

Abstract
The packaging of eukaryotic genomes into chromatin has wide-ranging influences on all DNA-templated processes, from DNA repair to transcriptional regulation. The repeating subunit of chromatin is the nucleosome, which comprises 147 base pairs of DNA wrapped around an octamer of proteins. Positioning of nucleosomes relative to underlying DNA is a key factor in the regulation of gene transcription by chromatin, as DNA sequences between nucleosomes are more accessible to regulatory factors than are DNA sequences within nucleosomes. Here, I describe protocols for mapping nucleosome positions across the yeast genome.
Introduction
The yeast genome, like that of all eukaryotes, is packaged into a nucleoprotein complex known as chromatin. The repeating subunit of chromatin is the nucleosome, which consists of 147 base pairs of DNA wrapped around an octamer of basic histone proteins. The positioning of nucleosomes on underlying DNA has consequences for gene regulation – nucleosomal occlusion of protein binding sites is generally thought to inhibit protein binding. Nucleosomal positioning has also recently been shown to affect sequence evolution, with point mutations fixing at lower rates in linker DNA relative to nucleosomal DNA (1, 2). Though there are clearly DNA sequences that favor and disfavor nucleosomal incorporation (3–9), essentially any DNA sequence can wrap around the histone octamer to form a nucleosome. Furthermore, protein complexes such as Isw2 regulate gene expression by moving nucleosomes away from their thermodynamically-favored positions (10, 11). Nucleosome positioning affects signal processing during gene regulation – nucleosomal occlusion of key promoter sequences has been shown to change the regulatory logic of the β-interferon promoter from an OR to an AND gate for three regulatory inputs (12), and to separate signaling threshold from dynamic range at the PHO5 promoter in yeast (13). Thus, understanding where nucleosomes are located in the genome therefore has implications for a wide range of interesting and important aspects of genome biology.
A number of features distinguish nucleosomal DNA from the linker DNA that intervenes between adjacent nucleosomes. Most notably, nucleosomal DNA is relatively protected from cleavage by a variety of nucleases. The most common nuclease used for assaying nucleosome positions in micrococcal nuclease, which has little (but measureable) sequence preference on naked DNA, but has a dramatic preference for linker DNA over nucleosomal DNA. Thus, identification of DNA protected from micrococcal nuclease digestion has been a mainstay for mapping of nucleosome positions in vivo.
In the past few years protocols have been developed enabling the mapping of nucleosome positions across the entire genome (14–19). I will not describe mapping methods, such as chromatin immunoprecipitation with an anti-histone antibody (20), or formaldehyde-assisted isolation of regulatory elements (21), that do not have single-nucleosome resolution. The current state-of-the-art protocols for nucleosome mapping all rely on protection from micrococcal nuclease. Isolated DNA is characterized by tiling microarray or by “deep” sequencing, providing whole-genome, high-resolution data on the packaging state of the yeast genome. The protocols described were developed and validated in S. cerevisiae, but we have used them successfully with over 10 different Ascomycete species (not shown), so they can be applied broadly.
Isolation of mononucleosomal DNA
Solutions (make beforehand):
-
Buffer Z:
1 M Sorbitol
50 mM Tris-HCl pH 7.4
-
NP Buffer:
1 M Sorbitol
50 mM NaCl
10 mM Tris pH 7.4
5 mM MgCl2
1 mM CaCl2
-
Glycine:
2.5 M Glycine
-
Spermidine:
250 mM in water. ~500 μL aliquots can be stored at −20 C, and can be repeatedly freeze/thawed.
-
MNase:
We always obtain MNase from Worthington. MNase is resuspended at 20 units/μL in 10 mM Tris 7.4, and stored in ~50 μL aliquots @ −80 C – can be freeze/thawed several (at least 2–3) times, store in –20 C after first use of an aliquot.
-
Proteinase K:
20 μg/μL. ~500 μL aliquots can be stored at −20 C, and repeatedly freeze/thawed.
