

Abstract
Class II viral fusion proteins are present on the envelope of flaviviruses and togaviruses, viruses that often cause tropical and subtropical diseases. These proteins use a second membrane protein as a molecular chaperone to assist their folding and to ensure proper function during viral assembly, maturation, and infection. Recent progress in structural studies of dengue viruses has revealed how the chaperone pre-membrane (prM) protein guides viral maturation and how pH is sensed in both the maturation and infection processes. Drastic conformation changes and reorganization of these viral membrane proteins occur during the transition from their metastable to stable structural states in a unidirectional, entropy-driven process.
Keywords: cryo-electron microscopy, flavivirus, togavirus, bio-threat agent, enveloped viruses, structures
Viral fusion proteins
An enveloped virus has a lipid bilayer membrane that encloses its capsid with genetic materials. This lipid bilayer is derived from its progenitor host cell and is comparable in physical and chemical properties to the host cell membranes. In order for the virus to infect its host, the two membranes have to fuse, allowing contents of the virus and the cell to mix. This fusion process is mediated by viral fusion proteins and host receptor molecules. The physical aspects of this process have been studied by fluorescence imaging of model systems [1].
Viral fusion proteins are transmembrane glycoproteins decorating viral envelopes [2]. They mediate membrane fusion under a specific condition, either receptor binding or fusogenic pH. At this condition, a portion of the fusion protein pops up, exposing and inserting its fusion peptide into its target membrane [3]. Then it undergoes a conformational change to bring the target membrane to the proximity of the viral membrane, leading to fusion. Viral fusion proteins are divided into three classes, classes 1, 2, and 3, based on their structural and functional characteristics (Figure 1).
Figure 1.
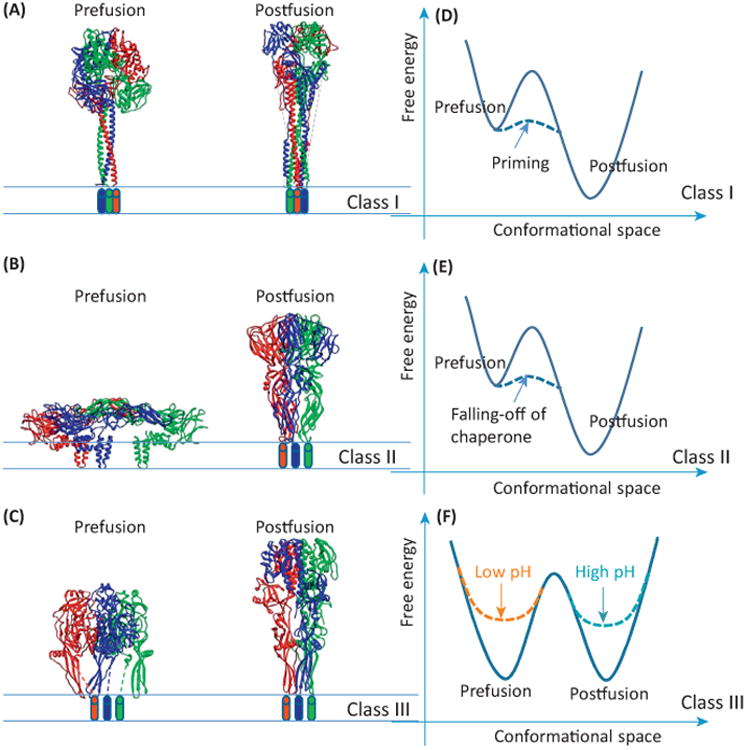
The three classes of viral fusion proteins. (A–C) Side views of the viral fusion proteins in their prefusion and postfusion conformations for (A) class I, (B) class II, and (C) class III. The atomic models are shown as ribbons and the three monomers of each model are colored differently. Cylinders represent transmembrane helices whose structures are unknown. (D–F) Free energy landscapes of the three classes of viral fusion proteins for (D) class I, (E) class II, and (F) class III. Unbroken lines: landscapes of original proteins or assemblies; broken lines: landscapes of proteins after specific event as marked. Coordinates in these three panels are for illustrative purpose only and are arbitrary.
