by flow cytometry of blood leukocytes (data not shown). Treg depletion was unable to restore the TH17 response in MyD88T-KO mice, consistent with a requirement for IL-1 signaling in the differentiation of these cells. However, in the absence of Tregs, proliferation and secretion of IFN-γ by restimulated CD4+ T cells was restored in MyD88T-KO mice (Figure 4A–B, Supplementary Figure 9 A–B). We obtained similar results after immunization of Treg-depleted MyD88T-KO mice and wild-type controls with OVA + CpG DNA (Supplementary Figure 7C–D, Supplementary Figure 9C–D).
Figure 4. Induction of a TH1 response requires T cell-specific MyD88 signaling in naïve or effector CD4+ T cells in order to overcome Treg-mediated suppression.
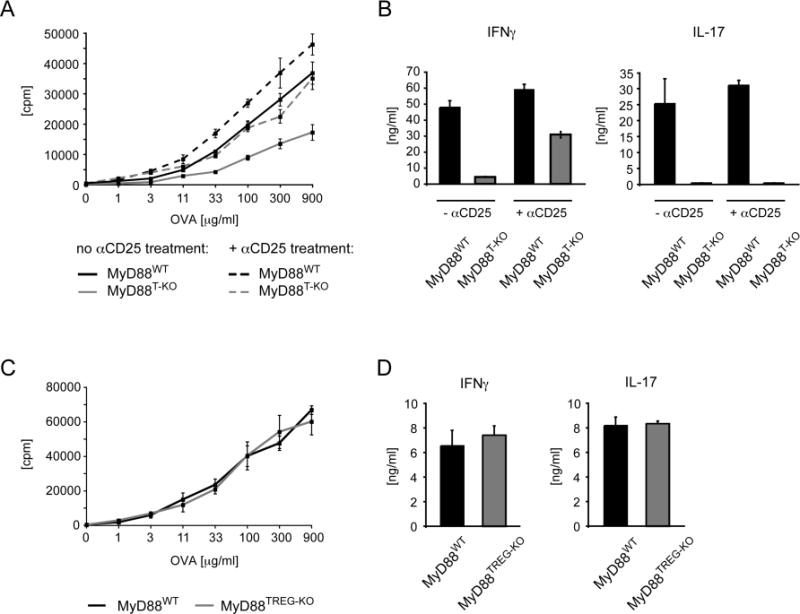
(A, B) Restoration of the TH1 response after transient depletion of CD25+ Tregs in MyD88T-KO mice. MyD88T-KO and control mice were immunized with OVA + LPS in IFA in the footpads following the injection of an αCD25 antibody 3 days earlier in order to deplete CD25+ Tregs. CD4+ T cells were isolated 7 days after immunization and restimulated with OVA in vitro in the presence of irradiated splenocytes. Proliferation (A) and cytokine secretion (B) of the CD4+ T cells were measured 3 days later. A representative experiment is shown. (C, D) CD4+ T cell response in MyD88TREG-KO mice and wild-type controls. The mice were immunized with OVA + LPS in IFA, the proliferation (C) and cytokine secretion (D) was measured as described before. A representative experiment is shown.
While these results suggested that IL-1 signaling in CD4+ T cells may function to overcome Treg-mediated suppression, they did not distinguish between the effects of IL-1 on the various CD4+ T cell populations. Since IL-1 may be acting on Tregs directly (Mercer et al.; O’Sullivan et al., 2006), we generated mice in which MyD88 is specifically ablated in FoxP3+ Tregs, using the FoxP3-cre allele (MyD88TREG-KO mice). Immunization of MyD88TREG-KO mice with OVA and LPS in IFA revealed that MyD88TREG-KO mice mounted TH1 and TH17 responses that were indistinguishable from wild-type controls (Figure 4C–D). Thus, these results indicate that IL-1 does not act on Tregs, but instead is required in naïve or effector CD4+ T cells in order to render these cells refractory to Treg-mediated suppression.
IL-1 signaling does not cause an increased frequency of induced Tregs
The results so far implied a requirement for IL-1 in naïve or effector CD4+ T cells to overcome suppression by “naturally-occurring” Tregs (nTregs). However, TLR-induced cytokines such as IL-6 have been shown to affect the balance between the frequencies of TH17 cells and TGF-β-induced Tregs (iTregs) and this mechanism can result in an increased frequency of iTregs under conditions where the development of TH17 cells is genetically compromised. A similar function has recently been suggested for IL-1 (Chung et al., 2009). The absence of a TH17 response in MyD88T-KO mice may therefore lead to an increased frequency of iTregs, which could provide an alternative explanation for the impaired TH1 response in MyD88T-KO mice. Thus, we investigated whether the frequency of iTreg is increased in MyD88T-KO mice. First, we determined by flow cytometry whether FoxP3+ Tregs are present at increased numbers in the lymph nodes of unimmunized MyD88T-KO mice. We did not observe any difference between MyD88T-KO mice and wild-type controls (Figure 5A). Next, we analyzed whether the frequency of Tregs is altered in MyD88T-KO mice after immunization with OVA and LPS in IFA and subsequent restimulation in vitro. The frequency of these cells remained unchanged in MyD88T-KO mice (Figure 5B). In order to measure the frequency of Tregs more directly, we also analyzed their presence in the draining lymph nodes of immunized mice. While we noticed a modest tendency of a higher frequency of Tregs in the draining lymph nodes of some immunized MyD88T-KO mice, we did not detect significant differences between these mice and wild-type controls (Figure 5C).
Figure 5. Frequency of Tregs in MyD88T-KO mice.
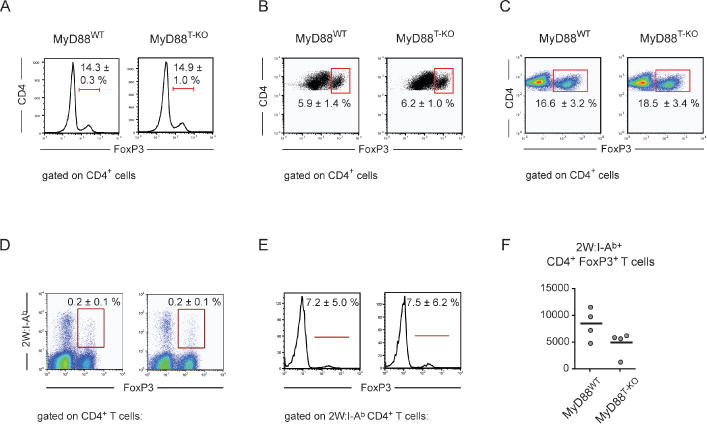
(A) Frequency of FoxP3+ Tregs as percentage of CD4+ T cells in unimmunized wild-type and MyD88T-KO mice. A representative of 3 independent experiments is shown. (B) Frequency of FoxP3+ Tregs after immunization with OVA + LPS in IFA and subsequent restimulation of isolated CD4+ T cells with OVA in the presence of irradiated splenocytes. An experiment representing 3 independent experiments is shown. (C) Frequency of FoxP3+ Tregs in wild-type and MyD88T-KO mice in the draining lymph nodes 7 days after immunization with OVA + LPS in IFA. A representative of three experiments is shown. (D, E) Frequency of antigen-specific FoxP3+ 2W:I-A(b)+ Tregs in the draining lymph nodes 7 days after immunization with 2W peptide + LPS in IFA, shown as percentage of all CD4+ T cells (D) or percentage of all 2W:I-A(b)+ CD4+ T cells (E). A representative of 3 experiments is shown. (F) Absolute numbers of FoxP3+ 2W:I-A(b)+ Tregs in the draining lymph nodes 7 days after immunization with 2W peptide + LPS in IFA.