-
6X Orange G loading buffer:
60% glycerol
1.5 mg/mL Orange G (Sigma O-1625)
Day 0:
Inoculate a 2–5 ml culture of YPD (or any other media) with the cells of interest – typically S288C derivatives BY4741 (MATa) or BY4742 (MATalpha).
Day 1:
Late PM: Count # cells per mL, calculate inoculation (BY4741 has a generation time of ~90 min in YPD) to have proper OD the following AM.
Inoculate 444 mL YPD in a 2 L flask, grow shaking 200 rpm at 28 degrees. We would typically inoculate ~7 μL of saturated BY4741 into this 444 mL culture at 7 pm for a midlog OD the next day around 11 am.
We prefer to use shaking water bath incubators when possible, since water bath incubation is less susceptible to temperature variability than incubation in air incubators. We have found a ~2-fold difference in doubling time between different clamp positions in a typical shaking incubator using blown air for heating (cultures near the fan generally grow faster, and are likely warmer), while no such difference is seen between different positions in a shaking water bath.
Day 2:
Check to make sure all water baths are at the correct temp and have enough water in them.
Grow yeast culture to desired OD600 (usually ~0.8).
Add 24 mL formaldehyde (37% as purchased) – to a final of 2%. We have found little difference between 1% and 2% formaldehyde in nucleosome positioning results, but we find better yields from experiments with 2%. This may be related to the spheroplasting step, but whatever the reason, we find optimal nucleosomal yields with 2%. Also, we always use formaldehyde within a month of purchase.
Incubate 30 minutes, 28 C/shaking. In general, whatever the growth condition was before fixation, fix cells in those conditions.
Pour culture into a 1 Liter centrifuge bottle containing 24 mL 2.5 M glycine (to a final of ~125 mM) to react with remaining formaldehyde. If going straight to spheroplasting (ie no waiting for other cultures) the glycine can be omitted.
Spin (4000 rpm, SLA-3000 rotor, 5 min). Pour off supernatant and resuspend cell pellet in ~50 mls ddH2O in a 50 mL conical. Pellet cells again (table top Sorvall, 3700 rpm, 2 min).
Cells can now be left on ice as long as necessary (we have tested up to 3 hours, with no change in positioning data) for other cells to “catch up”, etc.
If necessary make Buffer Z.
Make zymolyase solution (10 mg/ml in Buffer Z). Zymolyase isn’t particularly soluble at this concentration, so shake well right before adding to yeast to evenly resuspend. We obtain zymolyase from Seigaku, distributed by Cape Cod Associates in the US. Zymolyase solution lasts roughly a week, so should be made up fresh more or less every experiment.
Resuspend each cell pellet (from 450 mls of cells) in 39 mL Buffer Z. Add 28 μl β-mercaptoethanol (14.3 M, final conc. 10 mM) to each conical. Cap conical, and vortex cells to resuspend – it’s important to get cell pellet fully resuspended. Add 1 mL Zymolyase (10 mg/mL). Incubate at 28 degrees, shaking, for 30–35 minutes.
Note that the spheroplasting step is somewhat dependent on growth conditions, and on the strain/species. For example, S. cerevisiae in stationary phase are harder to spheroplast than midlog yeast. Success in this step can be assessed directly by microscopy (spheroplasts will lyse in water, intact yeast will not), but will also be readily apparent when nucleosomal DNA is isolated – unspheroplasted cells will result in a band of undigested large fragments of genomic DNA. If a small fraction of cells are not spheroplasted, this does not seem to be a problem beyond lost yield, as mononucleosomal DNA is gel-purified away from this genomic contamination in any case. But we prefer to have >90% spheroplasted cells for experiments we care about (ie non-troubleshooting).
Meanwhile, add sensitive components to NP buffer: to 5 mLs NP buffer add 10 μl 250 mM spermidine (final conc. 500 μM), 3.5 μl of β-mercaptoethanol diluted 1:10 in water (final conc. 1 mM), and 37.5 μl 10% NP-40 (final conc 0.075%).
During spheroplast spin (below), aliquot micrococcal nuclease (MNase) to 3 or 4 eppendorf tubes. Put a dot of MNase on the side of the tube (halfway down tube).
