

Abstract
Described are the synthesis and application of α-t-butyldimethylsilyl-α-methoxyacetaldehyde as a formal methoxyvinyl cation equivalent. Addition of Grignard reagents to the title aldehyde, followed by treatment of the intermediate β-hydroxysilanes with KH, gives good yields of large Z-methoxyvinylated products. Assuming a Peterson-like elimination mechanism, one can infer that the Grignard addition proceeds with high syn selectivity. These results are consistent with a chelation control model involving coordination to the α-methoxy group in the title aldehyde rather than an alternative stereoelectronic Felkin-Anh-type model. It must be noted that a steric Felkin-Anh model also accounts for the observed stereochemistry. All told, the title reagent can be employed to efficiently append a Z-configured methoxyvinyl group to an appropriate R-M species, in two steps.
Keywords: methoxyvinylation, chelation control, Peterson elimination, α-silyl-α-methoxyacetaldehyde, steric Felkin-Anh model
The methoxyvinyl group is a functionality of importance both as a component of bioactive natural products and as an essential element of a number of synthetic reagents. Figure 1 illustrates several examples this functional group in both such contexts. Specifically, in an amino acid context, E-methoxyvinylglycine (also known as 2-amino-4-methoxy-3-butenoic acid = AMB; Figure 1, a) is biosynthesized by the opportunistic pathogen, Pseudomonas aeroginosa. Recently, it has been demonstrated that E-AMB strongly inhibits the growth of Erwinia amylovora, the causal agent of the devastating orchard crop disease known as fire blight.1 On the other hand, synthetic Z-methoxyvinylglycine (b) is known to serve as an inhibitor of methionine adenosyltransferase,2 the enzyme catalyzing the biosynthesis of S-adenosylmethionine as the first step in the important transulfuration pathway. Antibiotic scaffolds bearing terminal methoxyvinyl groups include the terpenoid natural product d from the plant source, S. megistophylla3 and the quinolone e, isolated from a Eupenicillium fungus.4 Significantly, the latter shows good activity against S. aureus. Finally, compound c, though a synthetic intermediate toward benzoazepinoisoquinolones, itself displays significant antitumor activity. On the other hand, compounds f and g bear methoxyvinyl groups as useful components of synthons being exploited for asymmetric methoxyallylation5 and inverse-electron demand Diels-Alder chemistry,6 respectively.
Figure 1.

Naturally occurring and synthetically useful compounds bearing terminal methoxyvinyl groups. a - E-methoxyvinylglycine (natural; PLP enzyme inhibitor); b Z-methoxyvinylglycine (synthetic; SAM synthetase inhibitor); c isoquinolone anti-tumor agent; d fungal antibiotic eupenicinical B; e megistoquinone II antibiotic (natural); f asymmetric methoxyallylating agent (synthetic); g inverse electron demand dienophile (synthetic)
Interest in the Z-methoxyvinyl group, in particular, in our own research program is also stimulated by the finding that the Z-fluorovinyl group, among a range of vinylic functionalities,7 serves as a useful trigger for the mechanism-based inactivation of PLP-dependent enzymes.8 In synthesizing such vinylic PLP enzyme inhibitors initially, we had developed an approach whereby ethylene oxide, when combined with phenylselenolate anion, serves as a formal vinyl cation equivalent.9 It was therefore of interest for us to develop a complementary “methoxyvinyl cation equivalent” strategy here. Inspired by the pioneering work of Hudrlik10 that leverages Peterson olefination-type reactivity, we set out to construct an α-silyl-α-methoxy acetaldehyde suitable for condensation with appropriate nucleophiles. As is illustrated in Scheme 1, the desired methoxyvinyl cation equivalent could be assembled in a half dozen straightforward steps from propargyl alcohol. The key operation involves introduction of Lewis acid-catalyzed opening of epoxide 3 α-to silicon, regiochemistry that is well precedented.11 Compound 4 proved a convenient stage at which store the reagent. The title aldehyde 5 could then be unmasked oxidatively with Pb(OAc)4 very cleanly, consistent with the observations of Chauret et al. in related systems.12
Scheme 1. Synthesis of the title aldehyde 5.