All currently known class II viral fusion proteins belong to viruses in the families of Flaviviridae and Togaviridae and many of these viruses cause tropical diseases. These two families of viruses share considerable similarities and their viral fusion proteins closely resemble each other. During fusion, class II fusion proteins are all organized as trimers [4,5]. They all depend on a second membrane protein to fold [6], mature, and to cause fusion [7]. Therefore, this second protein acts as a chaperone for its partner, the viral fusion protein. Although the chaperone proteins in different viruses differ in size and shape, and in the way they are organized with their viral fusion proteins, these proteins perform similar roles and functions.
Dengue virus (DENV) is a prevalent mosquito-borne flavivirus that is endemic across most tropical and subtropical regions. DENV undergoes a maturation process that includes the formation of E (envelope) dimers [8], the cleavage of the prM chaperone, and the shedding of pr [9]. Recently, atomic models of the mature virion and an engineered combination protein that contains pr and the ectodomains of E and M were reported by cryo-electron microscopy (cryo-EM) [7] and X-ray crystallography [10], respectively. Cryo-EM structures of the spikey immature viruses were also published together with a pseudo-atomic model by fitting the X-ray structure into the cryo-EM envelope [11]. In this review, we will describe current understanding of class II fusion proteins, their chaperone proteins, and the interaction between the two during viral maturation and infection.
Metastability of viral fusion proteins
Proteins exist in biological systems as three-dimensional (3D) entities in order to perform their specific functions. Synthesized as a linear polymer, a protein has to find its way into its properly folded 3D structure in order to be functional. The process of folding can be thought of as exploring energy landscapes for low energy wells; at the bottom of this landscape lies its native, stable structure (the folding funnel model) [12]. However, this simplistic funnel model is not suitable for fusion proteins.
To enable fusion, a fusion protein has to have two stable, naturally-occurring structural states, a prefusion and a postfusion state. The protein first adopts the prefusion structural state, which is at a local energy minimum. Because this local minimum is higher in energy on the landscape than the bottom of the folding funnel, the prefusion structure is a spring-loaded structure. This model deviates from the funnel model because some native structural states (e.g., the prefusion state) of a fusion protein are not at the global minimum at the bottom of the folding funnel. Thus, during folding, a fusion protein has to find its way to arrest at a higher energy level other than going down directly to the bottom of the funnel. It only reaches the bottom of the funnel upon completion of fusion.
Because the prefusion state is not at the global energy minimum, this state is a less stable structure than the corresponding postfusion state. This form is locally stabilized by the energy barrier that is around the local minimum. At physiological conditions, this barrier is big enough to prevent a viral fusion protein in the prefusion form from getting into the lower energy, postfusion state. To improve infection efficiency, many viral fusion proteins undergo a ‘priming’ process that lowers the energy barrier.
Of the three classes of fusion proteins, each uses a different strategy to control the transition between the prefusion and postfusion states (Figure 1 and Table 1). Class I viral fusion proteins employ a ‘hidden knife’ strategy (Figure 1A,D). A protein of this class is first expressed in a continuous polypeptide chain. The chain then undergoes proteolytical cleavage to produce two new proteins: the functional fusion protein and a second protein. The cleavage occurs at the fusogenic helix, which becomes the N terminus of the functional fusion protein. During fusion, the first new protein either sheds off or gives way to the second protein, whose N-terminal helix inserts into its target host membrane and folds back to the second protein itself, forming a six-helix bundle. In this way, the target membrane is brought into proximity with the viral membrane, leading to membrane fusion [2]. Class III viral fusion proteins employ a ‘reversible form’ strategy (Figure 1C,F). A protein of this class can change its form in response to pH. At low pH, it changes to its postfusion form; at neutral pH, it changes to its prefusion form [13,14]. Because this kind of protein has two stable states, which are reversible between each other, class III fusion proteins can be described more closely as a tunable bistable system. The strategy employed by class II fusion proteins is described in detail below.