Suggested concentration range (1 flask of midlog cells, 444 total mls, OD600 = 0.8): 3, 6, and 10 μL of MNase. If doing four titration steps, 1, 1.8, 3.5, and 6 μL would be our starting MNase levels (since spheroplasts will be more dilute if dividing into four aliquots). Information from the first titration will guide further titrations.
After zymolyase digestion, pellet cells (4900 rpm in a Sorvall RC3 centrifuge, or ~7000 G 10 min, 4 C), or if absolutely necessary on tabletop Sorvall, 3700 rpm, 10 min. Aspirate supernatant – be very careful, as spheroplast pellets are fluffy and will suck into the aspirator. We generally try to get almost all the supernatant while losing a small amount of the spheroplasts (a small amount of supernatant will not affect the MNase reaction). Resuspend cells in ~ 2mL (600 μl for 3 titration steps + 200 μl) NP buffer by pipetting up and down several times. Make sure pellet is resuspended.
Add 600 μl of cells to each eppendorf already carrying micrococcal nuclease. Add the cells to the tubes directly over the spot of nuclease, and try to be even and quick between tubes. I generally start with the most dilute sample and use the same p1000 tip to add to each successive tube to minimize tip changing times.
As soon as all the cells have been added, start the timer, close the tubes, invert them once to mix, and incubate at 37 degrees (water bath) for 20 minutes.
About 5 minutes before the reaction ends, make 5X STOP buffer – to make 1 mL add 400 λ water, 500 λ 10% SDS, and 100 λ 0.5 M EDTA. STOP buffer needs to be made fresh every time since it precipitates during storage.
Stop the reaction after 20 minutes by adding 150 μL of (5X) STOP to each digestion.
Add 6 λ proteinase K (20 μg/μL). Invert tubes to mix. Incubate at 65 degrees over night to digest proteins and to reverse formaldehyde crosslinks.
Day 3:
Remove tubes from 65 degrees.
Phenol:chloroform extract once. We do this as below to maximize DNA yield, but any P:C extraction should work here.
Add ~1 vol (800 μl) Phenol:Chloroform:IAA, mix, centrifuge 5’ in Eppendorf “heavy” phase lock tubes.
Phase lock tubes – spin @ 16000×g for 30 sec. to bring down gel. Add phenol/aqueous mix, invert repeatedly to form homogeneous solution (do not vortex). Spin @ 16000×g for 5 minutes. Use P200 pipette to recover DNA-containing aqueous layer above phase lock gel.
Add 1/10 vol (75 μl) 3 M sodium acetate pH 5.5.
Add 1 vol isopropanol (fill to top) to precipitate DNA, freeze @ −20C for 30 min., spin at max speed in microcentrifuge for 10 minutes. Aspirate supernatant – pellet may be loose, wash with 500 μL ice-cold 70% ethanol, spin 5 more minutes. Aspirate – be esp. careful about pellet in this step. Air dry.
Resuspend pellet in 60 λ NEB buffer 2 (dilute commercial stock 1:10!), incubate pellet @ 37 C for 1 hr (or leave pellet at 4 C overnight).
Add 1 λ DNase-free RNase (10 μg/μl stock, Roche), incubate at 37 C, 1 hour
Run products on 1.8% agarose gel to find which part of titration worked the best. Loading ~ 1 μL is usually sufficient. Use 6x OrangeG loading buffer for all nucleosomal DNA, as the dye front in standard DNA loading buffer runs right on top of the mononucleosomal band in this percentage gel.
Take gel images – see Figure 1. Beware of the bright fuzzy band that runs with the Orange dye front – it’s not mononucleosomes!
Figure 1.