As can be seen from Table 1, upon condensing 5 with aryl Grignard reagents or lithium phenylacetylide, smooth aldehyde addition occurs and that addition is quite diastereoselective (from 5:1 to 20:1) as judged by crude NMR (see Figure 2 for a view of a representative NMR, for 6d, in this case). Note that it was found particularly convenient to utilize Rieke magnesium (Mg*)13 to generate the Grignard reagents at low T, e.g. leading to 6e-g and 6j. In some cases, the intermediate β-silyl alcohols 6 were isolated and purified chromatographically. However, yields were generally better if the crude β-silyl alcohol 6, after workup, was treated with KH in THF to generate the methoxyvinyl compound directly. This efficiency of this two step addition/elimination protocol for the rapid methoxyvinylation of a metalated carbon nucleophile is documented in Table 2. Note that the diastereomeric ratio seen in the aldehyde addition adducts is reflected in the Z:E ratio seen in the elimination products, providing strong evidence in support of a stereospecific mechanism for the elimination, as expected for Peterson-type chemistry.
Table 1. Representative additions to methoxyvinyl cation equivalent 5.

| ||||
|---|---|---|---|---|
|
| ||||
| Entry | R | Metal/Solvent | dr | Yield 6 (%)a |
| a |

|
Li/THF | 20:1 | 90a |
| b |

|
Mg/THF | 6:1 | 80a |
| c |

|
Mg/THF | 20:1 | 75 |
| d |

|
Mg/THF | 10:1 | 96a |
| e |

|
Mg*/THF | 6:1 | 69b |
| f |

|
Mg*/THF | 8:1 | 76b |
| g |

|
Mg*/THF | 6:1 | 81b |
| h |

|
Mg/THF | 5:1 | 91 |
| i |

|
Mg/THF | 12:1 | 72 |
| j |

|
Mg*/THF | 20:1 | 63b |
Diastereoselectivity determined by 1H-NMR.
Yield estimated by NMR vs. CH3CN as internal standard;
Note: Rieke magnesium (Mg*) was used to make the Grignard reagents in these cases.
Figure 2.

1H NMR (700 MHz, CDCl3) spectra for the formation of both addition compounds 6d and elimination products 7d. The diasteromeric ratio remains constant (@ 10:1) suggestive of a stereospecific syn Peterson-type elimination wherein the predominant syn-addition product gives the Z-alkene and the minor anti-addn product gives the minor E-alkene.
Table 2. Representative examples of the two step addition/elimination protocol.

| ||||
|---|---|---|---|---|
|
| ||||
| Entry | R | Metal/Solvent | Z:E | Yield 7 (%) |
| a |
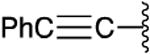
|
Li/THF | 20:1 | 83 |
| b |