Table 1. Strategies employed by the three classes of fusion proteins.
| Folding and arresting at prefusion state | TGN evasion | Priming | Fusion trigger | |
|---|---|---|---|---|
| Class I | Fusion protein expressed in a continuous chain | Non-fusogenic continuous fusion protein | Cleavage of fusion protein | pH trigger or receptor trigger |
| Class II | Coexpression and co-folding with chaperone | Part of the chaperone latches to the fusion protein and prevents fusion | Cleavage of chaperone at the holder loop | pH-triggered releasing of chaperone allows fusion protein to change into its fusogenic form |
| Class III | Protein exists in two forms | Acidic pH promotes non-fusogenic, postfusion form | Switching to prefusion form | pH trigger |
Maturation of DENV glycoprotein E, a class II fusion protein
Class II fusion proteins respond to the challenge of metastability by employing a second chaperone protein to assist in its function (Figure 1B,E). In flaviviruses like DENV, this second protein is prM [9]. Together with the viral fusion protein, E, these two membrane proteins are critical to the viral life cycle. They are expressed in a polyprotein that is cleaved to yield prM and E (among many other proteins). prM initially binds to E in the ‘spiky immature’ form (Figure 2A) [8,11] in the neutral pH of the endoplasmic reticulum. They undergo a maturation process that includes the formation of E dimers in the low pH environment of the trans-Golgi network (TGN) [8] (to form the low pH immature, or ‘smooth immature’ virus) (Figure 2B), the cleavage of the pr portion from prM (to produce M), and the shedding of pr when the virus is finally released in the neutral pH in the extracellular space, yielding the ‘smooth mature’ virus (Figure 2C) [7]. This maturation process was recapitulated by a crystal structure of an engineered protein that contains pr, the N-terminal 20-amino acid segment of M (M1–20) and the ectodomain of E (the portion outside the membrane) [10], along with the low resolution cryo-EM structures of both the spiky and the smooth immature viruses (12.5 Å and 25 Å, respectively) [8,15,16]. The crystal structure of the engineered protein was then fit into the 25 Å resolution cryo-EM structure of the ‘smooth immature’ virus [8] to explain how pr stabilizes E dimer at the low pH encountered in TGN. The prefusion dimer of the ectodomain of E at neutral pH, which was thought to resemble that in the ‘smooth mature’ virus, was also determined crystallographically [16,17]. During infection, following endocytosis and exposure to low pH in the late endosome, these E dimers dissociate to form fusogenic trimers that mediate fusion with the endosomal membrane. The structure of the ectodomain of E as a trimer (after fusion) was captured in the crystal structure of the low pH postfusion form [4]. Unfortunately, the crystals used for these structural determinations [4,10,16,17] were obtained outside the context of the whole virion and only contained the ectodomain of E. The structure of the whole virion has only been determined to intermediate resolution [18,19].
Figure 2.
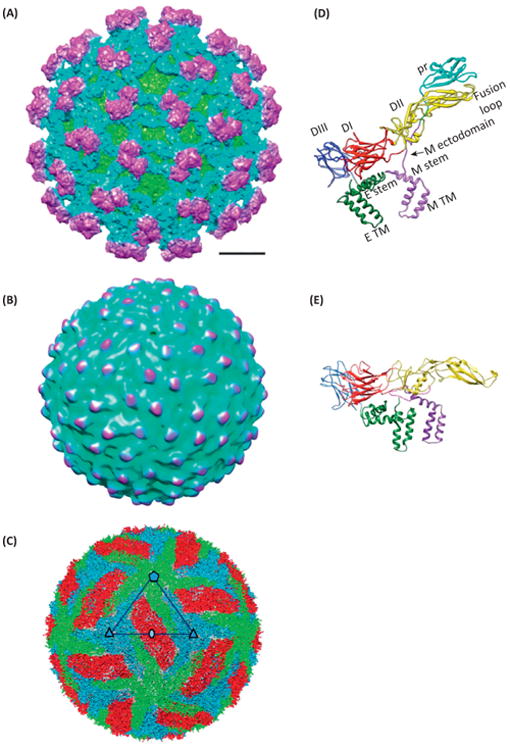
Structure of dengue virus (DENV) in different maturation states. (A–C) Cryo-electron microscopy (Cryo-EM) structure of DENV in its (A) spiky immature, (B) smooth immature, and (C) smooth mature states. Panels (A) and (B) are colored by radii of the particles. In panel (C), each of the three copies of envelope protein (E) and membrane protein (M) in one asymmetric unit is given a different color. (D) Atomic model of E and prM in the spiky immature state. DI, DII, and DIII: domains I, II, and III of E. TM: transmembrane domain. (E) Atomic model of E and M in the smooth mature state. The molecular ribbons in these two panels are colored by domains, namely DI of E in red, DII in yellow, DIII in blue, TM of E in green, pr in cyan, and M in purple. Panels (A) and (D) are adapted from [11], panels (C) and (E) from [7], with permissions from the publishers and authors. Panel (B) is rendered from Electron Microscopy Data Bank (EMDB) entry EMD-5006 [8].
