

Abstract

DAPK1 and ZIPK (also called DAPK3) are closely related serine/threonine protein kinases that regulate programmed cell death and phosphorylation of non-muscle and smooth muscle myosin. We have developed a fluorescence linked enzyme chemoproteomic strategy (FLECS) for the rapid identification of inhibitors for any element of the purinome and identified a selective pyrazolo[3,4-d]pyrimidinone (HS38) that inhibits DAPK1 and ZIPK in an ATP-competitive manner at nanomolar concentrations. In cellular studies, HS38 decreased RLC20 phosphorylation. In ex vivo studies, HS38 decreased contractile force generated in mouse aorta and rabbit ileum, and calyculin A stimulated arterial muscle by decreasing RLC20 and MYPT1 phosphorylation. The inhibitor also promoted relaxation in Ca2+-sensitized vessels. A close structural analogue (HS43) with 5-fold lower affinity for ZIPK produced no effect on cells or tissues. These findings are consistent with a mechanism of action wherein HS38 specifically targets ZIPK in smooth muscle. The discovery of HS38 provides a lead scaffold for the development of therapeutic agents for smooth muscle related disorders and a chemical means to probe the function of DAPK1 and ZIPK across species.
The Death Associated Protein Kinase (DAPK) family comprises three closely related serine/threonine kinases: DAPK1, DAPK2 (also called DRP-1), and Zipper-interacting Protein Kinase or ZIPK (also called DAPK3). In vivo they mediate cell death through transmission of apoptotic and autophagic signals1,2 and highly regulate both non-muscle and smooth muscle (SM) myosin phosphorylation.3 DAPK1 and ZIPK are attractive drug targets for the attenuation of ischemia-reperfusion induced tissue injury4−7 and for smooth muscle related disorders.3,8
DAPK1 was originally identified as a positive mediator of interferon-induced programmed cell death. Inhibition of the DAPK gene reduces the susceptibility of HeLa cells to apoptosis.9 This finding and subsequent reports that all three members of the kinase family display tumor and metastasis suppressor properties2,10,11 sparked significant interest in the structure, function, and physiological roles of the DAPKs and their relation to human disease.1 DAPK1 and ZIPK also serve as negative regulators of late stage inflammatory gene expression in response to interferon γ, another possible contributing factor to the onset of cancer.12
DAPKs also promote apoptotic cell death from ischemia-reperfusion events and acute brain injury in both kidney and brain tissue. Significant effort has been directed toward the discovery of DAPK inhibitors that can prevent cell death under these circumstances. Deletion of the kinase domain from DAPK1 reduces tubular cell apoptosis following renal ischemia-reperfusion events.5 In neuronal cells, DAPK is present in a deactivated, phosphorylated, and DANGER-associated state13 and becomes rapidly dephosphorylated and activated in response to cerebral ischemia.6
We have focused on the role of ZIPK in the regulation of both non-muscle and SM myosin phosphorylation.3,14 In SM, ZIPK positively regulates contractile activity by phosphorylating both the targeting subunit of myosin light chain phosphatase (MYPT1) and regulatory myosin light chain RLC20), promoting Ca2+ sensitization in response to hormones and agonists.15−17 Because Ca2+ sensitization is a possible cause of diseases associated with SM dysfunction, including hypertension, bronchial asthma, preterm labor, irritable bowel syndrome, and erectile dysfunction, ZIPK is an attractive target for the development of therapeutics for these disorders.3,8
Genetic models of ZIPK knockout have yet to be developed and may be complicated by the finding that in certain rodent species (mouse and rat) the kinase exhibits up to 40 nonconserved substitutions in its C-terminal domain. Several of the substituted sites are regulated by phosphorylation, and their mutation profoundly alters the subcellular localization of the kinase.18 However, the evolutionary reasons for these substitutions are not clear, since the kinase is otherwise highly conserved from Platypus to man.19 We have therefore focused on developing inhibitors of DAPK1 and ZIPK to serve as therapeutic agents and to help delineate the role of the kinases across species.
To discover potent and selective inhibitors of ZIPK, we developed FLECS, an expansion of proteome mining in which inhibitors of a fluorescently tagged target protein can be rapidly screened against a background of the entire purinome. Proteome mining is a well-established competitive equilibrium-based screen in which hundreds of purine-utilizing proteins can be assayed simultaneously to distinguish intrinsically more selective chemical starting points compared with those derived by more conventional small molecule screens.20,21 Proteome mining formed the basis of the chemoproteomic strategy used to discover SNX5422, a highly selective inhibitor of Hsp90.22 FLECS expands upon this original chemoproteomic strategy by utilizing a fluorescence-linked enzyme target, allowing drug candidates to be screened against specific protein targets without purification from crude cell lysates and allowing for rapid data collection with a fluorescence plate reader. Here we report the use of FLECS to discover a potent, selective, and ATP-competitive inhibitor HS38) of DAPK1 (Kd = 300 nM) and ZIPK (Kd = 280 nM). The physiological effects of HS38 smooth muscle cells and tissues were investigated.