These experiments so far looked at the overall frequency of Tregs and did not reveal any significant differences between MyD88T-KO and wild-type mice. However, it remained possible that the lack of IL-1 signaling in the T cells of MyD88T-KO mice alters the frequency of antigen-specific Tregs. We therefore immunized MyD88T-KO mice and wild-type controls with 2W peptide and LPS in IFA and measured the frequency of 2W:I-Ab+ FoxP3+ Tregs in the draining lymph nodes 7 days later by flow cytometry. Neither the frequency of these cells as a percentage of total CD4+ T cells, nor as percentage of antigen-specific 2W:I-Ab+ CD4+ T cells was changed in MyD88T-KO mice compared to wild-type mice (Figure 5D–E). Consistent with of the overall reduced size of the lymph nodes of immunized MyD88T-KO mice, we also observed a trend towards lower absolute numbers of 2W:I-Ab+ FoxP3+ Tregs in MyD88T-KO mice compared to wild-type mice (Figure 1E and 5F). Thus, we conclude that T cell-intrinsic MyD88 signaling does not significantly affect the frequency of induced Tregs, at least under the conditions used in this study.
Impaired memory CD4+ T cell response in MyD88T-KO mice
The absence of Tregs prior to the immunization enables MyD88T-KO mice to generate a TH1 response. Thus, we asked whether this response is able to produce memory T cells at a frequency similar to wild-type mice. We immunized MyD88T-KO mice and wild-type controls with 2W peptide + LPS in IFA in the presence or absence of Tregs and isolated the popliteal and inguinal lymph nodes 30–60 days later. Staining of the antigen-specific CD4+ T cells for the activation markers CD44 and CD62L revealed that by this time almost about half of 2W:I-Ab+ CD4+ T cells of both MyD88T-KO mice and wild- type controls represented CD44+ CD62L+ memory T cells, irrespective of whether or not Tregs were present during the immunization (Figure 6A). There was no difference between MyD88T-KO and wild-type control mice. Likewise, we noticed no difference after staining of antigen-specific CD4+ T cells for CXCR5, Ly-6C, and PD-1. The lymph nodes of both MyD88T-KO mice and wild-type controls harbored equal frequencies of Ly-6C+ CXCR5− PD-1− TH1 effector memory cells, Ly-6C− CXCR5+ PD-1− central memory CD4+ T cells, and Ly-6C− CXCR5+ PD-1+ TFH cells (Figure 6B). These results suggest that in MyD88T-KO mice, antigen-specific CD4+ T cell become antigen-experienced and form memory cells at similar frequencies as wild-type mice, even under conditions where Tregs are present.
Figure 6. Memory CD4+ T cell response in MyD88T-KO mice.

(A, B) Frequency of 2W:I-A(b)+ memory CD4+ T cells in MyD88T-KO mice and wild-type controls. The frequency of 2W:I-A(b)+ CD4+ T cells and 2W:I-A(b)+ memory CD4+ T cells was determined 40–60 days after immunization with 2W peptide + LPS in IFA. The mice were immunized either in the presence (− αCD25) or transient absence (+ αCD25) of Tregs during the primary immunization. (A). Frequency of 2W:I-A(b)+ CD4+ T cells as a percentage of total CD4+ T cells (upper panels) and frequency of 2W:I-A(b)+ CD44+ CD62L+ memory CD4+ T cells as a percentage of total 2W:I-A(b)+ CD4+ T cells (lower panels). Values represent mean ± standard error. (B). Frequency of 2W:I-A(b)+ CXCR5− PD-1− effector memory CD4+ T, 2W:I-A(b)+ CXCR5+ PD-1− central memory CD4+ T cells, and 2W:I-A(b)+ CXCR5+ PD-1+ T follicular helper (TFH) cells as a percentage of total 2W:I-A(b)+ CD4+ T cells. A representative experiment is shown. (C, D). CD4+ memory T cell response after transient depletion of CD25+ Tregs prior to both the primary and secondary immunization. MyD88T-KO mice and control mice were injected with an αCD25 antibody 3 days prior to the primary immunization with OVA + LPS in IFA. After 60 days, Tregs were transiently depleted again and the mice were challenged 3 days later with OVA + LPS in IFA. CD4+ T cells were isolated 7 days later and restimulated with OVA in vitro in the presence of irradiated splenocytes. The proliferation measured by 3H-thymidine incorporation (C) and cytokine secretion measured by ELISA are shown (D). The data reflect representative results from 2 independent experiments involving a total of 4 mice per genotype.
We then asked whether the CD4+ memory T cell response is functionally impaired. To this end, we immunized MyD88T-KO mice and wild-type controls with OVA + LPS in IFA in either the presence or absence of Tregs and re-immunized these mice 60 days after the primary immunization with OVA + LPS in IFA, again in either the presence of absence of Tregs. We isolated CD4+ T cells from the draining lymph nodes 7 days later and restimulated the cells with OVA in vitro as described before. As expected, wild- type CD4+ T cells proliferated robustly after restimulation and secreted IFNγ and IL-17, irrespective of whether or not Tregs were present during both the primary and secondary immunization. In contrast, CD4+ T cells from MyD88T-KO mice failed to expand to wild-type levels and did not secrete large amounts of cytokines. Surprisingly, this was also the case under conditions in which Tregs were absent during both the primary and secondary immunization (Figure 6C and D, Supplementary Figure 10). Therefore, while memory CD4+ T cells do not appear to be dependent on IL-1 for their generation, they do require this cytokine in order to become functionally competent, suggesting a dependence on IL-1 that goes beyond a requirement for IL-1 to overcome Treg- mediated suppression.