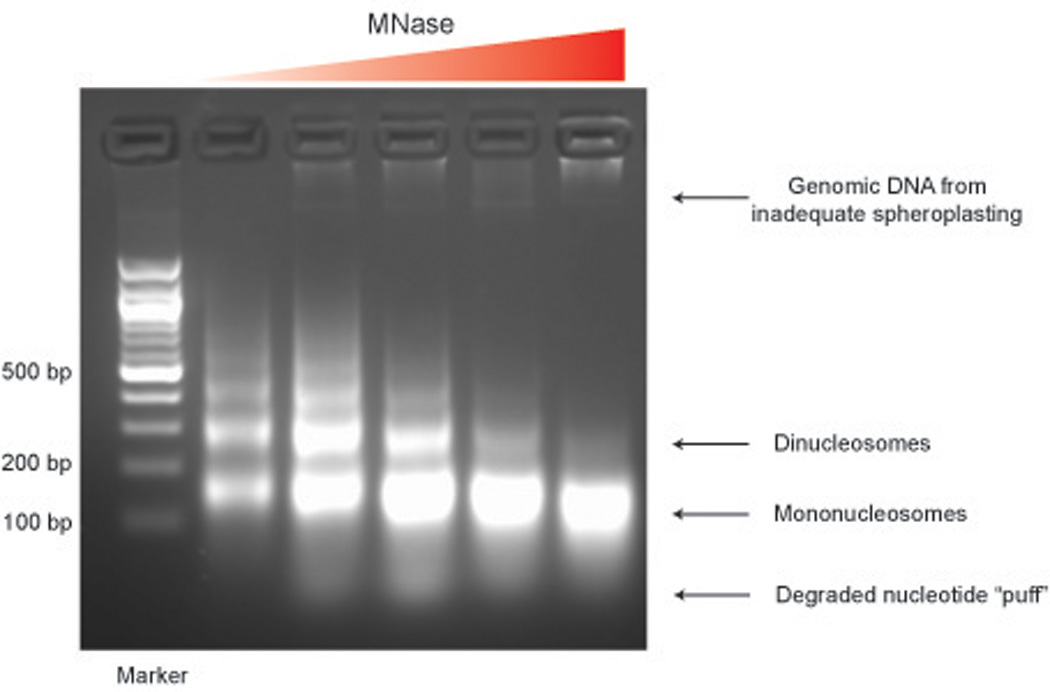
Take best titration (~80% mononucleosomal DNA with ~20% dinucleosome, and a ghost of trinucleosome – see below) and load entire fraction onto ~3 lanes of a fresh 1.8% gel. Using a brand new razor blade, gel purify mononucleosomal band away from dinucleosomes, and away from “puff” of degraded DNA/RNA (See Figure 1). As always with gel purification, trim band of excess agarose, but also attempt to minimize UV exposure of DNA.
If there’s a lot of degraded DNA (running <100 bp), first purify the DNA with a Qiagen cleanup column to remove the small pieces. Otherwise, the gel may not run correctly (bands will not separate). Resuspend entire samples (combined titrations) into 45 ul of elution buffer – will fit into two lanes.
Use BioRad Freeze-N-Squeeze tubes to purify DNA from gel. We use this method as typical gel purification kits get abysmal yields from DNA in this size range. Mince band, freeze for 5 min @ −20 C, spin @ 13000×g for 3 min. We typically add another 50–100 μL of TE on top of the squished gel in the tube and spin again to increase yield, but this is not necessary.
To clean up DNA, add 1 volume Phenol:Chloroform:IAA, vortex, remove the aqueous phase to a new tube. Repeat (P:C extract twice).
Add 1 μl of glycogen (20 mg/ml, from Mytilus edulis, Sigma G1767) to assist in precipitation. Add 1/10 volume of 3M Na-Acetate pH 5.5, 2.5–3 volumes 100% EtOH. Incubate at –20 C for 20–30 min., spin @ max speed for 10 min., gently aspirate off supernatant, being careful not to disturb pellet.
Wash with 500 μl of ice-cold 70% EtOH to remove salt.
Spin max speed for 5–10 minute. Aspirate supernatant, and air dry pellet.
Resuspend pellet in 25 μl of H2O. Pellet may not always completely dissolve – possibly due to glycogen? In that case, brief spin and take supernatant above slurry.
Run 1 μl of mononucleosome prep on a 1.8% gel after cleanup to confirm the DNA is there, clean, etc.