Recently, in situ cryo-EM structures of both spiky immature (Figure 2D) and smooth mature (Figure 2E) DENV particles provided atomic descriptions of E–M interactions and how pH triggers these structural transitions. Zhang et al. solved the structure of dengue virus 2 (DENV2) in its mature form to near atomic resolution (3.5 Å) [7]. This study overcame the challenge posed by imperfection of the cryo-EM sample (in terms of both damaged particles and partially mature particles [20]) by employing a recently developed, multipath Monte Carlo simulated annealing based method, which presumably can eliminate ‘bad’ particles from the dataset [21]. This structure revealed that in a mature DENV, M works as a latch to hold the spring-loaded E protein from prematurely rising (Figure 2E). This latch has a pH sensor that releases the latch in the acidic pH of a late endosome. This latch is presumably loaded in the TGN when prM is proteolytically cut into pr and M and is fastened when in the physiological pH outside a cell. Kostyuchenko et al. described cryo-EM structures of dengue virus 1 (DENV1) for both its mature (at 4.5 Å) and immature (∼6 Å) forms [11]. Surface charges of the E proteins on the DENV1 and DENV2 structures showed that DENV2 has more positively charged residues, consistent with the distinct antigenicity across different types of DENV. Models of the prM, M and E are deduced by taking advantage of existing atomic structures (Figure 2D). Direct comparison of the models for both the immature and mature particles has permitted the determination of the start and end positions of the E and M proteins during virus maturation. The structures of the immature and mature virions confirms a dramatic conformational rearrangement of the surface proteins during maturation and shows the intermolecular interactions between the viral proteins that stabilize specific structural features necessary for various stages of the viral lifecycle. It suggests how pr could prevent dimeric interactions between E protein monomers, thus the reversible pH-sensitive structural changes of the immature virus [11].
The current model is that prM acts as a pull-string in response to low pH in the TGN [7,10]. The 6.5 Å structure of the spiky immature DENV defined the structure of the pull-string as an 84-Å extended loop lining along the rim of E [11]. Presumably, exposure to the low pH in the TGN induces folding of part of this extended loop onto the folded part of pr, resulting in the shortening of the pull-string [7]. Consequently, E is pulled down towards the viral envelope and becomes spring-loaded in the smooth immature particle (Figure 2B), where E becomes dimers [8]. The pr then keeps the newly formed dimers of E in its mature position by binding to both copies (Figure 3B) [8] and it prevents premature rising of E at the acidic pH of TGN [22]. It is also suggested that the stem region of M (Figure 2D) participates in keeping this E dimer in its mature position as well [23]. In this scenario, the pop-up conformation of E is presumably in a lower energy state, which is likely to be attributable to higher entropy associated with more statistically accessible microscopic states when more surface areas are exposed to water molecules and when the ectodomains are in a more flexible configuration. At the same time, M finds its position in the mature DENV. However, in the absence of an atomic resolution structural description of the smooth immature particle, how the pull-string is folded onto the structured part of pr and how this conformational change can be triggered by pH remains unknown.
Figure 3.