IL-1 and IL-6 induce different gene expression programs
The phenotype of MyD88T-KO mice resembled that of mice with a T cell-specific ablation of IL-6 signaling (IL6RαT-KO mice) as both IL-1 and IL-6 are essential for the generation of a TH1 response in the presence of Tregs, but not essential in the absence of Tregs (Nish et al., manuscript submitted). It had been previously suggested that IL-6 induces the IL-1R and thus increases the sensitivity of CD4+ T cells to IL-1 (Chung et al., 2009). We therefore asked whether IL-1 regulates a subset of the IL-6-dependent gene expression program or whether these two cytokines regulate distinct gene expression programs with little overlap. We isolated 2W:I-Ab+ CD4+ T cells from the draining lymph nodes of immunized MyD88T-KO, IL6RαT-KO, and wild-type mice and analyzed polyA- containing RNAs by high-throughput sequencing (RNA-Seq). Comparison of the gene expression profiles of MyD88-, and IL6Rα-deficient CD4+ T cells mice with that of wild- type controls revealed that IL-1 and IL-6 modify the expression levels of distinct sets of genes that have only a few genes in common with each other (Figure 7A). A principal component analysis (PCA) confirmed this further as the vectors for both MyD88T-KO and IL6RαT-KO mice were not only distinct from that of wild-type controls but also different from each other (Figure 7B). We therefore conclude that IL-1 and IL-6 regulate different processes that are both required to achieve an optimal CD4+ T cell response.
Figure 7. Gene expression analysis of MyD88T-KO and IL6RαT-KO mice compared to wild-type controls.

(A) RNASeq of antigen-specific 2W:I-A(b)+ CD4+ T cells from the draining lymph nodes of MyD88T-KO, IL6RαT-KO, and wild-type control mice that were isolated by flow cytometry 7 days after immunization with 2W peptide + LPS in IFA. (B) Principal component analysis MyD88T-KO, IL6RαT-KO, and wild-type control mice.
Discussion
Here, we have shown that T cell-intrinsic MyD88 activation delivers an important signal for the induction of CD4+ T cell responses in vivo. While T cell-specific TLR2 signaling can augment the generation of TH17 responses (Reynolds et al., 2010), we did not observe a major role for T cell-specific TLR4 or TLR9 activation in the generation of either TH1 or TH17 responses, at least not under the conditions used in our experiments. We also did not observe a significant impact of IL-18 signaling on the generation of the CD4+ T cell response. Consistent with this, we found that the expression of the IL-18 receptor is reduced in antigen-specific CD4+ T cells after immunization. IL-18 is usually seen as a cytokine that promotes TH1 responses. However, TH1 responses do not universally depend on this cytokine as TH1 cells can be generated in absence of IL-18 signaling in certain experimental settings (Haring and Harty, 2009; Su et al., 2005). Instead, we found that the impairment of the CD4+ T cell response in MyD88T-KO mice was due to the lack of IL-1 signaling. The role of IL-1 in the differentiation of TH17 cells has been appreciated in recent years and our results confirm the central role for this cytokine in the generation of this T cell subset. Importantly, however, we found that IL-1 also plays an important role in the induction of TH1 responses.
MyD88 and, by extension, TLR activation in T cells had been previously suggested to deliver a survival signal to CD4+ T cells, in part via the upregulation of BCLXL (Gelman et al., 2006; Rahman et al., 2008). Likewise, IL-1 had also been implicated in the provision of a survival signal to CD4+ T cells (Ben-Sasson et al., 2009). The absence of such a signal could have explained the phenotype that we observed in MyD88T-KO mice. However, we found that antigen-specific MyD88-deficient CD4+ T cells proliferated normally and did not exhibit an increased tendency to undergo apoptosis. Consistent with this observation, antigen-specific CD4+ T cells were present at a similar frequency in MyD88T-KO mice as in wild-type controls. Thus, the provision of a survival signal per se is unlikely to be a major function of IL-1 in this context.
Instead, we could restore the TH1 response in MyD88T-KO mice in the absence of Tregs, whereas the TH17 response remained defective, presumably because of the additional requirement of IL-1 for the differentiation of TH17 cells. IL-1 signaling had been previously suggested to compromise the suppressive capability of Tregs (Mercer et al.; O’Sullivan et al., 2006). The normal CD4+ T cell response in MyD88TREG-KO mice suggests that this is not the case and points towards a function for IL-1 in either the naïve or effector CD4+ T cell compartment. The reciprocal relationship between developing TH17 cells and Tregs leads to an increase in iTregs in the absence of the former cells. This is the case in mice that are deficient in IL-6 signaling, at least under some conditions (Bettelli et al., 2006; Korn et al., 2008). A similar function has also been suggested for IL-1 (Chung et al., 2009). As MyD88T-KO mice fail to generate TH17 cells, an increase of iTregs after immunization could have explained the impaired TH1 response in these animals. We used multiple different approaches in order to detect an increase in the frequency of iTregs in MyD88T-KO mice compared to wild-type controls and were unable to find significant evidence for an enhanced generation of these cells in MyD88T-KO mice. It is therefore likely that IL-1 acts on naïve or effector CD4+ T cells in order to render them refractory to the suppressive effects of naturally-occurring nTregs.
The basic phenotype of MyD88T-KO mice, namely that antigen-specific CD4+ T cells of MyD88T-KO mice undergo normal clonal expansion, yet fail to differentiate into TH1 effector cells, is reminiscent of that observed in mixed bone marrow chimeras, which harbor both MyD88-deficient and MHC class II-deficient APCs (Sporri and Reis e Sousa, 2005). In these experiments, the separation of TLR activation and antigen-presentation into distinct APCs is sufficient to support the bystander maturation of the MyD88-deficent APCs by the MyD88-proficient but MHC class II-deficient APCs. Antigen-specific CD4+ T cells activated under such conditions clonally expand, but do not differentiate into TH1 cells (Sporri and Reis e Sousa, 2005). In light of our finding that IL-1 is required to render CD4+ T cells refractory to Treg-mediated suppression, it will be therefore interesting to determine whether the requirement for TLR activation and antigen-presentation in the same APC can be circumvented in the absence of Tregs.