RESULTS AND DISCUSSION
Discovery of HS38
FLECS can be utilized for the rapid and general discovery of ATP-competitive inhibitors of any fluorescence linked purine utilizing protein (Figure 1). Using this approach a potent and selective inhibitor of DAPK1 and ZIPK, (2-((1-(3-chlorophenyl)-4-oxo-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)thio)propanamide (HS38), was identified from screening a strategically chosen small molecule library against full length ZIPK fused at its N-terminus to green fluorescent protein (GFP-ZIPK). The 3379 member library was chosen from commercially available small molecules on the basis of selection criteria that simultaneously maximized structural diversity and similarity to known inhibitors of purine binding while minimizing chemical liabilities (e.g., electrophilic centers, labile moieties).22 FLECS is currently being used by multiple collaborating investigators as a discovery platform for inhibitors of ATP binding proteins (Supplementary Figure 1). The assay probes the ability of drug candidates to disrupt binding between elements of the purinome and immobilized ATP.23 Because drug candidates must competitively elute protein targets from local concentrations of immobilized ATP that are similar to those found in cells (~10 mM), positive hits from this screening method are predisposed to be physiologically active. Because the fluorescent linked target protein can be assayed in the presence of multiple elements of the purinome, FLECS can simultaneously probe the relative potency and selectivity of each drug candidate. In effect, the same eluents that are analyzed for fluorescence can then be analyzed by SDS-PAGE with silver stain or Western blotted to confirm target elution or to visualize off target protein elution.
Figure 1.

Schematic representing the fluorescence linked enzyme chemoproteomic strategy (FLECS). (A) (1) γ-Linked ATP sepharose beads were mixed with crude mammalian cell lysate containing GFPZIPK. (2) Charged beads were distributed into 96-well filter plates. (3) Drug candidates or ATP solutions were added to each well. (4) Eluates were separated into a filter plate by centrifugation. (5) The fluorescence of each eluate was determined, and a fluorescence histogram was generated. All wells containing >5000 fluorescence counts (2.5 × background) were considered to contain potential hits. Soluble ATP was used as a positive control. (6) Eluate from each hitcontaining well was Western blotted for GFP-ZIPK, and hits were refined on the basis of band intensity. (B) Summary of screen results and structure of HS38.
For the current screening effort, GFP-ZIPK was expressed in mammalian cells (HEK293). Crude lysates were loaded directly onto γ-linked ATP Sepharose beads without further purification. In 96-well format, drug candidate, ATP (positive control), or 10% DMSO (negative control) in buffer solutions were added to the charged beads, and the resulting eluates were collected by filtration and analyzed for fluorescence (full data is provided in Supplementary Data set 1 and also represented as a heat map in Supplementary Figure 2). The Z′ factor, calculated using average fluorescence data from the positive and negative controls (see Supporting Information Methods), for this screen was 0.53, which is above the threshold (0.5) for an excellent assay.24 HS38 competitively eluted GFP-ZIPK, resulting in >4-fold increase in fluorescence above background (Figure 1A5). Significant elution of GFP-ZIPK by 15 members of the library was confirmed by Western blot analysis. Of these 15 hits, HS38 was the most potent (Figure 1A6).
Potency and Selectivity of HS38
Although HS38 is a novel chemical entity, other thiol-substituted pyrazolo[3,4-d]pyrimidines have been investigated as anti-inflammatory agents25 and as potential anticancer agents that inhibit c-Src, Abl kinase, and EGFR-TK.26−28 In order to determine whether HS38 inhibits these or other clinically significant kinases, the molecule was evaluated for selectivity against a panel of purified protein kinases representing all gene family members within the human kinome (Figure 2 and Supplementary Figure 3) (data was generated by the International Centre for Kinase Profiling, University of Dundee).29 HS38 was highly specific for DAPK1 (IC50 = 200 nM, 89% inhibition at 10 µM) and the closely related PIM3 kinase (IC50 = 200 nM, 76% inhibition at 10 µM). HS38 displayed no activity against Src or Abl kinase (0% inhibition at 10 µM) and little activity against EGFR-TK (41% inhibition at 10 µM), suggesting that structural components of HS38 at the thioether and aryl regions around the pyrazolo[3,4-d]pyrimidinone core are important for DAPK and PIM3 affinity and are sufficient to distinguish the selectivity profile of HS38 from other inhibitors.
Figure 2.

Selectivity profiling of HS38 by the International Centre for Kinase Profiling. (A) Kinome dendrogram showing selectivity of HS38. Circles signify residual % enzyme activity in the presence of HS38 (10 µM). Dendrogram was created using the RBC Kinase Activity Mapper (Reaction Biology Corp.). (B) Kinase inhibition isotherms generated from radioactive (33P-ATP) filter-binding assay of HS38 against eight kinases. HS38 was most potent against DAPK1 and PIM3 (mean, n = 2).
The results of the initial kinome profile were validated in duplicate against eight kinases (Figure 2B). HS38 was most potent against DAPK1 and PIM3, ~10-fold less potent against IRAK4 and PIM1, ~100-fold less potent against PIM2 and SM myosin light chain kinase (smMLCK), and completely inactive against ROCK2 (Table 1). These two latter kinases are significant in the context of SM contractility because Ca2+ sensitization is linked to increased RLC20 phosphorylation by smMLCK, by ZIPK itself, or by ROCK-induced deactivation of MYPT1 either directly or indirectly by activation of ZIPK.3 Therefore it was important to establish that our inhibitor not act upon ROCK or smMLCK. This being the case, it is unlikely that activity of HS38 on SM contractility would be due to off-target inhibition of ROCK or smMLCK.
Table 1.