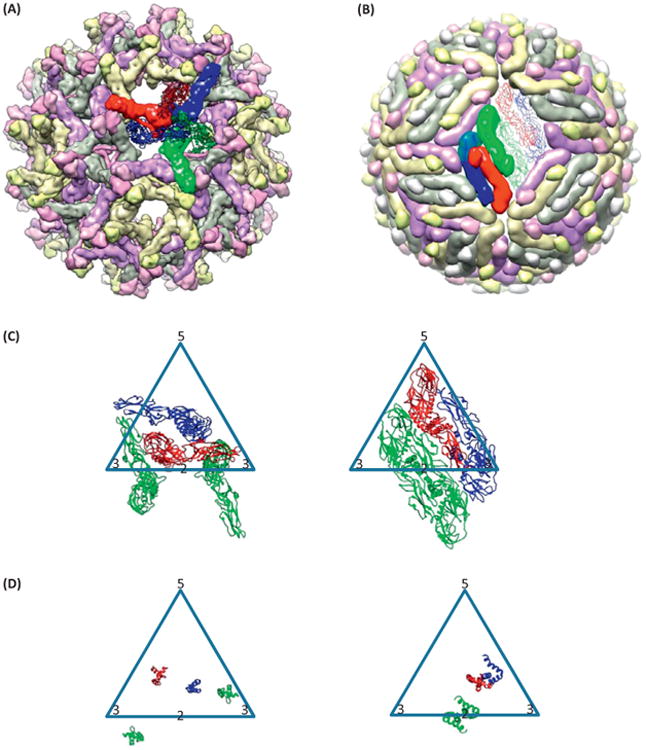
Maturation of dengue virus (DENV). (A,B) Multiscale model for the (A) spiky immature [Protein Data Bank (PDB) 4B03] and (B) smooth immature (PDB 3C6R) states of DENV. (C,D) The transition of conformation of (C) envelope protein (E) and (D) membrane protein (M) between spiky immature (left) and smooth immature (right) states in one icosahedral triangle formed by a fivefold axis (‘5’) and two threefold axes (‘3’), in which the twofold axis (‘2’) lies on an edge of this triangle. Purple, yellow, and gray colors of the subunits in (A) and (B) denote symmetry-related subunits. Red, blue, and green colors denote the three copies of E and M in one representative asymmetric unit [with the green copy doubled in (C) and (D) to show the dimer].
The choreography of the M and E subunits during transition from the spiky immature to the smooth immature state remains a subject of controversy. Zhang et al. described how E subunits would rearrange during this transition [16]. Their model naturally uses the proteins related by the fivefold symmetry axis to make dimers in the smooth immature state (Figure 3C,D). The transmembrane domains of both E and M move laterally across up to about 60 Å (Figure 3F–G). By contrast, Kostyuchenko et al. suggested a different manner of grouping [11]. The advantage of the first model is that there is no dramatic movement to form the dimer at the icosahedral fivefold axis. The advantage of the second model is that the transmembrane domains undergo minimal lateral movement during this transition. However, in both models, no pathway has been proposed for the movement of proteins. Mechanistic models for such movements must address possible steric clashes between E sub-units during transition, and the observation that the regions of the virion mature in patches non-concertedly might offer some clues [20].
Maturation of the class II fusion proteins in alphaviruses
Maturation of an alphavirus highly resembles its counterpart in flaviviruses. In alphaviruses, the fusion protein E1 and its chaperone p62 are coexpressed in a polyprotein and fold together [24]. It is thought that p62 assists the folding of E1, which cannot fold without the help of p62 [24]. E1 and p62 form a complex, which in turn forms trimeric spikes. Immature complexes go to the TGN, where p62 is cleaved by furin to yield E2 and E3 [25], and the complex matures. E3 may or may not dissociate from E2 after maturation [25]. During infection, E2 is thought to fall off from E1 before fusion can be induced [26]. This notion is supported by the trimeric structure of the postfusion form of E1 [5] and the fact that E2 partially falls off from E1 at acidic pH [26]. The maturation process of class II fusion proteins in the Flaviviridae and Togaviridae virus families both require a second membrane protein and cleavage by furin. However, one important difference between these two families is the site of budding. In alphaviruses, a new virion buds from the plasma membrane; by contrast, a new flavivirus virion buds into the endoplasmic reticulum [27].