This DNA can now be labeled for microarray analysis, or ligated to linkers to generate a deep sequencing library.
Variation in titration level used for nucleosome purification
For years we have used nucleosomal DNA from a titration step with ~10–20% dinucleosome. The rationale here is that overdigestion of mononucleosomal DNA leads to a slow, progressive trimming of nucleosomes, so the digestion level of a titration step with only mononucleosomal DNA is hard to judge, and hard to reproduce. Using deep sequencing, we have more recently characterized the nucleosomal populations from over and underdigested chromatin as well – see Figure 2.
Figure 2.

Nucleosome maps from the three titration steps broadly agree, with differences between them largely, though not entirely, being changes in occupancy rather than positions of nucleosomes. For example, the first and last nucleosomes in coding regions are often relatively-highly occupied in the underdigested map, presumably thanks to increased accessibility to MNase of exposed DNA adjacent to nucleosome-depleted promoters. Moreover, so-called “nucleosome-free regions”, especially the longer ones characteristic of very highly-expressed genes, tend to be partially occupied in underdigested chromatin, suggesting that these regions are in fact occupied by loosely-bound histones that are readily digested away.
What does this mean for mapping studies? Most importantly, when comparing two samples it is crucial to record the gels from which the nucleosomal DNA was isolated. Artifacts are a potential pitfall in data analysis – an accidental difference in digestion levels between wild-type and some mutant would give the false impression that promoter nucleosome occupancy was affected by the mutation of interest. Another way to determine whether chromatin has been digested to similar extents after sequencing is to average nucleosomal data for all genes by the transcriptional start site (Figure 2B), since in this view under- and over-digested chromatin have characteristic patterns that differ somewhat from our preferred titration level.
Finally, it is worth noting that this variation in occupancy at different digestion levels could potentially be utilized as a structural probe for chromatin in vivo, and some experimenters may find mapping over a range of digestion levels a valuable tool in their studies.
Labeling of mononucleosomal DNA for tiling microarray analysis
Note that this protocol describes labeling and hybridization on “home-made” tiling microarrays. For commercial microarrays, labeling and hybridization should be carried out according to manufacturer’s protocols. This is particularly true for Affymetrix microarrays. On the other hand, we have used this protocol quite successfully with Nimblegen and Agilent microarrays, with differences only occurring after cleanup of the labeled material. After cleanup, we add the blocking and hybridization solutions recommended by the manufacturer, then carry out hybridization as described in their protocols.
Solutions (make beforehand):
-
2.5X random primer mix (can be made, or can use the buffer from Invitrogen’s “BioPrime Klenow labeling kit):
125 mM Tris pH 6.8
12.5 mM MgCl2
25 mM β-mercaptoethanol
750 μg/mL random octamers
-
10X dNTP mix:
1.2 mM dATP, dGTP, dTTP
0.6 mM dCTP
10 mM Tris pH 8.0, 1 mM EDTA
-
tRNA:
5 mg/mL in water
Store aliquots at −80 C.
-
PolyA RNA:
5 mg/mL in water
Store aliquots at −80 C
Protocol:
Add 2–3 μg of nucleosomal DNA to a 0.5 mL eppendorf, and bring the volume up to 21 μL. In another tube, do the same with 2–3 μg of sheared genomic DNA.
Add 20 μL 2.5X random primer mix.
Boil 5 min (we use a heat block at 95 C with water in the tube holders), place on ice.
After 5 min on ice, add:
5 μL dNTP mix
3 μL Cy3-dCTP (to nucleosomes) or Cy5-dCTP (to gDNA) – the colors can be swapped, but we typically run experiments in this direction.
Add 1 μL High concentration Klenow
Place tubes at 37 C for one hour
Add another 1 μL of Klenow
Incubate another hour at 37 C
Stop reactions with 5 μL 0.5 M EDTA
Mix the two colors, add 400 μL TE (pH 8.0 or 7.4) to stopped reactions, and add to Microcon 30 filter.
Spin 10–11 min at 10,000 rpm in microcentrifuge, until ~30–50 μL remain, Solution should be darkly colored.