The generation of a TH1 response in MyD88T-KO mice in the absence of Tregs allowed us to determine the role of MyD88 signaling in memory T cells. Surprisingly, MyD88T-KO mice generated antigen-specific CD4+ memory T cells at wild-type levels, even in presence of Tregs. Based on the expression of CXCR5 and PD-1 as well as Ly-6C, memory TH1 cells can be further divided into distinct subsets that represent effector and central memory TH1 cells, respectively (Marshall et al., 2011; Pepper et al., 2011). While the generation of memory CD4+ T cells defined by these markers was also not affected in MyD88T-KO mice, CD4+ memory T cells were impaired in their ability to differentiate into IFNγ-secreting T cells after secondary immunization, even under conditions where Tregs were absent during both the primary and secondary immunization. In the context of a TH1 response, the IL-1 signal seems therefore to carry additional information that goes beyond the release of naïve or effector CD4+ T cells from Treg-mediated suppression. One possibility could be that during the primary immunization, CD4+ T cells fail to fully differentiate into memory CD4+ T cells and thus are present after the primary immunization in a functionally compromised state. Alternatively, the requirement for IL-1 signaling during a secondary TH1 response even in the absence of Tregs may reflect activation requirements that are unique for memory CD4+ T cells. Indeed, it has been suggested that the expansion of memory CD4+ T cells is sensitive to IL-1 signaling after secondary challenge with antigen (Luqman et al., 1992). This aspect may be an intrinsic feature of memory CD4+ T cells that is independent of their control by Tregs and may thus explain the lack of a memory CD4+ T cell response in MyD88T-KO mice even after transient depletion of Tregs during the secondary challenge. Further experiments will be necessary to distinguish between these possibilities.
Our study addressed the function of CD4+ T cell-specific MyD88 signaling in the context of immunizations using TLR ligands as adjuvants. IL-1 induced under these conditions is required for the induction of a TH1 response. However, it is certainly possible that other cytokines or membrane-bound signals carry similar information and are also releasing CD4+ T cell from Treg-mediated suppression, particularly as a consequence of the activation of other PRR systems. In this regard, it is interesting to note that IL-4 and IL-15 have been proposed to operate in this manner (Ben Ahmed et al., 2009; Thornton et al., 2004). Thus, cytokines induced by different types of infections may function in a way analogous to IL-1 by making naïve or effector T cells refractory to Treg-mediated suppression. Therefore, it remains to be seen to what extent IL-1 is required to overcome Treg-mediated suppression upon live infections.
IL-6 had been previously implicated in the release of CD4+ T cells from Treg-mediated suppression (Pasare and Medzhitov, 2003). The observation that MyD88T-KO and IL6RαT-KO mice share a similar overall phenotype supports the notion that that IL-1 and IL-6 both cooperate in this process in vivo (Nish et al., submitted). IL-6 has been reported to induce the expression of the IL-1R on CD4+ T cells (Chung et al., 2009). Our own gene expression analysis on antigen-specific CD4+ T cells from IL6RαT-KO mice also suggests that these cells express reduced levels of the IL-1R, thus raising the possibility that IL-6 serves as a permissive signal for IL-1 signaling. However, several lines of evidence point to a distinct function of these cytokines in CD4+ T cell response. First, the phenotype of IL6RT-KO mice tends to be stronger than that of MyD88T-KO mice and we would expect the opposite to be true if IL-6 was merely a permissive signal. Moreover, we noticed subtle but important differences in the CD4+ T cell response between these strains as the frequency of antigen-specific CD4+ T cells was reduced in IL6RαT-KO mice but not in MyD88T-KO mice. Finally, the comparison of the gene expression profiles of antigen-specific CD4+ T cells from IL6RαT-KO mice and MyD88T-KO mice revealed little overlap in the differentially expressed genes. It therefore seems more likely that IL-1 and IL-6 integrate different aspects of the immunological challenge. This notion is particularly interesting in light of the unique mechanism involved in IL-1 secretion, namely the requirement for inflammasome activation (Strowig et al., 2012). Inflammasome activation requires both a TLR signal and a second signal that can be triggered by microbial virulence activities (Brodsky and Monack, 2009). Thus, IL-1 can “report” microbial virulence activity to CD4+ T cells (Brodsky et al., 2010). IL-1 also signals the presence of live infections, at least under some circumstances (Sander et al., 2011). The critical role of IL-1 in CD4+ T cell responses may therefore reflect its unique expression requirements: while TLR activation reports on microbial origin of antigens, IL-1 expression may indicate the nature or the status of microbial threat (live versus dead, innocuous versus virulent). While antigen origin can be either microbial or not, virulence is a graded function (pathogen can be more or less virulent) as is the viability at the level of a microbial population. Interestingly, CD4+ T cell susceptibility to Treg-mediated suppression also appears to be a graded phenomenon and can be shifted in either direction by varying the conditions of T cell activation (Shevach, 2009). Therefore, the level of IL-1 production may calibrate CD4+ T cell responses by controlling susceptibility of CD4+ T cells to suppression by Tregs. Notably in this regard, the commonly used adjuvants alum and mineral oil (in Freund’s adjuvants), may function as mimics of microbial virulence or viability signals that normally induce inflammasome activation, while microbial TLR and other PRR ligands instruct the microbial origin of the admixed antigens. Consistent with this notion, at least some adjuvants are known to activate inflammasomes (Lambrecht et al., 2009). Both types of signals must be present in vaccine formulations for the efficient induction of CD4+ T cell responses.
Material and Methods
Generation of the conditional MyD88 allele
The targeting vector was based on a derivative of the pEZ-flox vector that contained a FRT-flanked neoR cassette and a diphtheria toxin gene for negative selection (Schenten et al., 2002). PCR fragments containing the appropriate restriction sites were amplified from a C57BL/6-derived BAC encoding MyD88 and inserted into the targeting vector. The culture and transfections of C57BL/6-derived Bruce4 embryonic stem (ES) cells has been published previously. Correctly targeted ES cell clones were identified by Southern hybridization of EcoRI-digested genomic DNA with probe A, which was PCR amplified from a MyD88-encoding BAC using primers 5′-ACTAAACCCGGGGATGGGGGTG and 5′CTTCCCTTAAGTCTCCTGCCAGCC. Co-integration of the 2nd loxP site was confirmed by Southern hybridization of HinDIII-digested ES cell DNA using probe C. That probe was amplified using primers 5′-GACAAACGCCGGAACTTTTCGATGG and 5′-AGAAGTACAAAGTACAAACACGAGCCC. Deletion of the neoR cassette was achieved in vitro by transfection of a FLIP recombinase encoding plasmid and confirmed by Southern blotting of EcoRI-digested ES cell DNA using probe A. A correctly targeted ES cell clones was injected C57BL/6 albino-derived blastocysts, which were implanted into pseudo-pregnant C57BL/6 foster mice in order to generate chimeric mice and achieve germline transmission. Mice carrying the floxed MyD88 allele (MyD88FL mice) were viable and born at Mendelian ratios.