The study of Chikungunya virus (CHIKV) glycoproteins has provided much insight into the maturation process of an alphavirus [25]. However, how the viral fusion proteins of alphaviruses tolerate the low pH of the TGN after the furin cleavage, without fusion with the TGN membrane, remains unknown; furin cleavage occurs in the TGN. We provide three possible answers to this question: first, the full separation of E2 from E1 is a kinetics-driven step, thus the E1–E2 dimer does not have enough time after the cleavage to fully separate, rendering free E1, which is necessary for fusion. Second, the acidity of the TGN may not be enough for the E2 to fully separate from E1, which is a critical step to allow fusion to occur. The third, and most plausible explanation, is that there is a histidine on the E3 side of the furin site. This histidine may form a salt bridge with any of the neighboring acidic residues (e.g., Asp4, Asp59, Asp60) on E2, resembling the Asp63/65–His244 bridge between pr and E in DENV [10]. Such a salt bridge would maintain the tethering function of E3 as if there is a linker, at the low pH in TGN, whereas such linkage would be abolished upon neutral pH upon exiting its progenitor host. If this is true, then the maturing mechanism in alphaviruses and that in flaviviruses would have a substantial resemblance. The difficulty of this explanation is that some alphaviruses do not shed E3 upon exit from their hosts unlike flaviviruses, where all the prs leave the viruses upon exit from their hosts.
A common schema for class II viral fusion proteins: chaperones
Given the close resemblance between viruses of the family Flaviviridae and those of the family Togaviridae in the organization and interaction of their membrane proteins, their maturation processes, and triggering mechanisms, we propose here the chaperone theory for class II viral fusion proteins. In addition, the term ‘viral fusion protein chaperone protein’ (or, in short, chaperone fusion protein) is proposed for the auxiliary membrane proteins, which accompany viral fusion proteins, such as DENV prM and CHIKV p62, assisting the corresponding viral fusion protein in its folding, maturation, and fusion. We theorize that a chaperone fusion protein is needed by every class II viral fusion protein.
Maturation is a unidirectional and entropy-driven process
Maturation of DENV can be viewed as the streamlined process performed by a molecular machine. The most salient aspect of such a process is its unidirectionality (i.e., free energy change, ΔG, less than zero), because a virion cannot be permitted to go back to the immature state for productive virus maturation. Therefore, each step of this process needs to be carried out in an orderly fashion when the virus goes through the secretion pathway from the endoplasmic reticulum, through the Golgi network, to the outside of the cell. In this pathway, the virus cycles through neutral pH, acidic pH, and neutral pH; a process in which the net enthalpy change is probably close to zero. (Not counting the enthalpy change due to the cleavage of the chaperone protein.) According to the laws of thermodynamics, the free energy change (ΔG = ΔH − T ΔS) must be less than zero for a spontaneous process. Because the change in enthalpy (ΔH) is close to zero, the maturation process of class II viral fusion protein has to be ‘entropy-driven’, i.e., ΔS>0, to ensure unidirectionality. During DENV maturation, there are a few entropy-driven steps, big and small in entropy difference, that provide the unidirectionality of the process.
First, the cleavage of the prM protein into pr and M gives rise to a huge entropy increase. The loose ends of both new proteins have much larger number of microscopic states than their precursor. In a sense, after the cut, new proteins can never find their way back to the precursor state. Imagine that a spiky immature dengue virion goes from neutral pH to low pH and back to neutral pH again. The second neutral pH would otherwise presumably, bring this virion back to the immature state. The entropy increase generated by the cleavage of prM compensates the free energy increase generated by loading M into its pocket at low pH, preparing its position for latching E in physiological pH.