Add another 400 μL TE.
Add 100 μg yeast tRNA (Sigma)
Add 20 μg polyA RNA
Spin 12–13 minutes at 10,000 rpm, until volume is less than 40 μL
Recover labeled DNA by inverting Microcon filter in a fresh collection tube, and spinning 10,000 rpm for 1 minute
Measure volume of recovered DNA and transfer to a 0.5 mL eppendorf tube
Bring volume up to 40 μL with water
Add 8.5 uL 20X SSC
Add 1.5 uL 10% SDS (make sure not to add any more SDS than this – we typically touch the edge of the pipette tip against a clean plastic surface to wipe away any SDS stuck to the outside of the pipette tip).
Boil hybridization mix for two minutes (we use a heat block at 95 C with water in the wells to ensure good heat transfer).
Remove tubes and let them sit at room temperature (in the dark, either in foil or in a drawer) for 10–15 minutes
Give the tube a quick spin, apply to microarray, and hybridize for 12–16 hours at 65 C.
Wash microarray and scan.
Generation of nucleosomal DNA libraries for deep sequencing
Over the past few years, ultra-high throughput “deep” sequencing methods have been effectively used for the analysis of mononucleosomal samples. As of this writing, two major commercial machines have been used for deep sequencing – 454 (Roche) and Solexa (Illumina). We have experience with Solexa sequencing and so present a protocol for Solexa library construction, but use of other sequencing methodologies will simply require creating libraries per the instructions of the manufacturer. We use Solexa because provides several million reads 36 bases in length – 36 bases is long enough to uniquely identify the vast majority of sequences in the yeast genome, and given that there are ~60,000 nucleosomes in yeast, 3 million reads provides 50X coverage of each nucleosome, allowing occupancy changes to be assessed between conditions. Solexa machine upgrades allow paired-end reads, and longer reads, and neither of these requires a change in the basic protocol.
Start with DNA from MNase titration, after RNase treatment, but prior to gel purification.
Clean up MNase titration with Qiagen MinElute (elute in 50 μL EB).
Treat DNA with Alkaline Phosphatase (CIP – NEB M0290L) for 1 hour at 37C.
Gel purify mononucleosomal DNA on 1.8% agarose gel, as described above. Use BioRad Freeze-N-Squeeze tubes to purify DNA from gel. Mince band, freeze for 5 min @ −20 C, spin @ 13000×g for 3 min. We typically add another 50–100 μL of TE on top of the squished gel in the tube and spin again to increase yield, but this is not necessary.
Repair DNA ends using End It DNA End-Repair Kit (Epicentre Biotechnologies ER0720):
| DNA | 150 ng |
| END IT buffer | 5 μL |
| dNTP mix (2.5mM) | 5 μL |
| ATP (10mM) | 5 μL |
| END IT enzyme mix | 1 μL |
| Q/S to 50 μL |
Incubate at room temperature for 1 hour
Clean up reaction with Qiagen MinElute column, elute in 30 μL EB
Klenow exo- (Epicenter Biotechnologies KL06041K):
| DNA | 30 μL |
| Klenow buffer | 5 μL |
| dATP (10mM) | 1 μL |
| Water | 14 μL |
| Klenow exo- | 1 μL |
Incubate at room temperature for 45 minutes
Clean up reaction with Qiagen MinElute column, elute in 20 μL EB
Dry DNA down to about 10 μL in a Speed Vac, then bring to exactly 10 μL with water.
Ligate Illumina adapters to polished mononucleosomal DNA using Fast Link DNA Ligation Kit (Epicenter Biotechnologies LK0750H):
| DNA | 10 μL |
| Fast-Link Ligation Buffer | 1.5 μL |
| ATP (10mM) | 0.75 μL |
| Ligase | 1 μL |
| Genomic adapters | 2 μL |
Incubate at room temperature for 1 hour, then add:
| Water | 7.5 μL |
| Fast-Link Ligation Buffer | 1 μL |
| ATP (10mM) | 0.5 μL |
| Ligase | 1 μL |
Incubate at 16 C overnight.