Mice
In order to ablate MyD88 function specifically in T cells, MyD88FL mice were intercrossed with mice carrying a CD4-cre allele (Lee et al., 2001). MyD88FL/FL; CD4-cre mice were designated MyD88T-KO mice, while MyD88FL/FL or C57BL/6 mice were collectively termed MyD88WT mice. For some experiments, CD4-cre mice were used as controls in order to monitor for negative effects of Cre activity. MyD88TREG-KO mice carrying a specific deletion of MyD88 in FoxP3+ Tregs were generated by intercrossing MyD88FL mice with mice carrying a FoxP3-cre allele (Rubtsov et al., 2008). IL-1RKO mice, IL-18RKO mice, and TCRβKO and TCRδKO compound mutant mice were purchased from the Jackson Laboratory (Glaccum et al., 1997; Hoshino et al., 1999). TLR9KO mice and TLR2KO and TLR4KO compound mutant mice were maintained at the Yale School of Medicine. All mice were kept on a C57BL/6 genetic background. The mice were housed in the Yale Animal Resources Center (YARC) and the experiments were done with approval by the Yale University Institutional Animal Care and Use Committee (IACUC).
Immunizations
The mice were immunized in both feet with either 100 μg/mouse Ovalbumin (OVA, Sigma) or 100 μg/mouse 2W1S peptide (EAWGALANWAVDSA, Genscript) plus 10 μg/mouse LPS (Sigma) emulsified in incomplete Freund’s adjuvant (IFA) as carrier (Sigma). For some experiments, LPS was replaced with either 20 μg/mouse CpG DNA 1826 (Keck Biotechnology Resource Laboratory, Yale Medical School), 50 μg/mouse polyI:C (Invivogen), 20 μg/mouse Peptidoglycan (Invivogen), or 150 μg/mouse heat-inactivated M. tuberculosis (Sigma). The effects of contaminating endotoxin in the OVA preparations were controlled by using endotoxin-free OVA (Hyglos/Biovendor) in key experiments. CD4+ memory responses were induced similarly with the exception that one foot was used for the primary immunization, while the other foot was used for the secondary immunization.
Antibodies and other reagents
All standard antibodies for flow cytometry were purchased from either BD Biosciences or Ebiosciences. The αHelios antibody (clone 22F6) was obtained from Biolegend, FITC-PNA was purchased from Biovector, and the α-activated caspase-3 antibody was obtained from R&D Systems. PE-conjugated 2W:I-Ab tetramers were a gift from Marc Jenkins (University of Minnesota, Minneapolis, MN). BrdU incorporation was measured using the BrdU staining kit from BD Bioscences. The α-CD25 antibody PC61 was purified from hybridoma cells adapted to serum-free medium and grown in Cellline 2-compartment bioreactors (BD Biosciences). MyD88 protein was detected by Western blotting using a goat α-MyD88 antibody (R&D Systems).
T cell assay
CD4+ T cells were isolated from the popliteal and inguinal lymph nodes 7 days after immunization by magnetic cell sorting using αCD4 beads (Miltenyi Biotech). The cells were seeded in round-bottom 96-well plates at a concentration of 100,00 cells/well in the presence of 300,000 irradiated splenocytes in the presence of serial dilutions of OVA, starting at 900 μg/ml. Proliferation was measured on day 4 following the addition of 3H-thymidine for the last 24 hours and the secretion of IFNγ and IL-17 was measured by ELISA or intracellular flow cytometry.
Treg depletion
Tregs were depleted in vivo with 150 μg/mouse of an αCD25 antibody (clone PC61) by intravenous injection 3 days prior to immunization. Depletion of Tregs was confirmed on the day of immunization by flow cytometry upon staining of CD4+ T cells from the blood with an αCD25 antibody (clone 7D4).
Bone marrow chimeras
Bone marrow isolated from TCRβKO and TCRδKO compound mutant mice and mixed in a ratio of 3:1 with bone marrow isolated from C57BL/6 wild-type mice, IL-1RKO mice, IL-18RKO mice, TLR9KO mice, or TLR2KO and TLR4KO compound mutant mice. 10 × 106 total cells/mouse were injected into Rag2KO mice that had been lethally irradiated with 900 rad 1 day prior to the bone marrow transfer. The reconstitution of the hematopoetic system was confirmed after 6 weeks by flow cytometry.
RNAseq
Antigen-specific CD4+ T cells were isolated by FACS from a total of 15–20 immunized mice per genotype. RNA was isolated from approximately 100,000 cells per genotype using the RNeasy isolation kit (Qiagen) and processed and sequenced by the Yale Center for Genome Analysis (YCGA).
Statistical Analysis
Data were analyzed with a Student’s t-test using Prism4 (Graphpad Software). In some case with abnormal distribution, the data were also analyzed with a Mann-Whitney test using the same software. Unless not mentioned specifically, data are shown as mean ± SD. P values: *, p ≤ 0.05; **, p ≤ 0.005; ***, p ≤ 0.0005; ns., not significant. PCA analysis of gene expression data was performed using R (2.15.3).
Supplementary Material
Supplementary Figure 1. Generation of MyD88T-KO mice. (A). Targeting scheme of the MyD88 locus. Exons 3–5 of MyD88, encoding the TLR domain, were flanked by loxP sites, thus rendering these exons susceptible to Cre-mediated recombination. Shown are the wild-type locus, the targeting vector, the targeted locus before and after removal of the FRT-flanked neor cassette upon FLIP-mediated recombination, and the deleted locus are shown. Black boxes, exons; region of homology, bold lines; shaded triangles, loxP sites; shaded ovals, FRT sites; shaded boxes, selection markers of the targeting vector (DTA, fragment A of diphtheria toxin; neor, neomycin resistance); RI, EcoRI sites, and H, HinDIII sites. The lengths of various DNA fragments following restriction digest with either EcoRI or HinDIII using Southern probes A and C are indicated. (B, C). Southern blot analysis of genomic DNA from correctly targeted ES cells. Southern blot of EcoRI-digested genomic DNA using probe A confirms correct targeting of the MyD88 locus (B), whereas Southern blot analysis of HinDIII-digested genomic DNA using probe C confirms the co-integration of the 2nd loxP site (C). (D). Cre-mediated deletion of MyD88 protein in T cells from MyD88Fl/FL; CD4-cre mice (MyD88T-KO mice), determined by Western blot analysis of T and B cells from MyD88T-KO and wild-type controls.
Supplementary Figure 2. ature of 2W:IA(b)+ CD4+ T cells from MyD88T-KO and wild-type control mice. Frequency of antigen-specific 2W:I-A(b)+ CXCR5+ PD-1+ TFH cells in the draining lymph nodes of MyD88T-KO and wild-type control mice on day 7 after immunization with 2W peptide + LPS in IFA.