IC50 Values (Mean, n = 2) and Residual % Activity Derived from Kinase Inhibition Isotherms (Figure 2B)
| kinase | IC50 (nM) | % activity (10 µM HS38) |
|---|---|---|
| DAPK1 | 200 | 11 |
| PIM3 | 200 | 24 |
| IRAK4 | 1,400 | 20 |
| PIM1 | 1,700 | 23 |
| DYRK2 | 1,800 | 21 |
| PIM2 | 18,000 | 64 |
| smMLCK | 19,000 | 71 |
| ROCK2 | >200,000 | 91 |
The first reported small molecule inhibitor of the DAPK family was based on an aminopyridazine scaffold.7 Although its potency was in the micromolar range, a single injection of this inhibitor attenuated brain tissue damage when administered before or 6 h after ischemic injury.30 These therapeutic effects were validated in oxygen glucose deprivation and middle cerebral artery occlusion models.6 A second set of benzylidene oxazolone inhibitors of both DAPK1 and ZIPK was reported. Although they displayed potencies ranging from 250 to 580 nM, the electrophilic and amine reactive nature of benzylidine oxazolones raises concerns about their stability in a biological milieu.31,32 A third class of mixed inhibitors of ZIPK, PIM1, and PIM3 kinases displays nanomolar potencies against all three enzymes; however, these oxo-β-carboline derivatives also display similar potencies against a large number of other nonrelated protein kinases including CaMK2, CK1α, PKN2, ROCK2, and PKA.33 HS38 displayed similar or greater potency versus DAPK1 and, other than activity against PIM3, did not display any of the off-target liabilities of other published DAPK inhibitors (<10% activity against CaMK2, CK1α, PKN2, ROCK2, and PKA).
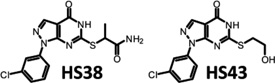
Affinity of HS38 and HS43 for DAPKs and PIMKs
The analogue HS43 was synthesized and used as a negative control (see Supporting Information Methods). Titration curves were generated from the systematic elution of GFP-ZIPK and PIM3 with C-terminal GFP tag (PIM3-GFP) from γ-linked ATP Sepharose media with solution phase ATP (positive control), HS38, HS43, and the commercially available nonselective kinase inhibitor ML-7 (Supplementary Figure 4). Sigmoidal isotherms generated by elution of GFP-ZIPK and PIM3-GFP with soluble ATP confirm that both were bound to the ATP media through noncovalent association with their ATP binding pockets. HS38 elution of GFP-ZIPK produced a sigmoidal curve, suggesting ATP competitive binding. HS43 eluted far less GFP-ZIPK, and ML-7 failed to elute GFP-ZIPK. In contrast, HS38, HS43, and ML-7 eluted PIM3-GFP from ATP affinity media with similar efficacy.
Dissociation constants for HS38 and HS43 against all DAP and PIM kinases were determined (Table 2) using an active-site dependent competition binding assay (KINOMEscan).34 Kd values were generally consistent with IC50 values reported in Table 1. HS43 was ~5-fold less potent than HS38 against DAPK1 and DAPK3. Both compounds displayed high affinity toward DAPK2; however, DAPK2 is not implicated in smooth muscle contractility because it lacks the C-terminal regulatory domain (present on full length ZIPK), which is essential for regulating and targeting its activity in muscle tissue.18 Kd values were similar for both compounds against the PIMKs.
Table 2.
Kd Values Obtained from KINOMEscan for HS38 and HS43 against All PIMKs and DAPKs
 | ||
|---|---|---|
| kinase | HS38 Kd (nM) | HS43 Kd (nM) |
| DAPK1 | 300 | 1500 |
| DAPK2 | 79 | 320 |
| DAPK3 | 280 | 1500 |
| PIM1 | 1800 | 1800 |
| PIM2 | 6500 | 3200 |
| PIM3 | 810 | 1600 |
Effects of HS38 and HS43 on Human Cells
Phosphorylation of RLC20 by ZIPK is a key mechanism by which ZIPK regulates SM contractility. In order to confirm that HS38 can inhibit ZIPK in living cells, the relative percent phosphorylation of RLC20 in human coronary artery smooth muscle (CA-VSMC) and aortic smooth muscle cells was investigated. Incubation of CA-VSMC cells with HS38 resulted in a 21−23% decrease in RCL20 phosphorylation under both serum-free and 5% FBS conditions (Figure 3A). Under the same conditions the phosphorylation state of T855 of MYPT1, which is not a target of ZIPK,3 and Par-4, a modulator of vascular smooth muscle contractility,35 are not significantly affected by HS38. These results are consistent with the notion that inhibition of ZIPK by HS38 mediates RLC20 phosphorylation.
Figure 3.

Effects of HS38 and HS43 on phosphorylation in cells. (A) Human coronary artery smooth muscle cells (CA-VSMCs, passage 12) were serum-starved overnight, incubated for 40 min with HS38 (10 µM) or vehicle control (DMSO), and treated or not with 5% FBS for 2 min prior to lysis in SDS-PAGE buffer and Western blotting with anti-pT18/S19-RLC20, anti-pT855-MYPT1, or anti-T155-Par-4. Phosphorylated bands were quantified by scanning densitometry and normalized to the loading control (SM22). Phosphorylation levels in the presence of HS38 are expressed as a percentage of control (absence of ZIPK inhibitor). Values represent means ± SEM for n = 6 or 7 separate treatments. *Significantly different from control treatment (Student’s t test, p < 0.05). (B) Serum-starved aortic SM cells were treated with S1P (0.25 µM) and HS38 (±50 µM) or HS43 (±50 µM) for 30 min. HS38 significantly decreased the relative percent RLC20 phosphorylation at the basal (unstimulated) state (p < 0.0002, n = 8) and S1P stimulated state (p < 0.0009, n = 8), while HS43 did not significantly affect phosphorylation.