|
Mg/THF | 6:1 | 72 |
| c |

|
Mg/THF | 20:1 | 55 |
| d |

|
Mg/THF | 10:1 | 86a |
| e |

|
Mg*/THF | 6:1 | 65b |
| f |

|
Mg*/THF | 8:1 | 68b |
| g |

|
Mg*/THF | 6:1 | 70b |
| h |

|
Mg*/THF | 5:1 | 86b |
| i |

|
Mg*/THF | 12:1 | 65 |
| j |

|
Mg*/THF | 20:1 | 53b |
E:Z-ratio determined by 1H-NMR.
Yield estimated by NMR vs. CH3CN as internal standard.;
Note: Rieke magnesium (Mg*) was used in these examples.
The two-step addition/elimination protocol lends itself well to examination by 1H NMR as is illustrated in Figure 2. Thus, addition of p-fluorophenylmagnesium bromide to the title aldehyde results in a 10:1 ratio of aldehyde addition adducts. Subsequent treatment with KH in THF at 0 °C delivers a 10:1 product distribution of Z- to E-methoxyvinylated arenes, with J values of 7.0 Hz and 12.6 Hz, respectively. Notice that the chemical shift windows for addition adducts and elimination products are well resolved, allowing for convenient mapping of the transformation of individual β-silyl-β-methoxy-alcohol adducts onto their derivative methoxyvinyl elimination products by 1H NMR. A particularly interesting example is represented by the formation of the dienyl methyl ether from α-bromostyrene, utilizing Mg* to generate the corresponding vinyl Grignard reagent, and employing the two step procedure.
Given the definitive identification of the Z-methoxyvinyl product as the major elimination product in each case under classic Peterson elimination conditions (KH, THF), it is presumed that the elimination occurs in a stereospecific syn-fashion, via the intermediacy of an pentacoordinate ate-addition complex at silicon (Figure 3).14 Logically then, it follows that the initial additions of aryl Grignard or lithiated acetylide nucleophiles to the title α-methoxy-α-silyl aldehyde must proceed in a highly syn-selective manner. The prevailing models for diastereoselection in the nucleophilic additions to chiral α-substituted aldehydes are presented in Figure 3. When an α-substituent bearing an electron-withdrawing heteroatomic substituent X is present, a Felkin-Anh model15 in which stereoelectronic effects predominate is often posited. There are two views of the nature of that stereoelectronic interaction in the transition state. If the transition state is late, then it is argued, the develop C-Nu σ-bonding orbital overlaps with the C-X σ*-anti-bonding orbital. On the other hand, for early transition state, the Cieplak picture holds that C-X σ-bonding orbital overlaps with the C-Nu σ*-anti-bonding orbital associated with the newly forming bond.16 Both of these stereoelectronic models require that the incoming nucleophile approach the carbonyl from an angle opposite to the C-X bond, albeit at the Bürgi-Dunitz angle. In the case at hand, this would mean approach opposite to the C-OMe bond, at that face of the carbonyl presenting the H-substituent, rather than the large TBS-substituent, thereby giving the anti-diastereomeric addition product, as shown in Figure 3. In all cases, this is the minor product, indicating that the stereoelectronic Felkin-Anh model does not apply here.
Figure 3.

Comparison of the predictions made by the stereoelectronic Felkin-Anh model, the Cornforth-Evans model and by the chelation control model for the addition of RMgBr to the title methoxyvinyl cation equivalent, to form adduct 6. Experimental results indicate that there is generally high syn selectivity in these additions suggesting that chelation contributions by the α-methoxy group in the electrophile outweigh stereoelectronic effects here.
Evans17 has recently put forth a modification of the traditional Cornforth model that is in keeping with widely accepted view that nucleophilic attack must proceed at the Bürgi-Dunitz18 angle. The Cornforth model19 postulates that for such electronegative substituents X, dipole-dipole interactions predominate. That is to say, in this view, there is a strong predilection for the C-X dipole to oppose the C=O dipole in the lowest energy transition state. Attack would then occur from the least hindered trajectory, i.e. opposite the TBS substituent here, subject to the aforementioned dipolar constraints. Once again, this model predicts the anti-product here and therefore cannot describe the predominant transition state.
On the other hand, a chelation control model,20 in which the α-methoxy substituent and the aldehyde carbonyl are bridged by a chelating MgX or Li metal ion, is consistent with the observed stereochemistry here, in that attack from the least hindered face in this model would give the syn-product. It is important to note, however, that these results are also consistent with a steric-Felkin-Anh model,21 in which the sterics of the TBS group are postulated to override favorable stereoelectronic interactions involving the C-OMe bond.
As it stands, the methodology reported herein serves as a nice complement to existing methods for the installation of an alkoxyvinyl group. These include Horner-Wittig olefinations with Ar2P(O)CRH(OMe) reagents,22 as well as Julia olefination approaches with ArSO2CH2(OMe) reagents.23 Finally, as is shown in Figure 4, this methoxyvinyl cation approach, though employing a Peterson-type elimination step following the condensation reaction, represents a different bond disconnection from traditional Peterson olefination approaches,14, 24 some of which have been developed for enol ether synthesis.25 What differentiates the methodology reported herein from all of these approaches is, indeed, the bond disconnection, whereby the title reagent serves as a true methoxyvinyl cation equivalent, supplying the methoxy group and both carbons of the π-bond for convergent condensation with a nucleophile of choice. As such, we believe that this reagent has the potential for broad application in synthesis.
Figure 4.