Supplementary Figure 3. Release of IL-1 by irradiated APCs in vitro does not account for defective CD4+ T cell response in MyD88T-KO mice. (A) Release of IL-1α and IL-1β by irradiated APCs. Splenocytes were cultured at a high density (4–5 fold higher than in standard CD4+ T cell assays) in the presence of CD4+ T cell from immunized mice and OVA for 3 days. Splenocytes stimulated with LPS and ATP were used as positive control. nd, not detected. (B) Proliferation of CD4+ T cells from MyD88T-KO mice and wild-type controls after restimulation in the presence of irradiated splenocytes from Caspase-1-deficient mice. CD4+ T cells were isolated from the draining lymph nodes 7 days after immunization in the feet with OVA + LPS in IFA and re-stimulated with OVA in the presence of irradiated splenocytes. Proliferation was measured 3 days later by 3H-thymidine incorporation. (C). Cytokine secretion of CD4+ T cells following restimulation with OVA in the presence of Caspase-1-deficient APCs. Left panels, representative experiment; right panels, statistical summary of all experiments.
Supplementary Figure 4. CD4+ T cell response in MyD88T-KO and wild-type control mice following protein immunization with several TLR stimulants. (A) CD4+ T cell response following immunization with OVA + CpG DNA in IFA. Proliferation and IFNγ secretion of CD4+ T cells isolated after 7 days and restimulated with OVA for 3 days in the presence of irradiated splenocytes was measured by 3H-thymidine incorporation and ELISA, respectively. (B). CD4+ T cell response following immunization with OVA + polyIC in IFA. Proliferation and IFNγ secretion was measured as described before. (C). CD4+ T cell response following immunization with OVA in CFA. For all experiments, a representative experiment is shown.
Supplementary Figure 5. Cellular composition of the draining lymph nodes after immunization. Following immunization of MyD88T-KO and wild-type control mice with either OVA or 2W peptide plus LPS in IFA in the footpads, the draining inguinal and popliteal lymph nodes were isolated on day 7 post immunization and the cells stained for the following cell populations by flow cytometry: (A). T and B cells. (B). CD4+ and CD8+ T cells (C). F4/80+ macrophages and CD11c+ dendritic cells (D). F4/80+ Gr1+ macrophages and F4/80− Gr1+ neutrophils. A representative experiment is shown.
Supplementary Figure 6. Statistical representation of the CD4+ T cell response following immunization with OVA + LPS in IFA in bone marrow chimeras carrying a wild-type, TLR2/TLR4-, IL1R-, or IL18R-deficient T cell compartment. CD4+ T cell response of bone marrow chimeras harboring bone marrow of TCRβKO mice mixed with bone marrow of either TLR2KO/TLR4KO, IL-1RKO, IL-18RKO or wild-type control mice. The mice were immunized with OVA + CpG DNA in IFA. Proliferation and IFNγ secretion of isolated CD4+ T cells restimulated with OVA in vitro in the presence of irradiated splenocytes were measured by 3H-thymidine incorporation (A) and ELISA (B), respectively. Shown are the combined data of at least three independent experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Supplementary Figure 7. Requirement for T cell-specific IL-1 signaling in CD4+ T cells in order to overcome Treg-mediated suppression following immunization with OVA + CpG DNA in IFA. (A, B). CD4+ T cell response of bone marrow chimeras harboring bone marrow of TCRβKO mice mixed with bone marrow of either IL-1RKO, TLR9KO or wild-type control mice. The mice were immunized with OVA + CpG DNA in IFA. Proliferation and IFNγ secretion of isolated CD4+ T cells restimulated with OVA in vitro in the presence of irradiated splenocytes were measured by 3H-thymidine incorporation (A) and ELISA (B), respectively. (C, D). Restoration of the TH1 response in MyD88T-KO mice immunized with OVA + CpG DNA in IFA after prior transient depletion of CD25+ Tregs. Proliferation (C) and IFNγ secretion (D) of isolated CD4+ T cells after restimulation were measured as described before.
Supplementary Figure 8. Statistical representation of the CD4+ T cell response following immunization with OVA + CpG DNA in IFA in bone marrow chimeras carrying a wild-type, TLR9-, or IL1R-deficient T cell compartment. Proliferation (A) and IFNγ secretion (B) of restimulated CD4+ T cells as in Supplementary Figure 8A, B. Shown are the combined data of all experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Supplementary Figure 9. Statistical representation of the CD4+ T cell response in the presence or absence of Tregs. (A, B) Statistical representation of CD4+ T cell response following immunization with OVA + LPS in IFA. When indicated, Tregs were transiently depleted by administrating an αCD25 antibody three days prior to the immunization. CD4+ T cells were isolated from the draining lymph nodes 7 days after the immunization and re-stimulated with 900 μg/ml OVA in vitro in the presence of irradiated splenocytes. Proliferation was measured three days later by 3H-thymidine incorporation (A) and cytokine secretion of CD4+ T cells was measured by ELISA (B). (C, D) Statistical representation of the CD4+ T cell response following immunization with OVA + CpG DNA in IFA. Shown are the proliferation (C) and cytokine secretion (D) after restimulation of CD4+ T cells with OVA in vitro. (A–D) Shown are the combined data of at least three independent experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Supplementary Figure 10. Statistical representation of the CD4+ T cell memory response following immunization with OVA + LPS in IFA. (A, B) Statistical representation of the CD4+ T cell response following immunization with OVA + LPS in IFA. Mice were immunized again at least 30 days after the primary immunization. When indicated, Tregs were transiently depleted by administrating an αCD25 antibody three days prior to both the primary and secondary immunization. CD4+ T cells were isolated from the draining lymph nodes 7 days after the secondary immunization and re-stimulated with 900 μg/ml OVA in vitro in the presence of irradiated splenocytes. Proliferation was measured three days later by 3H-thymidine incorporation (A) and cytokine secretion of CD4+ T cells was measured by ELISA (B). Shown are the combined data of all experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Highlights.
Ablation of MyD88 in T cells impairs both Th1 and Th17 response
Defect is due to abrogated IL-1 signaling
Th1 response is recovered in the absence of Tregs.
IL-1 acts on naïve or effector CD4+ T cells and not on Tregs.
Acknowledgments
We would like to thank Marc Jenkins for reagents, Alexander Rudensky and Richard Flavell for mouse strains, Jorge Henao-Mejia for technical advice, Sophie Cronin and Charles Annicelli for expert mouse work, Gouzel Tokmoulina for cell sorting, Scott Pope for bioinformatic expertise and discussion, and Noah Palm and Jelena Bezbradica for discussion and critical reading of the manuscript. This work was funded by the Howard Hughes Medical Institute and the NIH through grant AI055502. D. S. was supported by a training grant of the NIH and then by an Irvington Fellowship of the Cancer Research Institute. S.N. was a recipient of a NIH training grant.