Additionally, incubation of human aortic SM cells in HS38 significantly reduced relative RLC20 phosphorylation in both the basal and sphingosine 1-phosphate (S1P) activated states. HS43, a close structural analogue with similar PIMK and significantly less ZIPK affinity, showed no effect (Figure 3B and Supplementary Figure 5).36 This result suggests RLC20 phosphorylation is directly mediated by the action of HS38 on ZIPK and not another off-target kinase.
Effects of HS38 on SM Contractility
Muscles derived from mouse and rabbit represent the evolutionary sequence diversities of ZIPK. Aortic tissue was excised from wild-type mice and mounted on a myograph for contractility studies. When administered before contraction, HS38 decreased the maximum contractile force achieved from application of the γ1-adrenergic receptor agonist, phenylephrine (PE), in a reversible manner. HS38 also decreased force maintenance when administered after PE-induced contraction (Figure 4).
Figure 4.

HS38 reduced contractile forces generated by intact mouse aorta. (A) Typical myograph force trace showing the effect of HS38 on intact mouse aorta contractility. (B) The maximum contractile force of PE-induced contraction was reduced by approximately 30% when HS38 (10 µM) was added 5 min prior to PE addition and by approximately 40% when added 10 min after PE addition.
In addition to its effect on mouse aortic SM, HS38 relaxed carbachol-induced,37 Ca2+-sensitized force and decreased RLC20 and MYPT1 phosphorylation in permeabilized rabbit ileum (Figure 5A,B). HS38 induced an approximate 30% decrease in carbachol-induced force exerted by γ-toxin permeabilized rabbit ileum in the presence of Ca2+ (pCa 6.5). Western blot analysis of RLC20 and MYPT1 from these tissues showed that HS38 significantly reduced phosphorylation levels of both.
Figure 5.

HS38 reduced the contractile force, RLC20 phosphorylation, and MYPT1 phosphorylation in Ca2+-sensitized rabbit ileum (A, B) and decreased the kinetics of Ca2+ independent force development (C, D). (A) Typical force trace and graphical representation showing the effect of HS38 (50 µM) on carbachol-induced, Ca2+-sensitized force in α-toxin permeabilized rabbit ileum. HS38 induced an approximate 30% decrease in the plateaued force at 20 min (mean ± SE, n = 7). (B) Western blot analysis of RLC20 and MYPT1 Thr696 phosphorylation following treatment with HS38 (50 µM). HS38 significantly reduced both RLC20 and MYPT phosphorylation levels associated with sensitized force maintenance in ileum samples (mean ± SE, n = 7). (C) Typical force trace showing the effect of HS38 on Ca2+-independent force production in rabbit ileum smooth muscle. (D) HS38 markedly decreased the kinetics of force development at pCa = 9.0 in response to phosphatase inhibition by MC-LR (10 µM). (E) Both the lag time to the onset of force (p < 0.05, n = 3) and the rate of force development (t1/2) (p < 0.02, n = 3) were increased following the addition of MC-LR in the presence of HS38 (mean ± SE, n = 3).
The rate of Ca2+-independent (non-smMLCK-mediated) force production in permeabilized rabbit ileum was also affected by HS38 (Figure 5C−E). The kinetics of force development in response to the phosphatase inhibitor microcystin-LR (MC-LR) were significantly affected by HS38. Both lag time to the onset of force and rate of force development were increased following the addition of MC-LR in the presence of HS38. The finding that H38 promotes Ca2+ desensitization in muscles from both rabbit and mouse supports the hypothesis that the functions of ZIPK are evolutionarily conserved across species, even if the primary sequences are not entirely conserved within the C-terminal domain. Unlike mouse or rat, rabbit ZIPK does not contain the same sequence variations and is identical to human and all other mammalian species sequenced.
Application of HS38 to rat caudal arterial smooth muscle strips prior to calyculin A stimulation more than doubled the time required to reach 50% of maximal contraction (t1/2 max) without significantly affecting the maximum contractile force (Figure 6A−C). HS43 did not significantly affect either t1/2 max or maximum contractile force. Treatment of rat caudal arterial smooth muscle tissues with HS38 caused significant reduction in RLC20 phosphorylation (Figure 6D,E), while the negative control HS43 did not affect phosphorylation levels. These results are consistent with direct mediation of RLC20 phosphorylation by the action of HS38 on ZIPK and not another off-target kinase.
Figure 6.

(A) Representative contractile responses of intact rat caudal arterial smooth muscle strips to calyculin A (CLa, 0.5 mM) in the presence of: vehicle control (DMSO), HS38 (100 µM), and HS43 (100 µM). (B) Time (s) required to reach 50% of maximal contraction after application of CLa was calculated. (C) Maximal contractile force developed with CLa exposure (0.5 mM, 3 h) was expressed as a percent of an initial reference contraction to KCl. (D) RLC20 phosphorylation was analyzed by Phos-tag SDS-PAGE with detection of unphosphorylated, mono (1P)-, di (2P)-, and tri (3P)-phosphorylated forms by Western blotting with anti-panRLC20. (E) RLC20 bands were quantified by scanning densitometry, and the data are expressed as phosphorylation stoichiometry (mol Pi/mol RLC20; 30 min CLa exposure). *Significantly different from CLa (DMSO) treatment (ANOVA with Dunnett’s post hoc test, p < 0.05, n = 5).