Second, there is another entropy-driven step that is not as obvious. When the C-terminal, unstructured part of pr folds onto the structured part of it at low pH, this movement pulls the former part of pr out from its interaction with E. This potentially increases the entropy of pr in the high pH environment. When the virion exits the cell, the part of pr that had interacted with E can never find its way back to its former interaction with E. Thus, pr leaves E even when encountering a high pH.
pH sensing during infection
In contrast to the entropy-driven process of maturation, the process of infection, which includes the release of the E protein from its horizontal position, is mostly enthalpy driven. The virus is first endocytosed into the cell via the mediation of the cellular receptor dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) [28,29]. During the acidification of a late endosome, the histidines in the first20 amino acids of M and the binding pocket for this loop become charged due to addition of protons. This change in charge renders the binding between M and E energetically unfavorable [7]. Therefore, M dissociates from E, releasing the latch, and allows E to rise up and to trigger fusion.
Antiviral targets in class II fusion proteins
The characteristic features of viral fusion proteins can be their Achilles' heels. A class I viral fusion protein triggers membrane fusion by forming its distinctive six-helix bundle. Three helices with fusion peptides collapse onto another three helices of the same protein to form this bundle [2]. Peptides, such as enfuvirtide [30], can preemptively bind to the latter three helices and block the former three helices from binding to them. These peptides are found to be antivirals for infections by viruses displaying class I viral fusion proteins.
Class II viral fusion proteins, nevertheless, depend heavily on their chaperone fusion proteins for proper function. Thus, the interaction between these two proteins is a potential target for drugs that aim to cure viral infections with viruses displaying class II viral fusion proteins. DENV would provide a perfect case for the design of such drugs because the small chaperone fusion protein M interacts with the main fusion protein E by a very small interface [7]. For alphaviruses, such drug design would be unfeasible because the interaction between E1 and the massive E2 is too extensive [25,26] for a small molecule to block.
To design such a drug, one needs to put an analog in any of the three pockets of E that host M [7], preferably pocket 2, which is the most critical for viral function [7]. With the analog present, the interaction between M and E is disrupted and the viral lifecycle is blocked at two points. First, the immature virion cannot mature because the E–M interaction is necessary for maturation. Second, the analog could attack mature virions, displacing M from E, thus releasing the latch and leading to premature, futile rising of E (Figure 4).
Figure 4.
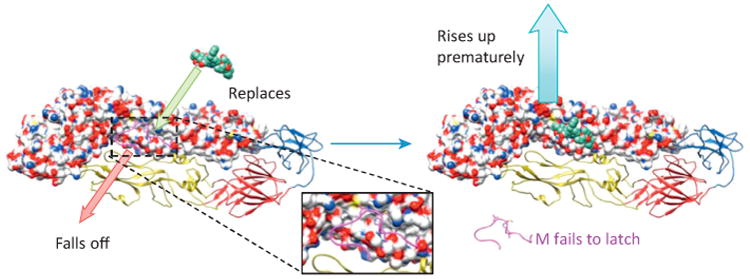
Illustration of how a small molecule analog may break dengue virus (DENV). A small molecule analog competes for the binding site in the envelope protein (E) and displaces the latch of the membrane protein (M). Subsequently, the release of the latch causes premature rising of E in DENV, thus disrupting infection.
Concluding remarks
In this review, we have discussed about the unique way that class II viral fusion proteins fulfill their task by employing a second membrane protein as a chaperone. This chaperone assists in the folding, maturation, and fusogenicity of the corresponding viral fusion protein. We described the characteristics of the chaperone in flaviviruses and alphaviruses. Given its characteristic and critical roles, a chaperone fusion protein and its interaction with its viral fusion protein are potential targets for novel antiviral drugs (Box 1). Recent insights about the interaction between class II fusion proteins and their chaperone fusion proteins will put us onto a better vantage point in our longstanding battle against the large group of viral pathogens from the Flaviviridae and Togaviridae virus families.
Box 1. Future directions.
To improve the resolution of the structures of both spiky and smooth immature dengue virions. This will allow better understanding of the ‘pull-string’ and thus the mechanism by which E gets to its position in the mature virion.