Footnotes
Contributions
D. S. generated the conditional MyD88 allele, designed and performed experiments, and wrote the manuscript. S. A. N. designed and performed experiments and wrote the manuscript; S. Y. did the genotyping and purification of antibodies, X. Y. and H. Z. helped with the bioinformatics analysis, H. K. L., I. B., L. P., and B. Y. helped with the design and execution of specific experiments; F. T. W. and J. C. B. provided key reagents, and R. M. provided overall intellectual direction and wrote the manuscript.
References
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abdeladhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. 2009;182:6763–6770. doi: 10.4049/jimmunol.0801792. [DOI] [PubMed] [Google Scholar]
- Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Monack D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin Immunol. 2009;21:199–207. doi: 10.1016/j.smim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns B, Maile R, Barnes CM, Frelinger JA, Meyer AA. Increased Toll-like receptor 4 expression on T cells may be a mechanism for enhanced T cell response late after burn injury. J Trauma. 2006;61:293–298. doi: 10.1097/01.ta.0000228969.46633.bb. discussion 298–299. [DOI] [PubMed] [Google Scholar]
- Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, Rudensky AY. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, Conduah D, Xu R, Abreu MT. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol. 2008;180:1886–1894. doi: 10.4049/jimmunol.180.3.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065–6073. doi: 10.4049/jimmunol.172.10.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, Livingston DJ, Peschon JJ, Morrissey PJ. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol. 1997;159:3364–3371. [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, Corr M, Katakura K, Eckman L, Lee J, Raz E. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. 2010;120:570–581. doi: 10.1172/JCI40055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, Paul W. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A. 2009;106:13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring JS, Harty JT. Interleukin-18-related genes are induced during the contraction phase but do not play major roles in regulating the dynamics or function of the T-cell response to Listeria monocytogenes infection. Infect Immun. 2009;77:1894–1903. doi: 10.1128/IAI.01315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Tsutsui H, Kawai T, Takeda K, Nakanishi K, Takeda Y, Akira S. Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol. 1999;162:5041–5044. [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2011;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, Vollmar P, Stritesky GL, Kaplan MH, Waisman A, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:18460–18465. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I, Wei S, Vatan L, Escara-Wilke J, Szeliga W, Keller ET, Zou W. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J Immunol. 2007;179:1423–1426. doi: 10.4049/jimmunol.179.3.1423. [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, Kool M, Willart MA, Hammad H. Mechanism of action of clinically approved adjuvants. Curr Opin Immunol. 2009;21:23–29. doi: 10.1016/j.coi.2009.01.004. [DOI] [PubMed] [Google Scholar]
- LaRosa DF, Gelman AE, Rahman AH, Zhang J, Turka LA, Walsh PT. CpG DNA inhibits CD4+CD25+ Treg suppression through direct MyD88-dependent costimulation of effector CD4+ T cells. Immunol Lett. 2007;108:183–188. doi: 10.1016/j.imlet.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luqman M, Greenbaum L, Lu D, Bottomly K. Differential effect of interleukin 1 on naive and memory CD4+ T cells. Eur J Immunol. 1992;22:95–100. doi: 10.1002/eji.1830220115. [DOI] [PubMed] [Google Scholar]
- Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity. 2011;35:633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer F, Kozhaya L, Unutmaz D. Expression and function of TNF and IL-1 receptors on human regulatory T cells. PLoS One. 5:e8639. doi: 10.1371/journal.pone.0008639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–213. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- O’Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, Kay TW, Thomas R. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol. 2006;176:7278–7287. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, Wherry EJ, Kaech SM, Turka LA. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J Immunol. 2008;181:3804–3810. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, Dong C. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32:692–702. doi: 10.1016/j.immuni.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenten D, Gerlach VL, Guo C, Velasco-Miguel S, Hladik CL, White CL, Friedberg EC, Rajewsky K, Esposito G. DNA polymerase kappa deficiency does not affect somatic hypermutation in mice. Eur J Immunol. 2002;32:3152–3160. doi: 10.1002/1521-4141(200211)32:11<3152::AID-IMMU3152>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Schenten D, Medzhitov R. The control of adaptive immune responses by the innate immune system. Adv Immunol. 2011;109:87–124. doi: 10.1016/B978-0-12-387664-5.00003-0. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102. doi: 10.1038/nri2691. [DOI] [PubMed] [Google Scholar]
- Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- Su SB, Silver PB, Grajewski RS, Agarwal RK, Tang J, Chan CC, Caspi RR. Essential role of the MyD88 pathway, but nonessential roles of TLRs 2, 4, and 9, in the adjuvant effect promoting Th1-mediated autoimmunity. J Immunol. 2005;175:6303–6310. doi: 10.4049/jimmunol.175.10.6303. [DOI] [PubMed] [Google Scholar]
- Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Robinson AW, Phillips RS. Expression of the IL-1 receptor discriminates Th2 from Th1 cloned CD4+ T cells specific for Plasmodium chabaudi. Immunology. 1994;81:216–221. [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol. 2004;34:366–376. doi: 10.1002/eji.200324455. [DOI] [PubMed] [Google Scholar]
- Tomita T, Kanai T, Fujii T, Nemoto Y, Okamoto R, Tsuchiya K, Totsuka T, Sakamoto N, Akira S, Watanabe M. MyD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J Immunol. 2008;180:5291–5299. doi: 10.4049/jimmunol.180.8.5291. [DOI] [PubMed] [Google Scholar]
- Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Generation of MyD88T-KO mice. (A). Targeting scheme of the MyD88 locus. Exons 3–5 of MyD88, encoding the TLR domain, were flanked by loxP sites, thus rendering these exons susceptible to Cre-mediated recombination. Shown are the wild-type locus, the targeting vector, the targeted locus before and after removal of the FRT-flanked neor cassette upon FLIP-mediated recombination, and the deleted locus are shown. Black boxes, exons; region of homology, bold lines; shaded triangles, loxP sites; shaded ovals, FRT sites; shaded boxes, selection markers of the targeting vector (DTA, fragment A of diphtheria toxin; neor, neomycin resistance); RI, EcoRI sites, and H, HinDIII sites. The lengths of various DNA fragments following restriction digest with either EcoRI or HinDIII using Southern probes A and C are indicated. (B, C). Southern blot analysis of genomic DNA from correctly targeted ES cells. Southern blot of EcoRI-digested genomic DNA using probe A confirms correct targeting of the MyD88 locus (B), whereas Southern blot analysis of HinDIII-digested genomic DNA using probe C confirms the co-integration of the 2nd loxP site (C). (D). Cre-mediated deletion of MyD88 protein in T cells from MyD88Fl/FL; CD4-cre mice (MyD88T-KO mice), determined by Western blot analysis of T and B cells from MyD88T-KO and wild-type controls.