Our results are consistent with current knowledge of the mechanisms by which ZIPK acts to increase Ca2+ sensitization and contractility. Although PIM kinases are close in proximity to DAP kinases on the CAMK stion of the dendrogram of human kinases,38 to date no studies have linked the PIMK family to SM contractility. Initial Western blot analysis failed to detect significant expression of PIM3 within rabbit ileum and mouse aortic SM tissues (Supplementary Figure 6), supporting the hypothesis that H38 acts through inhibition of ZIPK only. PIM3 kinase, which is inhibited by HS38 (IC50 = 200 nM), is a member of the PIM family of kinases (provirus integrating site for Moloney murine leukemia virus). PIM kinases play significant roles in tumorigenesis by preventing apoptosis and by promoting proliferation and survival of normal and cancerous cells. PIM3 phosphorylates a set of substrates that regulate apoptosis, cellular division, and metabolism.39 PIM1 kinase, which is inhibited by HS38 (IC50 = 1.7 µM), plays a critical role in SM cell proliferation40 and in the pathogenesis of pulmonary artery hypertension.41 However, this activity has been linked not to the effect of PIM1 on SM contractility, but to its ability to prevent apoptosis and promote proliferation of cardiac SM cells.
Because the PIM kinases are constitutively active and aberrantly expressed in numerous types of cancer, they are regarded as attractive targets for cancer therapy. Ongoing medicinal chemistry efforts are underway to develop a PIM-specific inhibitor based on our pyrazolo[3,4-d]pyrimidinone core as a potential cancer therapeutic and as a probe for the effects of PIM kinases on SM.
Conclusions
Over the past several decades, the pharmaceutical industry has allocated up to 30% of its research and development budget to targeting kinases.42 Numerous small molecule kinase inhibitors, which predominantly function by competing with ATP binding, have been granted FDA approval.43,44 We have developed a screening method called FLECS, for the rapid identification of inhibitors for all elements of the purinome, including protein kinases. FLECS was utilized to identify HS38, which as an unoptimized lead is a potent and selective inhibitor of DAPK1 and ZIPK that also targets the closely related PIM3 kinase. This is a significant first step in the development of pyrazolo[3,4-d]pyrimidinone based inhibitors that are highly potent and selective for members of the DAPK family or the PIM kinase family of enzymes. We plan to explore this scaffold through iterative synthetic effort to derive inhibitors that separately target DAPKs and PIMKs.
METHODS
Library Construction
A collection of 10,000 compounds was initially selected from commercially available compounds using a previously described set of filters.22 For a detailed description, see Supporting Information Methods.
Expression of GFP Fusion Proteins
GFP fusion constructs were transfected into HEK293 cells, and the resulting lysates were used for all subsequent screening and titration experiments. For a detailed description, see Supporting Information Methods.
Preparation of γ-Linked ATP Sepharose Media
Dry CNBr-Activated Sepharose 4B media (28.6 g, GE Healthcare) was equilibrated in HCl (1 mM, 333 mL) for 5−15 min, isolated by filtration, and then washed with HCl (1 mM, 600 mL) followed by H2O (333 mL). The media was combined with reaction mixture A (NaHCO3, 0.97 g; NaCl, 3.4 g; H2O, 115 mL; 1,4-dioxane, 29 mL; 1,10-diaminodecane, 3.6 g; ethanolamine, 3.6 mL) and shaken for 2 h. Meanwhile, reaction mixture B (H2O, 143 mL; ATP, disodium salt, 7 g; 1-methylimidazole, 5.2 mL; EDC, 12 g) was stirred for 1 h. Mixture A was removed by filtration, and the media was washed with HCl (1 mM, 600 mL) and then H2O (333 mL). Mixture B and media were combined and shaken for 24 h. The resulting ATP Sepharose media was isolated by filtration and washed with HCl (1 mM, 600 mL) and then H2O (333 mL). The media was stored at 4 °C in phosphate buffer (0.1 M, pH 7.4) containing NaN3 (3 mM).
Synthesis of (2-((1-(3-Chlorophenyl)-4-oxo-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)thio)propanamide (HS38) and 1-(3-Chlorophenyl)-6-((2-hydroxyethyl)thio)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (HS43)
See Supplementary Scheme 1. A full description is given in Supporting Information methods.
Drug Candidate Screening and Titrations
Crude lysates containing recombinant purinomic GFP fusion proteins were combined with ATP Sepharose media (1:1 slurry, >50,000 fluorescence counts per 50 µL of slurry) in lysis buffer (0.1% Triton; NaCl, 150 mM; MgCl2, 60 mM; Tris·HCl, pH 7.5, 25 mM; Microcystin, 1 µM; protease inhibitor tablet) for 0.5 h at 4 °C. The buffer was removed by filtration, and the media was washed with high salt wash buffer (Tris, 50 mM; NaCl, 1 M; MgCl2, 60 mM; DTT, 1 mM) (3 × resin volume) followed by low salt wash buffer (LSWB) Tris, 50 mM; NaCl, 150 mM; MgCl2, 60 mM; DTT, 1 mM) (3 × resin volume). LSWB (1 × resin volume) was then added to the resin, and the resulting 1:1 slurry was partitioned into a 96-well filter plate Corning 3504) (50 µL per well). Positive control: to each well was added 50 µL of ATP solution (2−200 mM in LSWB with 10% DMSO). Drug candidate screen: to each well was added 50 µL of drug candidate (900 µM in LSWB with 10% DMSO). Drug candidate titrations: to each well was added 50 µL of HS38 or ML-7 (Sigma Aldrich) solution (0.1−300 µM in LSWB with 10% DMSO). After 10 min of incubation at RT, the filtrates were isolated by centrifugation 1000 rpm, 2 min) into a black 96-well catch plates (Costar 3915). Fluorescence in each well was determined using a plate reader (Perkin-Elmer Victor X2 Multilabel Reader, lamp filter 485 nm, emission filter 535 nm).