Clean up reaction with Qiagen MinElute column, elute in 30 μL EB
Amplify library with Pfx polymerase (Invitrogen 11708–039):
| DNA | 30 μL |
| Pfx buffer | 10 μL |
| Illumina genomic primers (1.1 & 2.1) | 1 μL each |
| dNTPs (10mM) | 3 μL |
| MgSO4 (50mM) | 2 μL |
| Water | 53 μL |
| Pfx | 1 μL |
| Step number | temperature | Time |
| 1 | 94 | 2 min |
| 2 | 94 | 15 sec |
| 3 | 65 | 1 min |
| 4 | 68 | 30 sec |
| 5 | Go to step 2 | 18 times |
| 6 | 68 | 5 min |
| 7 | 4 | forever |
Gel-purify library on 1.5% agarose gel. Band should run ~250 base pairs. We sometimes see two bands here, and in our experience the smaller band corresponds to primer dimer, and if it is an issue it can be eliminated by gel-purifying DNA after the primer ligation step above, prior to PCR.
Freeze ‘N Squeeze purify mononucleosome band from gel as above.
Clone a small portion (~100 ng) of the library using TOPO cloning, and transform into BL21DE3 competent E. Coli.
The next day, isolate 20 colonies, and isolate plasmids by miniprep method of your choice. Send these 20 library inserts out for sequencing (or sequence by hand). Inserts should average ~120 bp for our typical MNase digestion level, should map to the yeast genome, and should contain no mitochondrial sequences. If we get more than 3 inserts which are simply primer dimers, we re-make the library from mononucleosomal DNA.
If library looks OK, submit 30 μL of 10 nM sample in EB to your Solexa operator.
References
- 1.Washietl S, Machne R, Goldman N. Trends Genet. 2008;24:583–587. doi: 10.1016/j.tig.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki S, Mello CC, Shimada A, Nakatani Y, Hashimoto S, Ogawa M, Matsushima K, Gu SG, Kasahara M, Ahsan B, Sasaki A, Saito T, Suzuki Y, Sugano S, Kohara Y, Takeda H, Fire A, Morishita S. Science. 2009;323:401–404. doi: 10.1126/science.1163183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ioshikhes I, Bolshoy A, Derenshteyn K, Borodovsky M, Trifonov EN. J Mol Biol. 1996;262:129–139. doi: 10.1006/jmbi.1996.0503. [DOI] [PubMed] [Google Scholar]
- 4.Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nat Genet. 2006;38:1210–1215. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- 5.Iyer V, Struhl K. Embo J. 1995;14:2570–2579. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Leproust EM, Hughes TR, Lieb JD, Widom J, Segal E. Nature. 2008 doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekinger EA, Moqtaderi Z, Struhl K. Mol Cell. 2005;18:735–748. doi: 10.1016/j.molcel.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Yuan GC, Liu JS. PLoS Comput Biol. 2008;4:e13. doi: 10.1371/journal.pcbi.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Nature. 2007;450:1031–1035. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- 11.Whitehouse I, Tsukiyama T. Nat Struct Mol Biol. 2006;13:633–640. doi: 10.1038/nsmb1111. [DOI] [PubMed] [Google Scholar]
- 12.Lomvardas S, Thanos D. Cell. 2002;110:261–271. doi: 10.1016/s0092-8674(02)00822-x. [DOI] [PubMed] [Google Scholar]
- 13.Lam FH, Steger DJ, O'Shea EK. Nature. 2008;453:246–250. doi: 10.1038/nature06867.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Nature. 2007;446:572–576. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- 15.Field Y, Kaplan N, Fondufe-Mittendorf Y, Moore IK, Sharon E, Lubling Y, Widom J, Segal E. PLoS Comput Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, Gilmour DS, Albert I, Pugh BF. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- 20.Bernstein BE, Liu CL, Humphrey EL, Perlstein EO, Schreiber SL. Genome Biol. 2004;5:R62. doi: 10.1186/gb-2004-5-9-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Nat Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]