Supplementary Figure 2. ature of 2W:IA(b)+ CD4+ T cells from MyD88T-KO and wild-type control mice. Frequency of antigen-specific 2W:I-A(b)+ CXCR5+ PD-1+ TFH cells in the draining lymph nodes of MyD88T-KO and wild-type control mice on day 7 after immunization with 2W peptide + LPS in IFA.
Supplementary Figure 3. Release of IL-1 by irradiated APCs in vitro does not account for defective CD4+ T cell response in MyD88T-KO mice. (A) Release of IL-1α and IL-1β by irradiated APCs. Splenocytes were cultured at a high density (4–5 fold higher than in standard CD4+ T cell assays) in the presence of CD4+ T cell from immunized mice and OVA for 3 days. Splenocytes stimulated with LPS and ATP were used as positive control. nd, not detected. (B) Proliferation of CD4+ T cells from MyD88T-KO mice and wild-type controls after restimulation in the presence of irradiated splenocytes from Caspase-1-deficient mice. CD4+ T cells were isolated from the draining lymph nodes 7 days after immunization in the feet with OVA + LPS in IFA and re-stimulated with OVA in the presence of irradiated splenocytes. Proliferation was measured 3 days later by 3H-thymidine incorporation. (C). Cytokine secretion of CD4+ T cells following restimulation with OVA in the presence of Caspase-1-deficient APCs. Left panels, representative experiment; right panels, statistical summary of all experiments.
Supplementary Figure 4. CD4+ T cell response in MyD88T-KO and wild-type control mice following protein immunization with several TLR stimulants. (A) CD4+ T cell response following immunization with OVA + CpG DNA in IFA. Proliferation and IFNγ secretion of CD4+ T cells isolated after 7 days and restimulated with OVA for 3 days in the presence of irradiated splenocytes was measured by 3H-thymidine incorporation and ELISA, respectively. (B). CD4+ T cell response following immunization with OVA + polyIC in IFA. Proliferation and IFNγ secretion was measured as described before. (C). CD4+ T cell response following immunization with OVA in CFA. For all experiments, a representative experiment is shown.
Supplementary Figure 5. Cellular composition of the draining lymph nodes after immunization. Following immunization of MyD88T-KO and wild-type control mice with either OVA or 2W peptide plus LPS in IFA in the footpads, the draining inguinal and popliteal lymph nodes were isolated on day 7 post immunization and the cells stained for the following cell populations by flow cytometry: (A). T and B cells. (B). CD4+ and CD8+ T cells (C). F4/80+ macrophages and CD11c+ dendritic cells (D). F4/80+ Gr1+ macrophages and F4/80− Gr1+ neutrophils. A representative experiment is shown.
Supplementary Figure 6. Statistical representation of the CD4+ T cell response following immunization with OVA + LPS in IFA in bone marrow chimeras carrying a wild-type, TLR2/TLR4-, IL1R-, or IL18R-deficient T cell compartment. CD4+ T cell response of bone marrow chimeras harboring bone marrow of TCRβKO mice mixed with bone marrow of either TLR2KO/TLR4KO, IL-1RKO, IL-18RKO or wild-type control mice. The mice were immunized with OVA + CpG DNA in IFA. Proliferation and IFNγ secretion of isolated CD4+ T cells restimulated with OVA in vitro in the presence of irradiated splenocytes were measured by 3H-thymidine incorporation (A) and ELISA (B), respectively. Shown are the combined data of at least three independent experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Supplementary Figure 7. Requirement for T cell-specific IL-1 signaling in CD4+ T cells in order to overcome Treg-mediated suppression following immunization with OVA + CpG DNA in IFA. (A, B). CD4+ T cell response of bone marrow chimeras harboring bone marrow of TCRβKO mice mixed with bone marrow of either IL-1RKO, TLR9KO or wild-type control mice. The mice were immunized with OVA + CpG DNA in IFA. Proliferation and IFNγ secretion of isolated CD4+ T cells restimulated with OVA in vitro in the presence of irradiated splenocytes were measured by 3H-thymidine incorporation (A) and ELISA (B), respectively. (C, D). Restoration of the TH1 response in MyD88T-KO mice immunized with OVA + CpG DNA in IFA after prior transient depletion of CD25+ Tregs. Proliferation (C) and IFNγ secretion (D) of isolated CD4+ T cells after restimulation were measured as described before.
Supplementary Figure 8. Statistical representation of the CD4+ T cell response following immunization with OVA + CpG DNA in IFA in bone marrow chimeras carrying a wild-type, TLR9-, or IL1R-deficient T cell compartment. Proliferation (A) and IFNγ secretion (B) of restimulated CD4+ T cells as in Supplementary Figure 8A, B. Shown are the combined data of all experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Supplementary Figure 9. Statistical representation of the CD4+ T cell response in the presence or absence of Tregs. (A, B) Statistical representation of CD4+ T cell response following immunization with OVA + LPS in IFA. When indicated, Tregs were transiently depleted by administrating an αCD25 antibody three days prior to the immunization. CD4+ T cells were isolated from the draining lymph nodes 7 days after the immunization and re-stimulated with 900 μg/ml OVA in vitro in the presence of irradiated splenocytes. Proliferation was measured three days later by 3H-thymidine incorporation (A) and cytokine secretion of CD4+ T cells was measured by ELISA (B). (C, D) Statistical representation of the CD4+ T cell response following immunization with OVA + CpG DNA in IFA. Shown are the proliferation (C) and cytokine secretion (D) after restimulation of CD4+ T cells with OVA in vitro. (A–D) Shown are the combined data of at least three independent experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.
Supplementary Figure 10. Statistical representation of the CD4+ T cell memory response following immunization with OVA + LPS in IFA. (A, B) Statistical representation of the CD4+ T cell response following immunization with OVA + LPS in IFA. Mice were immunized again at least 30 days after the primary immunization. When indicated, Tregs were transiently depleted by administrating an αCD25 antibody three days prior to both the primary and secondary immunization. CD4+ T cells were isolated from the draining lymph nodes 7 days after the secondary immunization and re-stimulated with 900 μg/ml OVA in vitro in the presence of irradiated splenocytes. Proliferation was measured three days later by 3H-thymidine incorporation (A) and cytokine secretion of CD4+ T cells was measured by ELISA (B). Shown are the combined data of all experiments. Each dot represents one mouse and the experiments were normalized to the first replicate of triplicates of the wild-type mice.