Kinome Profiling
HS38 was evaluated using a 33P-ATP filter-binding assay by the International Centre for Kinase Profiling University of Dundee) against 124 purified protein kinases using previously described methods.29
Mouse Aortic SM Cell Culture
The cell line was cultured in AmnioMax medium (Gibco) supplemented with 10% embryonic stem cell-qualified fetal bovine serum (Invitrogen) and penicillin-streptomycin (Invitrogen). Cultured cells were maintained at 37 °C in a humidified chamber supplemented with 5% CO2. Cells were seeded on 100 mm culture dishes. Prior to experimental conditions, cells were starved for 16 h with serum-free medium. Subconfluent, serum-starved smooth muscle cells were treated with sphingosine-1-phosphate (0.25 µM) for 5 min followed by treatment with HS38 (50 µM) or diluent (0.1% DMSO) for 30 min.
Treatment of Human Coronary Artery Smooth Muscle Cells
Human coronary artery vascular smooth muscle cells (CA-VSMC; Lonza Inc., no. CC-2583) were maintained in Smooth Muscle Basal Medium (SmBM; Lonza) supplemented with 0.5 mg mL−1 hEGF, 5 mg mL−1 insulin, 1 mg mL−1 hFGF, 50 mg mL−1 gentamicin/amphotericin-B, and 5% fetal bovine serum (FBS) and used at passage 12 for all experiments. Cells were serum-starved overnight prior to treatment with HS38.
Tissue Preparation and Force Measurements
All protocols and procedures for tissue harvest were carried out according to protocols approved by the Animal Care and Use Committees at the University of Virginia, Duke University, and the University of Calgary. For a detailed description, see Supporting Information Methods.
MYPT1 and RLC20 Phosphorylation
To examine the phosphorylation of MYPT1 and RLC20, rabbit ileum SM strips were treated as described above and previously reported.45−47 Following the stimulation protocols, muscle strips were immediately frozen by immersion in −80 °C acetone with trichloroacetic acid (10% w/v) and stored at −80 °C. Frozen strips were then washed in acetone, dried, and homogenized in sample buffer in a glass−glass, hand-operated homogenizer. Phosphorylation of MYPT1 Thr696 and of RLC20 was determined by SDS-PAGE and Western blotting.
For Western blots, tissues and cultured cells were lysed in 1% SDS, NaCl (300 mM), Tris-HCl (50 mM, pH 7.5), subjected to SDS-PAGE, transferred to polyvinylidene difluoride membrane (Millipore), and visualized using the Odyssey System (Li-Cor). For Odyssey imaging, the membranes were blocked with Odyssey Blocking Buffer and then subjected to anti-MYPT1 (1:2,000) (BD Transduction), phospho-MYPT1 (Thr696) (1:1,000) (Millipore), RLC20 (1:2000) (Sigma), and phospho-RLC20 (1:1000) (Cell Signaling Technology) antibodies. Antibodies were diluted in appropriate blocking buffer. The membranes were washed in TBS with 0.05% Tween 20. Primary antibodies were visualized using secondary antibodies conjugated to Alexa 680 (Invitrogen) or IRDye800 (Li-Cor). Western blots were visualized with a LAS4000 Imaging Station (GE Healthcare), ensuring that the representative signal occurred in the linear range. Quantification was performed by densitometry with ImageQuant TL software (GE Healthcare).
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by grants from the Mandel Foundation (to T.A.J.H.), in part by a grant (MOP-111262) from the Canadian Institutes of Health Research (to M.P.W.), and in part by the Heart & Stroke Foundation of Canada (to J.A.M.). M.P.W. is an Alberta Innovates-Health Solutions Scientist and Canada Research Chair in Vascular Smooth Muscle Research. J.A.M. is an Alberta Innovates-Health Solutions Senior Scholar and Canada Research Chair Vascular Smooth Muscle Research.
Footnotes
ASSOCIATED CONTENT
Supporting Information
This material is available free of charge via the Internet at http://pubs.acs.org.edu.
The authors declare no competing financial interest.