Comparison of bond constructions and stereochemical consequences of the methoxyvinylation approach reported here vs. the traditional Peterson olefination approach.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Professor Harry H. Wasserman, a true scholar, leader in the organic chemistry community and a real gentleman. HHW served as an inspiration to DBB during his postdoctoral time at Yale and beyond. We thank Reuben Rieke and Rieke Metals, Inc. for providing the Rieke magnesium (Mg*) utilized herein and for helpful discussion. The authors thank the NSF (1214019) for support. This research was facilitated by the IR/D (Individual Research and Development) program associated with DBB's appointment at the NSF. The authors thank the NIH (SIG-1-510-RR-06307) and NSF (CHE-0091975,MRI-0079750) for NMR instrumentation support and the NIH (RR016544) for facilities renovation.
Footnotes
Supplementary data: Experimental procedures, characterization data and copies of representative NMR spectra associated with this article can be found, in the online version, at http://
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Lee X, Reimmann C, Greub G, Sufrin J, Croxatto A. Microbes Infect. 2012;14:268–272. doi: 10.1016/j.micinf.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 2.(a) González B, Pajares MaA, Hermoso JA, Guillerm D, Guillerm G, Sanz-Aparicio J. J Mol Biol. 2003;331:407–416. doi: 10.1016/s0022-2836(03)00728-9. [DOI] [PubMed] [Google Scholar]; (b) Berkowitz DB, Charette BD, Karukurichi KR, McFadden JM. Tetrahedron: Asymmetry. 2006;17:869–882. doi: 10.1016/j.tetasy.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gonzalez B, Pajares MA, Hermoso JA, Alvarez L, Garrido F, Sufrin JR, Sanz-Aparicio J. J Mol Biol. 2000;300:363–75. doi: 10.1006/jmbi.2000.3858. [DOI] [PubMed] [Google Scholar]
- 3.Fokialakis N, Magiatis P, Chinou I, Mitaku S, Tillequin F. Chem Pharm Bull. 2002;50:413–414. doi: 10.1248/cpb.50.413. [DOI] [PubMed] [Google Scholar]
- 4.Li G, Kusari S, Lamshoft M, Schuffler A, Laatsch H, Spiteller M. J Nat Prod. 2014;77:2335–41. doi: 10.1021/np500111w. [DOI] [PubMed] [Google Scholar]
- 5.Munoz-Hernandez L, Soderquist JA. Org Lett. 2009;11:2571–2574. doi: 10.1021/ol900865y. [DOI] [PubMed] [Google Scholar]
- 6.(a) Stehl A, Seitz G, Schulz K. Tetrahedron. 2002;58:1343–1354. [Google Scholar]; (b) Che D, Wegge T, Stubbs MT, Seitz G, Meier H, Methfessel C. J Med Chem. 2001;44:47–57. doi: 10.1021/jm000949w. [DOI] [PubMed] [Google Scholar]
- 7.(a) Berkowitz DB, Chisowa E, McFadden JM. Tetrahedron. 2001;57:6329–6343. doi: 10.1016/S0040-4020(01)00499-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Berkowitz DB, McFadden JM, Sloss MK. J Org Chem. 2000;65:2907–2918. doi: 10.1021/jo9918091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Berkowitz DB, McFadden JM, Chisowa E, Semerad CL. J Am Chem Soc. 2000;122:11031–11032. doi: 10.1021/ja0055110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Berkowitz DB, Smith MK. Synthesis. 1996;1:39–41. doi: 10.1055/s-1996-4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Berkowitz DB, Karukurichi KR, de la Salud-Bea R, Maiti G, McFadden JM, Morris ML. ACS Symp Ser. 2009;1009:288–303. [Google Scholar]; (b) Berkowitz DB, Karukurichi KR, de la Salud-Bea R, Nelson DL, McCune CD. J Fluorine Chem. 2008;129:731–742. doi: 10.1016/j.jfluchem.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karukurichi KR, De la Salud-Bea R, Jahng WJ, Berkowitz DB. J Am Chem Soc. 2007;129:258–259. doi: 10.1021/ja067240k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Berkowitz DB, de la Salud-Bea R, Jahng WJ. Org Lett. 2004;6:1821–1824. doi: 10.1021/ol049422u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Berkowitz DB, Pedersen ML, Jahng WJ. Tet Lett. 1996;37:4309–4312. doi: 10.1016/0040-4039(96)00832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Berkowitz DB, Jahng WJ, Pedersen ML. Bioorg Med Chem Lett. 1996;6:2151–2156. doi: 10.1016/0960-894X(96)00366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pedersen ML, Berkowitz DB. J Org Chem. 1993;58:6966–75. [Google Scholar]