To discover small molecule analogs of M that bind to M's pocket in E. Such molecules will block the latching interaction between M and E, disrupting the virus, and serve as potential antidengue drugs.
Acknowledgments
We acknowledge grant support from the National Institute of Health (AI046420 and GM071940 to Z.H.Z.).
References
- 1.Lei G, MacDonald RC. Lipid bilayer vesicle fusion: intermediates captured by high-speed microfluorescence spectroscopy. Biophys J. 2003;85:1585–1599. doi: 10.1016/S0006-3495(03)74590-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gibbons DL, et al. Visualization of the target-membrane-inserted fusion protein of Semliki Forest virus by combined electron microscopy and crystallography. Cell. 2003;114:573–583. doi: 10.1016/s0092-8674(03)00683-4. [DOI] [PubMed] [Google Scholar]
- 4.Modis Y, et al. Structure of the dengue virus envelope protein after membrane fusion. Nature. 2004;427:313–319. doi: 10.1038/nature02165. [DOI] [PubMed] [Google Scholar]
- 5.Gibbons DL, et al. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 6.Vaney MC, Rey FA. Class II enveloped viruses. Cell Microbiol. 2011;13:1451–1459. doi: 10.1111/j.1462-5822.2011.01653.x. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, et al. Cryo-EM structure of the mature dengue virus at 3.5-Å resolution. Nat Struct Mol Biol. 2013;20:105–110. doi: 10.1038/nsmb.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu IM, et al. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science. 2008;319:1834–1837. doi: 10.1126/science.1153264. [DOI] [PubMed] [Google Scholar]
- 9.Mukhopadhyay S, et al. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 10.Li L, et al. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science. 2008;319:1830–1834. doi: 10.1126/science.1153263. [DOI] [PubMed] [Google Scholar]
- 11.Kostyuchenko VA, et al. Immature and mature dengue serotype 1 virus structures provide insight into the maturation process. J Virol. 2013;87:7700–7707. doi: 10.1128/JVI.00197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karplus M. Behind the folding funnel diagram. Nat Chem Biol. 2011;7:401–404. doi: 10.1038/nchembio.565. [DOI] [PubMed] [Google Scholar]
- 13.Roche S, Gaudin Y. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology. 2002;297:128–135. doi: 10.1006/viro.2002.1429. [DOI] [PubMed] [Google Scholar]
- 14.Connolly SA, et al. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol. 2011;9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, et al. Structures of immature flavivirus particles. EMBO J. 2003;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, et al. Conformational changes of the flavivirus E glycoprotein. Structure. 2004;12:1607–1618. doi: 10.1016/j.str.2004.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Modis Y, et al. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhn RJ, et al. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108:717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W, et al. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plevka P, et al. Maturation of flaviviruses starts from one or more icosahedrally independent nucleation centres. EMBO Rep. 2011;12:602–606. doi: 10.1038/embor.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, et al. Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using a multi-path simulated annealing optimization algorithm. J Struct Biol. 2007;160:11–27. doi: 10.1016/j.jsb.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu IM, et al. Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J Virol. 2009;83:12101–12107. doi: 10.1128/JVI.01637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, et al. The stem region of premembrane protein plays an important role in the virus surface protein rearrangement during dengue maturation. J Biol Chem. 2012;287:40525–40534. doi: 10.1074/jbc.M112.384446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kielian M. Structural biology: an alphavirus puzzle solved. Nature. 2010;468:645–646. doi: 10.1038/468645a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Voss JE, et al. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature. 2010;468:709–712. doi: 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 26.Li L, et al. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468:705–708. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol. 2006;4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lozach PY, et al. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J Biol Chem. 2005;280:23698–23708. doi: 10.1074/jbc.M504337200. [DOI] [PubMed] [Google Scholar]
- 29.Pokidysheva E, et al. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell. 2006;124:485–493. doi: 10.1016/j.cell.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 30.Lalezari JP, et al. A phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy. AIDS. 2003;17:691–698. doi: 10.1097/00002030-200303280-00007. [DOI] [PubMed] [Google Scholar]