REFERENCES
- 1.Bialik S, Kimchi A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006:189–210. doi: 10.1146/annurev.biochem.75.103004.142615. [DOI] [PubMed] [Google Scholar]
- 2.Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2:74–79. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
- 3.Haystead TAJ. ZIP kinase, a key regulator of myosin protein phosphatase 1. Cell. Signaling. 2005;17:1313–1322. doi: 10.1016/j.cellsig.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discovery. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kishino M, Yukawa K, Hoshino K, Kimura A, Shirasawa N, Otani H, Tanaka T, Owada-Makabe K, Tsubota Y, Maeda M, Ichinose M, Takeda K, Akira S, Mune M. Deletion of the kinase domain in death-associated protein kinase attenuates tubular cell apoptosis in renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2004;15:1826–1834. doi: 10.1097/01.asn.0000131527.59781.f2. [DOI] [PubMed] [Google Scholar]
- 6.Shamloo M, Soriano L, Wieloch T, Nikolich K, Urfer R, Oksenberg D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J. Biol. Chem. 2005;280:42290–42299. doi: 10.1074/jbc.M505804200. [DOI] [PubMed] [Google Scholar]
- 7.Velentza AV, Schumacher AM, Watterson DM. Structure, activity, regulation, and inhibitor discovery for a protein kinase associated with apoptosis and neuronal death. Pharmacol. Ther. 2002;93:217–224. doi: 10.1016/s0163-7258(02)00190-0. [DOI] [PubMed] [Google Scholar]
- 8.Ulke-Lemée A, MacDonald JA. Opportunities to target specific contractile abnormalities with smooth muscle protein kinase inhibitors. Pharmaceuticals. 2010;3:1739–1760. doi: 10.3390/ph3061739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feinstein E, Druck T, Kastury K, Berissi H, Goodart SA, Overhauser J, Kimchi A, Huebner K. Assignment of DAP1 and DAPK-genes that positively mediate programmed cell- death triggered by inf-gamma-to chromosome regions 5p12.2 and 9q34.1, respectively. Genomics. 1995;29:305–307. doi: 10.1006/geno.1995.1255. [DOI] [PubMed] [Google Scholar]
- 10.Brognard J, Zhang YW, Puto LA, Hunter T. Cancer-associated loss-of-function mutations implicate DAPK3 as a tumor-suppressing kinase. Cancer Res. 2011;71:3152–3161. doi: 10.1158/0008-5472.CAN-10-3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velentza AV, Schumacher AM, Weiss C, Egli M, Watterson DM. A protein kinase associated with apoptosis and tumor suppression - Structure, activity, and discovery of peptide substrates. J. Biol. Chem. 2001;276:38956–38965. doi: 10.1074/jbc.M104273200. [DOI] [PubMed] [Google Scholar]
- 12.Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK-ZIPK-L13a axis constitutes a negative- feedback module regulating inflammatory gene expression. Mol. Cell. 2008;32:371–382. doi: 10.1016/j.molcel.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang BN, Ahmad AS, Saleem S, Patterson RL, Hester L, Dore S, Snyder SH. Death-associated protein kinase- mediated cell death modulated by interaction with DANGER. J. Neurosci. 2010;30:93–98. doi: 10.1523/JNEUROSCI.3974-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler T, Paul J, Europe-Finner N, Smith R, Chan EC. Role of serine-threonine phosphoprotein phosphatases in smooth muscle contractility. Am. J. Physiol.-Cell Physiol. 2013;304:C485–C504. doi: 10.1152/ajpcell.00161.2012. [DOI] [PubMed] [Google Scholar]
- 15.Borman MA, MacDonald JA, Muranyi A, Hartshorne DJ, Haystead TAJ. Smooth muscle myosin phosphatase-associated kinase induces Ca2+ sensitization via myosin phosphatase inhibition. J. Biol. Chem. 2002;277:23441–23446. doi: 10.1074/jbc.M201597200. [DOI] [PubMed] [Google Scholar]
- 16.Ihara E, MacDonald JA. The regulation of smooth muscle contractility by zipper-interacting protein kinase. Can. J. Physiol. Pharmacol. 2007;85:79–87. doi: 10.1139/y06-103. [DOI] [PubMed] [Google Scholar]
- 17.MacDonald JA, Borman MA, Murányi A, Somlyo AV, Hartshorne DJ, Haystead TAJ. Identification of the endogenous smooth muscle myosin phosphatase-associated kinase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:2419–2424. doi: 10.1073/pnas.041331498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weitzel DH, Chambers J, Haystead TAJ. Phosphorylation-dependent control of ZIPK nuclear import is species specific. Cell. Signaling. 2011;23:297–303. doi: 10.1016/j.cellsig.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shoval Y, Pietrokovski S, Kimchi A. ZIPK: A unique case of murine-specific divergence of a conserved vertebrate gene. PloS Genet. 2007;3:1884–1893. doi: 10.1371/journal.pgen.0030180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haystead TAJ. The purinome, a complex mix of drug and toxicity targets. Curr. Top. Med. Chem. 2006;6:1117–1127. doi: 10.2174/156802606777812059. [DOI] [PubMed] [Google Scholar]
- 21.Moellering RE, Cravatt BF. How chemo- proteomics can enable drug discovery and development. Chem. Biol. 2012;19:11–22. doi: 10.1016/j.chembiol.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, Dubois LG, Freed TA, Silinski MAR, Barta TE, Hughes PF, Ommen A, Ma W, Smith ED, Spangenberg AW, Eaves J, Hanson GJ, Hinkley L, Jenks M, Lewis M, Otto J, Pronk GJ, Verleysen K, Haystead TA, Hall SE. Application of chemo- proteomics to drug discovery: identification of a clinical candidate targeting Hsp90. Chem. Biol. 2010;17:686–694. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Haystead CMM, Gregory P, Sturgill TW, Haystead TAJ. Gamma-phosphate-linked atp-sepharose for the affinity purification of protein-kinases–rapid purification to homogeneity of skeletal-muscle mitogen-activated protein-kinase kinase. Eur. J. Biochem. 1993;214:459–467. doi: 10.1111/j.1432-1033.1993.tb17942.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25.Russo F, Guccione S, Romeo G, Barretta GU, Pucci S, Caruso A, Amicoroxas M, Cutuli V. Pyrazolothiazolo- pyrimidine derivatives as a novel class of antiinflammatory or antinociceptive agents - synthesis, structural characterization and pharmacological evaluation. Eur. J. Med. Chem. 1993;28:363–376. [Google Scholar]