- 10.Hudrlik PF, Kulkarni AK. J Am Chem Soc. 1981;103:6251–6253. [Google Scholar]
- 11.(a) Yoshida Ji, Maekawa T, Morita Y, Isoe S. J Org Chem. 1992;57:1321–1322. [Google Scholar]; (b) Croudace MC, Schore NE. J Org Chem. 1981;46:5357–5363. [Google Scholar]; (c) Hudrlik PF, Hudrlik AM, Rona RJ, Misra RN, Withers GP. J Am Chem Soc. 1977;99:1993–1996. [Google Scholar]
- 12.Chauret DC, Chong JM, Ye Q. Tetrahedron: Asymmetry. 1999;10:3601–3614. [Google Scholar]
- 13.(a) Rieke RD. Aldrichimica Acta. 2000;33:52–60. [Google Scholar]; (b) Lee Js, Velarde-Ortiz R, Guijarro A, Wurst JR, Rieke RD. J Org Chem. 2000;65:5428–5430. doi: 10.1021/jo000413i. [DOI] [PubMed] [Google Scholar]
- 14.Peterson DJ. J Org Chem. 1967;33:780–784. [Google Scholar]
- 15.(a) Yang X, Liu P, Houk KN, Birman VB. Angew Chem Int Ed. 2012;51:9638–42. doi: 10.1002/anie.201203327. [DOI] [PubMed] [Google Scholar]; (b) Frenking G, Koehler KF, Reetz MT. Tetrahedron. 1993;49:3983–94. [Google Scholar]; (c) Cherest M, Felkin H, Prudent N. Tet Lett. 1968;18:2199–2204. [Google Scholar]; (d) Anh NT. Top Curr Chem. 1980;88:145–162. [Google Scholar]; (e) Anh NTE, Odile Nouv J Chim. 1977;1:61–70. [Google Scholar]
- 16.Cieplak AS. J Am Chem Soc. 1981;103:4540–4552. [Google Scholar]
- 17.Cee VJ, Cramer CJ, Evans DA. J Am Chem Soc. 2006;128:2920–2930. doi: 10.1021/ja0555670. [DOI] [PubMed] [Google Scholar]
- 18.Buergi HB, Dunitz JD. Acc Chem Res. 1983;16:153–61. [Google Scholar]
- 19.Cornforth JW, Cornforth RH, Mathew KK. J Chem Soc. 1959:112–127. [Google Scholar]
- 20.(a) Raffier L, Stanton GR, Walsh PJ. Org Lett. 2013;15:6174–6177. doi: 10.1021/ol4030259. [DOI] [PubMed] [Google Scholar]; (b) Stanton GR, Koz G, Walsh PJ. J Am Chem Soc. 2011;133:7969–7976. doi: 10.1021/ja201629d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stanton GR, Johnson CN, Walsh PJ. J Am Chem Soc. 2010;132:4399–4408. doi: 10.1021/ja910717p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lodge EP, Heathcock CH. J Am Chem Soc. 1987;109:3353–61. [Google Scholar]
- 22.(a) Mukai K, Urabe D, Kasuya S, Aoki N, Inoue M. Angew Chem Int Ed. 2013;52:5300–4. doi: 10.1002/anie.201302067. [DOI] [PubMed] [Google Scholar]; (b) Earnshaw C, Wallis CJ, Warren S. J Chem Soc. 1979:3099–3106. [Google Scholar]
- 23.Surprenant S, Chan WY, Berthelette C. Org Lett. 2003;5:4851–4854. doi: 10.1021/ol035918k. [DOI] [PubMed] [Google Scholar]
- 24.(a) Hamlin TA, Kelly CB, Cywar RM, Leadbeater NE. J Org Chem. 2014;79:1145–55. doi: 10.1021/jo402577n. [DOI] [PubMed] [Google Scholar]; (b) Barbero An, Blanco Y, García C, Pulido FJ. Synthesis. 2000;9:1223–1228. [Google Scholar]; (c) Welch JT, Lin J. Tet Lett. 1998;39:9613–9616. [Google Scholar]; (d) Welch JT, Lin J. Tetrahedron. 1996;52:291–304. [Google Scholar]
- 25.(a) Seto M, Morihira K, Katagiri S, Furkukawa T, Horiguchi Y, Kuwajima I. Chem Lett. 1993;22:133–136. [Google Scholar]; (b) Furkukawa T, Seto M, Horiguchi Y, Kuwajima I. Chem Lett. 1993;22:1279–1282. [Google Scholar]; (c) Magnus P, Roy G. J C S, Chem Commun. 1979:822–823. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.