- 26.Manetti F, Santucci A, Locatelli GA, Maga G, Spreafico A, Serchi T, Orlandini M, Bernardini G, Caradonna NP, Spallarossa A, Brullo C, Schenone S, Bruno O, Ranise A, Bondavalli F, Hoffmann O, Bologna M, Angelucci A, Botta M. Identification of a novel pyrazolo 3,4-d pyrimidine able to inhibit cell proliferation of a human osteogenic sarcoma in vitro and in a xenograft model in mice. J. Med. Chem. 2007;50:5579–5588. doi: 10.1021/jm061449r. [DOI] [PubMed] [Google Scholar]
- 27.Radi M, Brullo C, Crespan E, Tintori C, Musumeci F, Biava M, Schenone S, Dreassi E, Zamperini C, Maga G, Pagano D, Angelucci A, Bologna M, Botta M. Identification of potent c-Src inhibitors strongly affecting the proliferation of human neuroblastoma cells. Bioorg. Med. Chem. Lett. 2011;21:5928–5933. doi: 10.1016/j.bmcl.2011.07.079. [DOI] [PubMed] [Google Scholar]
- 28.Schenone S, Bruno O, Bondavalli F, Ranise A, Mosti L, Menozzi G, Fossa P, Manetti F, Morbidelli L, Trincavelli L, Martini C, Lucacchini A. Synthesis of 1-(2-chloro-2-phenylethyl)-6-methylthio-1H-pyrazolo 3,4-d pyrimidines 4-amino substituted and their biological evaluation. Eur. J. Med. Chem. 2004;39:153–160. doi: 10.1016/j.ejmech.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Velentza AV, Wainwright MS, Zasadzki M, Mirzoeva S, Schumacher AM, Haiech J, Focia PJ, Egli M, Watterson DM. An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorg. Med. Chem. Lett. 2003;13:3465–3470. doi: 10.1016/s0960-894x(03)00733-9. [DOI] [PubMed] [Google Scholar]
- 31.Okamoto M, Takayama K, Shimizu T, Muroya A, Furuya T. Structure-activity relationship of novel DAPK inhibitors identified by structure-based virtual screening. Bioorg. Med. Chem. 2010;18:2728–2734. doi: 10.1016/j.bmc.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 32.Betlakowska B, Banecki B, Czaplewski C, Lankiewicz L, Wiczk W. Reaction of 4-benzylidene-2-methyl-5-oxazolone with amines, Part 2: Influence of substituents in para-position in the phenyl ring and a substituent on amine nitrogen atom on the reaction kinetics. Int. J. Chem. Kinet. 2002;34:148–155. [Google Scholar]
- 33.Huber K, Brault L, Fedorov O, Gasser C, Filippakopoulos P, Bullock AN, Fabbro D, Trappe J, Schwaller J, Knapp S, Bracher F. 7,8-dichloro-1-oxo-beta-carbolines as a versatile scaffold for the development of potent and selective kinase inhibitors with unusual binding modes. J. Med. Chem. 2012;55:403–413. doi: 10.1021/jm201286z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wodicka LM, Ciceri P, Davis MI, Hunt JP, Floyd M, Salerno S, Hua XH, Ford JM, Armstrong RC, Zarrinkar PP, Treiber DK. Activation state-dependent binding of small molecule kinase inhibitors: Structural insights from biochemistry. Chem. Biol. 2010;17:1241–1249. doi: 10.1016/j.chembiol.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 35.Vetterkind S, Lee E, Sundberg E, Poythress RH, Tao TC, Preuss U, Morgan KG. Par-4: A new activator of myosin phosphatase. Mol. Biol. Cell. 2010;21:1214–1224. doi: 10.1091/mbc.E09-08-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watterson KR, Ratz PH, Spiegel S. The role of sphingosine-1-phosphate in smooth muscle contraction. Cell. Signaling. 2005;17:289–298. doi: 10.1016/j.cellsig.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 37.Somlyo AP, Somlyo AV. Signal-transduction and regulation in smooth-muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 38.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 39.Mukaida N, Wang YY, Li YY. Roles of Pim-3, a novel survival kinase, in tumorigenesis. Cancer Sci. 2011;102:1437–1442. doi: 10.1111/j.1349-7006.2011.01966.x. [DOI] [PubMed] [Google Scholar]
- 40.Katakami N, Kaneto H, Hao H, Umayahara Y, Fujitani Y, Sakamoto K, Gorogawa S, Yasuda T, Kawamori D, Kajimoto Y, Matsuhisa M, Yutani C, Hori M, Yamasaki Y. Role of pim-1 in smooth muscle cell proliferation. J. Biol. Chem. 2004;279:54742–54749. doi: 10.1074/jbc.M409140200. [DOI] [PubMed] [Google Scholar]
- 41.Paulin R, Courboulin A, Meloche J, Mainguy V, de la Roque ED, Saksouk N, Cote J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/PIM1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation. 2011;123:1205–U1163. doi: 10.1161/CIRCULATIONAHA.110.963314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen P. Protein kinases - the major drug targets of the twenty-first century? Nat. Rev. Drug Discovery. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 43.Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- 44.Cohen P, Alessi DR. Kinase drug discovery–What’s next in the field? ACS Chem. Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong MC, Kinter MT, Somlyo AV, Somlyo AP. Arachidonic-acid and diacylglycerol release associated with inhibition of myosin light-chain dephosphorylation in rabbit smooth- muscle. J. Physiol. (London U. K.) 1995;486:113–122. doi: 10.1113/jphysiol.1995.sp020795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled permeabilized smooth-muscle role of the phosphatidylinositol cascade, g-proteins, and modulation of the contractile response to Ca2+ J. Biol. Chem. 1989;264:5339–5342. [PubMed] [Google Scholar]
- 47.Kobayashi S, Gong MC, Somlyo AV, Somlyo AP. Ca2+ channel blockers distinguish between g-protein-coupled pharmacomechanical Ca2+ release and Ca2+ sensitization. Am. J. Physiol. 1991;260:C364–C370. doi: 10.1152/ajpcell.1991.260.2.C364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.